Co-reporter:Weiwei Li, Xinliang Yu, and Xueye Wang
The Journal of Physical Chemistry C 2017 Volume 121(Issue 3) pp:
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.jpcc.6b10228
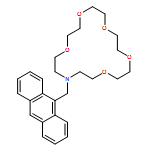
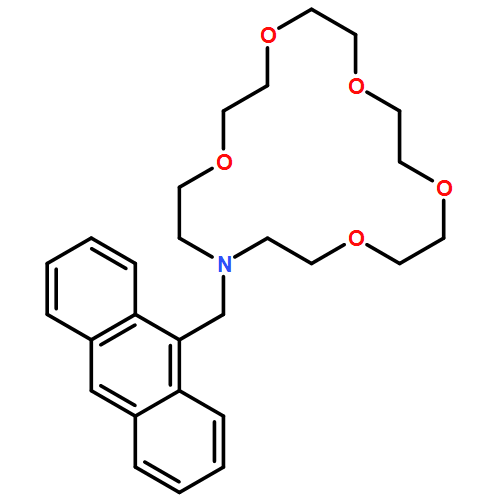
A novel molecular device (trans-azobenzene embedded N-(11-pyrenyl methyl)aza-21-crown-7) with double functional devices was designed on the basis of theoretical calculations. Pyrenyl methyl covalently bonded to aza-21-crown-7 at the nitrogen position interacting with a series of alkaline-earth metal cations (Mg2+, Ca2+, Sr2+, and Ba2+) was investigated. The fully optimized geometries and real frequency calculations were investigated using a computational strategy based on density functional theory at B3LYP/6-31G(d) level. Free ligand (L) and their metal cation complexes (L/M2+) were studied using mixed basis set (6-31G(d) for the atoms C, H, O, and N and LANL2DZ for alkaline-earth metal cations Mg2+, Ca2+, Sr2+, and Ba2+. The natural bond orbital analysis that is based on optimized geometric structures was used to explore the interaction of L/M2+ molecules. The absorption spectra of L and L/M2+, excitation energies, and absorption wavelength for their excited states were studied by time-dependent density functional theory with 6-31G(d) and LANL2DZ. A new type of molecular device is found, which has the selectivity to Ca2+ and the emission fluorescence of L/Ca2+ under the condition of illumination. This molecular device would serve as an allosteric switch and a fluorescence chemosensor.
Co-reporter:Na Liu, Xueye Wang, Yali Wan
Applied Surface Science 2015 Volume 328() pp:591-595
Publication Date(Web):15 February 2015
DOI:10.1016/j.apsusc.2014.12.079
Highlights
- •
The chemisorption of NO on Pt (1 1 1), Pt (1 0 0), Au/Pt (1 1 1) and Au/Pt (1 0 0) has been investigated.
- •
The adsorptive position of NO on metal surface has been discussed.
- •
The interaction between NO and metal surface is mainly NO 5σ/2π* and metal d-bands.
- •
Different compositions and facets can produce varied catalytic effects.
Co-reporter:Yali Wan;Na Liu
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 1) pp:25-30
Publication Date(Web):
DOI:10.1002/poc.3394
The phenyl acetylene and benzyl azide cycloaddition reaction in water in the presence of β-cyclodextrin (β-CD) as a phase transfer catalyst (PTC) can get a better yield in a shorter time. The interaction between β-CD and phenyl acetylene or benzyl azide plays an important role in this reaction. This paper studies the complexes of β-CD with phenyl acetylene and benzyl azide using density functional theory (DFT) method. In order to find out the orientations of guests in the cavity of β-CD, binding energy and deformation energy are investigated, and the calculated results are confirmed by 1H nuclear magnetic resonance (1HNMR). The data from single point energy indicate that the inclusion complexes can improve the solubilities of phenyl acetylene and benzyl azide in water. The 13C and 15N spectra show that the most obvious variation concentrates on C6 and C8 of phenyl acetylene and N15 of benzyl azide in complexes. Mulliken charge and frontier orbital are employed for revealing the charge distribution. The effect of β-CD is discussed in terms of the calculated parameters. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Na Liu, Xueye Wang, Yali Wan
Intermetallics 2015 Volume 66() pp:103-110
Publication Date(Web):November 2015
DOI:10.1016/j.intermet.2015.06.024
Co-reporter:Yali Wan;Na Liu
Journal of Molecular Modeling 2015 Volume 21( Issue 5) pp:
Publication Date(Web):2015 May
DOI:10.1007/s00894-015-2680-7
Phenacyl bromide as one starting material in multicomponent reactions (MCRs) with β-cyclodextrin (β-CD) as catalyst can get an excellent yield in short reaction times. The interaction of β-CD with phenacyl bromide plays an important role in this process. This paper studies the complex of β-CD with phenacyl bromide using density functional theory (DFT) method. Energy is investigated to find out the lowest energy of two possible complexation models. Hydrogen bonds are researched on the basis of natural bonding orbital (NBO) analysis. The relative position between phenacyl bromide and β-CD is confirmed by 1H nuclear magnetic resonance (1HNMR). The results of frontier molecular orbitals and charge distribution reveal that β-CD catalyst improves the reactivity and electrophilicity of phenacyl bromide, meanwhile, the carbonyl group of phenacyl bromide more easily gives a carbocationic intermediate in the presence of β-CD as catalyst. The reactivity of phenancyl bromide under β-CD as supramolecular catalysis is improved.
Co-reporter:Qin Wang;Ling Li
Journal of Physical Organic Chemistry 2014 Volume 27( Issue 7) pp:546-554
Publication Date(Web):
DOI:10.1002/poc.3300
Three derivatives of alkyl anthracene covalently bonded to aza-18-crown-6 at the nitrogen position, anthracene(CH2)n, (n = 1–3) which act as an on–off fluorogenic photoswitch have been theoretically studied using a computational strategy based on density functional theory at B3LYP/6-31 + G(d,p) method. The fully optimized geometries have been performed with real frequencies which indicate the minima states. The binding energies, enthalpies and Gibbs free energies have been calculated for aza-18-crown-6 (L) and their metal complexes. The natural bond orbital analysis is used to explore the interaction of host–guest molecules. The absorption spectra differences between L and their metal ligands, the excitation energies and absorption wavelength for their excited states have been studied by time-dependent density functional theory with the basis set 6-31 + G(d,p). These fluorescent sensors and switchers based on photoinduced electron transfer mechanism have been investigated. The PET process from aza-crown ether to fluorophore can be suppressed or completely blocked by the entry of alkali metal cations into the aza-crown ether-based receptor. Such a suppression of the PET process means that fluorescence intensity is enhanced. The binding selectivity studies of the aza-crown ether part of L indicate that the presence of the alkali metal cations Li+, Na+ and K+ play an important role in determining the internal charge transfer and the fluorescence properties of the complexes. In addition, the solvent effect has been investigated. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Ling Li, Mi Li, Xueye Wang, Qin Wang
Computational and Theoretical Chemistry 2014 Volume 1031() pp:40-49
Publication Date(Web):1 March 2014
DOI:10.1016/j.comptc.2013.12.028
•“Molecular Taekwondo” process of pyrene-armed calix[4]azacrowns have been studied through the fluorescence change.•Optimized structures and electronic properties have been performed at B3LYP/6-31G(d).•Metal-π and metal–metal ion repulsive interactions are main driving forces.•TD-DFT calculations on the emission spectra are investigated and discussed.New calixarene derivatives pyrene-armed calix[4]azacrowns L (L1 and L2) bearing a crown ether and an azacrown ether as two binding sites are calculated systemically using density functional theory (DFT). The optimized structures and electronic properties, such as HOMO and LUMO energies, band gaps of the free ligands L (L1 and L2), the complexes L/M+ (Ag+, K+ and Cs+) and the di-cation complexes L·Ag+·M+ have been performed at B3LYP/6-31G(d) and Lanl2DZ level. Natural bond orbital (NBO) analysis is discussed on the basic of the optimized geometric structures. The results demonstrate that metal-π and metal–metal ion repulsive interactions are main driving forces of the coordination and the influence of the solvent effect is significant. Moreover, the interesting “Molecular Taekwondo” processes evolving between Ag+–K+ and Ag+–Cs+ pairs have been studied through the fluorescence change. Time-dependent density functional theory (TD-DFT) calculations on the emission spectra of the ligand L1 and compounds L1/M+ are investigated and discussed, which further evidences the existence of the metal ion exchange process.Graphical abstract
Co-reporter:Benhua Luo, Xueye Wang, Yu Zhang, Yong Xia
Materials Chemistry and Physics 2012 Volume 133(2–3) pp:857-862
Publication Date(Web):16 April 2012
DOI:10.1016/j.matchemphys.2012.01.107
The structural, electronic and optical properties of Sr0.875Pb0.125HfO3 are investigated using the pseudo-potential plane wave method within the generalized gradient approximation (GGA) by first principles. The lattice constants obtained by minimization of the total energy are in agreement with the available experimental data. Dopant formation energy results show that Pb preferentially enters the Sr site in SrHfO3, which is in good agreement with experimental observations. The band structures indicate that both SrHfO3 and Sr0.875Pb0.125HfO3 are insulators. The density of states and charge density map indicate that bonding between Hf and O is mainly covalent, whereas bonding of SrHfO3 and PbHfO3 is ionic. In order to understand the optical properties of Sr0.875Pb0.125HfO3, the dielectric functions, loss function, refractive index, absorption coefficient and reflectivity are calculated for radiation up to 40 eV.Graphical abstractHighlights► Dopant formation energy results show that Pb enters the Sr site in SrHfO3. ► The bandgap of Sr0.875Pb0.125HfO3 is slightly wider compared with SrHfO3. ► Bonding between Hf and O is covalent whereas SrHfO3 and PbHfO3 are ionic. ► O 2p states and Hf 5d states play a major role in optical transitions.
Co-reporter:Yong Xia;Yu Zhang ;Benhua Luo
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 9) pp:778-786
Publication Date(Web):
DOI:10.1002/poc.2917
A novel fluorescent switchable chemosensor 1, which is composed of an anthracene-modified calix[4]crown in the 1,3-alternate conformation, was calculated by density functional theory and time-dependent density functional theory method. Geometries, molecular orbitals and binding thermal energies were evaluated at the restricted hybrid Becke's three-parameter exchange functional using 6-31 G(d) basis set and relativistic effective core potentials. The metal–ligand and cation–π interactions were investigated acting as two main types of driving force. Our calculations clearly show that solvent effects strongly influence cation selectivity, and K+ selectivity is recovered when even a few waters of hydration are considered. The calculations indicate that because of the photoinduced electron transfer effect, the addition of alkali metal ions have hardly any effect on the fluorescence of ligand 1 under neutral or basic conditions. Also, the high selectivity of ligand 1 for K+ and Rb+, under acidic conditions, the complexed metal ion can result in ammonium ion deprotonation, which leads to quenching of fluorescence of 1•H+. Copyright © 2012 John Wiley & Sons, Ltd.
Co-reporter:Yu Zhang;Benhua Luo ;Yong Xia
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 3) pp:222-229
Publication Date(Web):
DOI:10.1002/poc.1895
The binding interactions of bis-3-benzo-15-crown-5 ethers and bis-3-benzo-18-crown-6 ethers (neutral hosts) with a series of alkali metal cations Na+, K+, Rb+ and Cs+ (charged guests) were investigated using quantum chemical density functional theory. Different optimized structures, binding energies and various thermodynamic parameters of free crown ethers and their metal cation complexes were obtained based on the Becke, three-parameter, Lee–Yang–Parr functional using mixed basis set (C, H, O, Na+ and K+ using 6-31 g, and the heavier cation Rb+ and Cs+ using effective core potentials). Natural bond orbital analysis is conducted on the optimized geometric structures. The main types of driving force host–guest interactions are investigated. The electron donating O offers a lone pair of electrons to the contacting LP* (1-center valence antibond lone pair) orbitals of metal cations. The bis-3-benzocrown ethers are assumed to have sandwich-like conformations, considering the binding energies to gauge the exact interactions with alkali cations. It is found that there are two different types of complexes: one is a tight ion pair and the other is a separated ion pair. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Yong Xia;Yu Zhang;Benhua Luo;Yi Liu
Journal of Molecular Modeling 2012 Volume 18( Issue 6) pp:2291-2299
Publication Date(Web):2012 June
DOI:10.1007/s00894-011-1243-9
Theoretical studies of a new ion-pair receptor, meso-octamethylcalix[4]pyrrole (OMCP), and its interactions with the halide anions F−, Cl−, and Br− and the cesium halides CsF, CsCl, and CsBr have been performed. Geometries, binding energies, and binding enthalpies were evaluated with the restricted hybrid Becke three-parameter exchange functional (B3LYP) method using the 6-31+G(d) basis set and relativistic effective core potentials. The optimized geometric structures were used to perform natural bond orbital (NBO) analysis. The two typical types of hydrogen bonds, N–H…X− and C–H…X−, were investigated. The results indicate that hydrogen bonding interactions are dominant, and that the halide anions (F−, Cl−, and Br−) offer lone pair electrons to the σ*(N–H) or σ*(C–H) antibonding orbitals of OMCP. In addition, electrostatic interactions between the lone pair electrons of the halide anion and the LP* orbitals of Cs+ as well as cation–π interactions between the metal ion and π-orbitals of the pyrrole rings have important roles to play in the Cs+•OMCP•X− complexes. The current study further demonstrates that this easy-to-make OMCP host compound functions as not only an anion receptor but also an ion-pair receptor.
Co-reporter:Yu Zhang, Xueye Wang, Benhua Luo, Yong Xia
Journal of Organometallic Chemistry 2012 699() pp: 31-38
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.10.032
Co-reporter:Yuan Miao;Dan Ouyang
Journal of Molecular Modeling 2012 Volume 18( Issue 3) pp:963-972
Publication Date(Web):2012 March
DOI:10.1007/s00894-011-1132-2
A series of crown ethers containing the azobenzene moiety incorporated into crowns of various sizes [Cr(O6), Cr(O7) and Cr(O8)] and their corresponding alkali metal cation (Li+, Na+, K+, Rb+) complexes have been studied theoretically. The density functional theory (DFT) method was employed to elucidate the stereochemical structural natures and thermodynamic properties of all of the target molecules at the B3LYP/6-31 G(d) and LANL2DZ level for the cation Rb+. The fully optimized geometries had real frequencies, thus indicating their minimum-energy status. In addition, the bond lengths between the metal cation and oxygen atoms, atomic torsion angles and thermodynamic energies for complexes were studied. Natural bond orbital (NBO) analysis was used to explore the origin of the internal forces and the intermolecular interactions for the metal complexes. The calculated results show that the most significant interaction is that between the lone pair electrons of electron-donating oxygens in the cis-forms of azobenzene crown ethers (cis-ACEs) and the LP* (1-center valence antibond lone pair) orbitals of the alkali-metal cations (Li+, Na+, K+ and Rb+). The electronic spectra for the cis-ACEs [cis-Cr(O6), cis-Cr(O7) and cis-Cr(O8)] are obtained by the time-dependent density functional theory (TDDFT) at the B3LYP/6-31 G(d) level. The spectra of the cis-isomers show broad π → π* (S0 → S2) absorption bands at 310–340 nm but weaker n → π* (S0 → S1) bands at 480–490 nm. The calculated results are in good agreement with the experimental results.
Co-reporter:Ling Yi, Xueye Wang
Journal of Luminescence 2011 Volume 131(Issue 10) pp:2083-2088
Publication Date(Web):October 2011
DOI:10.1016/j.jlumin.2011.05.002
This paper studied poly[(3,6-di-tert-butyl-N-hexadecyl-1,8-carbazolylene) butadiynylene] (P1), butadiynylene-linked poly (3,6-carbazole) (P2) and butadiynylene-linked poly (2,7-carbazole) (P3) through the theoretical measurements with Gaussian 03 program package. To investigate the relationship between structures and properties of these multifunctional electroluminescent materials, their geometrical structures of ground and excited-states were optimized by B3LYP/6-31G (d) and CIS/6-31G (d) methods, respectively. The lowest excitation energies (Eg's), and the maximum absorption and emission wavelengths of these polymers were calculated by time-dependent density functional theory methods (TD-DFT). The important parameters for luminescent materials were also predicated including the ionization potentials (Ip's) and electron affinities (Ea's). The calculated results show that the highest-occupied molecular orbital (HOMO) energies lift about 0.27–0.49 eV compared to N,N′-bis(naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPB), suggesting the significant improved hole-accepting and transporting abilities. In addition, substitution of alkyne for carbazole resulted in a narrow band gap and a red shift of both the absorption and emission peaks. Through above calculations, it is evidenced that these polymers can be considered as candidates for excellent OLEDs with good hole-creating abilities and high blue-light emission.Highlights► We studied poly [(3,6-di-tert-butyl-N-hexadecyl-1,8-carbazolylene) butadiynylene] by theoretical method. ► The geometrical structures of ground and excited-states had been optimized by B3LYP/6-31G (d) and CIS/6-31G (d). ► The relationship between structures and properties of these multifunctional electroluminescent materials had been investigated. ► These molecules are excellent candidates for multifunctional OLED materials. ► The substitution of alkyne for carbazole results in a narrow band gap and a red shift of both the absorption and emission peaks.
Co-reporter:Yuan Miao; Dr. Xueye Wang;Xin Jin;Ling Yi ;Cuihuan Ren
Chinese Journal of Chemistry 2010 Volume 28( Issue 10) pp:1835-1843
Publication Date(Web):
DOI:10.1002/cjoc.201090307
Abstract
A series of ring-contracted (14-crown-5, 17-crown-6) and ring-enlarged (16-crown-5, 17-crown-5, 19-crown-6, 20-crown-6) crown ethers and their complexes with alkali-metal cations Na+ and K+ had been explored using density functional theory (DFT) at B3LYP/6-31G* level in order to reveal the effects of the methylene-chain length in a crown ether. The nucleophilicity of all crown ethers had been investigated by the Fukui functions. The quantum chemistry parameters, such as the energy gap (ΔE), the highest occupied molecular orbital energy (EHOMO) and the lowest unoccupied molecular orbital energy (ELUMO) for less-symmetrical crown ethers and symmetrical frameworks (15-crown-5, 18-crown-6) had been calculated. In addition, the thermodynamic energies of complexation reactions had also been studied. The results of the DFT calculations show that the methylene-chain length plays an important role in determining the structure characters of the crown ethers and also strongly influences the properties of the ethers. Some of the calculated results are in a good agreement with the experimental values.
Co-reporter:Cuihuan Ren, Xueye Wang, Yuan Miao, Ling Yi, Xin Jin, Yuanqiang Tan
Journal of Molecular Structure: THEOCHEM 2010 Volume 949(1–3) pp:96-100
Publication Date(Web):15 June 2010
DOI:10.1016/j.theochem.2010.03.014
Density functional theory calculation has been used to investigate the dissociative adsorption of hydrogen sulfide molecules on the Si(1 1 1)-7 × 7 surface. The reaction pathways for four hydrogen sulfide molecules dissociation over the adatom and rest atom sites are explored theoretically. The calculated results demonstrate that the initial dissociation of the first H2S molecule on both different sites to form the adsorbed SH species is no energy barrier. The elevated temperature can result in further SH bond dissociation, which is started by an SH insertion, and then followed by an H-transfer process. Subsequently, the second H2S attacks the adatom site (Sia), H atoms from the SH of the second H2S and surface SiaH are extracted to release the gas phase H2, and then the SH species from the second H2S occupies on the Sia site. Furthermore, the SH species inserts into SiSi backbond of the adatom, followed by an H-transfer process. In the same way, the SH species from the third and the fourth H2S molecule are also inserted into remnant SiSi backbonds of the adatom followed by an H-transfer process up to the formation of Si4+ state.
Co-reporter:Xinyu Peng, Xueye Wang, Liming Liu, Yanling Wang, Yuanqiang Tan
Chemical Physics 2009 Volume 359(1–3) pp:21-26
Publication Date(Web):18 May 2009
DOI:10.1016/j.chemphys.2009.02.010
Abstract
The chemisorption and decomposition of pyrrole (C4H4NH) on the reconstructed Si(1 1 1)7 × 7 surface have been investigated by means of hybrid density functional B3LYP method in combination with the cluster model approach. Three chemisorption mechanisms, N–H dissociative adsorption, [4 + 2] cycloaddition, C–H and N–H dissociative adsorption of pyrrole onto a rest atom–adatom pair site, have been considered comparatively. It is shown that the dissociaton of C4H4NH occurs readily on either the adatom site (Sia) or the rest atom site (Sir), giving rise to C4H4N and H adspecies, and forming NadsPa and NadsPr products respectively. The calculations showed that the Cα adsorption process is barrierless and favorable over the N adsorption one. Through compare and analysis the three chemisorption mechanisms, N–H dissociative adsorption on a rest atom–adatom pair of the Si(1 1 1)7 × 7 surface is the most favorable, whereas C–H and N–H dissociative adsorption on a rest atom–adatom pair is the most difficult.
Co-reporter:Yanling Wang, Xueye Wang, Liming Liu, Xinyu Peng
Journal of Molecular Structure: THEOCHEM 2009 Volume 896(1–3) pp:34-37
Publication Date(Web):28 February 2009
DOI:10.1016/j.theochem.2008.10.047
The thermal dehydrochlorination of model compounds for poly(vinyl chloride) (PVC) was investigated theoretically. To determine the reaction paths of the thermal dehydrochlorination, geometries, structures, and energies were evaluated for reactants, products, and transition states of the proposed paths by using the density functional theory (DFT) at the B3LYP/6-31G(d) level. On the basis of the experimental observations, three possible paths of the thermal dehydrochlorination for the model compounds of PVC have been postulated. The activation energies of path (2) and path (3) are 55.1 kcal/mol and 41.5 kcal/mol at the B3LYP/6-31G(d) level, respectively.
Co-reporter:Liming Liu;Yanling Wang;Xinyu Peng ;Yuexiang Mo
Journal of Polymer Science Part B: Polymer Physics 2009 Volume 47( Issue 7) pp:706-714
Publication Date(Web):
DOI:10.1002/polb.21678
Abstract
The polycarbazoles have been proved to efficiently suppress the keto defect emission. Three carbazole-based conjugated polymers, poly[9-methyl-3-(4-vinylstyryl)-9H-carbazole] (PBC), poly[9-methyl-3-(2-(5-vinylthiophen-2-yl)vinyl)-9H-carbazole] (PBT) and poly[9-methyl-3-(2-(5-vinylfuran-2-yl)vinyl)-9H-carbazole] (PBF), were investigated by quantum-chemical techniques, and gain a detailed understanding of the influence of carbazole units and the introduction of electron-donating on the electronic and optical properties. The electronic properties of the neutral molecules, HOMO-LUMO gaps (ΔE), in addition to ionization potential (Ip) and electron affinity (Ea), are studied using B3LYP density functional theory. The lowest excitation energies (Eg) and the absorption wavelength are studied using the time dependent density functional theory (TDDFT). The calculated results show that all three series of polymers have good planarity. And the highest-occupied molecular orbital (HOMO) energies lift about 0.36–0.61 eV and thus the IP decrease about 0.01–0.19 eV compared to polycarbazole, suggesting the significant improved hole-accepting and transporting abilities. By introducing the electron-donating 1,4-divinylphenylene or 2,5-divinylthiophene or 2,5-divinylfuran units in the backbone, and the lowest-unoccupied molecular orbital (LUMO) energies decrease 0.20–0.39 eV. In addition, PBC, PBT and PBF have longer maximal absorption wavelengths than polycarbazole. © 2009 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 47: 706–714, 2009
Co-reporter:Yanling Wang;Liming Liu;Xinyu Peng
Journal of Molecular Modeling 2009 Volume 15( Issue 9) pp:1043-1049
Publication Date(Web):2009 September
DOI:10.1007/s00894-009-0457-6
The thermal decomposition of model compounds for poly (dialkyl fumarate) was studied by using ab initio and density functional theory (DFT) calculations. To determine the most favorable reaction pathway of thermal decomposition, geometries, structures, and energies were evaluated for reactants, products, and transition states of the proposed pathways at the HF/6-31G(d) and B3LYP/6-31G(d) levels. Three possible paths (I, II and III) and subsequent reaction paths (IV and V) for the model compounds of poly (dialkyl fumarate) decomposition had been postulated. It has been found that the path (I) has the lowest activation energy 193.8 kJ mol−1 at B3LYP/6-31G(d) level and the path (I) is considered as the main path for the thermal decomposition of model compounds for poly (dialkyl fumarate).
Co-reporter:Liming Liu, Xueye Wang, Yanling Wang, Xinyu Peng
Journal of Molecular Structure: THEOCHEM 2008 Volume 868(1–3) pp:82-86
Publication Date(Web):15 November 2008
DOI:10.1016/j.theochem.2008.08.003
2,2′-difuryl-4,4′-(N,N,N′,N′-tetraphenyl) diaminobiphenyl (FurylBz-Ph4) and 5,5′-bis(4-N,N′- diphenylaminophenyl)-2,2′-bifuryl (PFDA-Ph4) are new light-emitting materials, which are capable of transporting holes. A series of new compounds whose structures were similar to that of FurylBz-Ph4 or PFDA-Ph4, were designed and calculated. The total energies, HOMO and LUMO orbital energies of these compounds at neutral, cationic and anionic states were obtained based on the optimised geometrical structures. The ionization potential (Ip) and electron affinity (Ea) were generated by means of density functional theory (DFT) calculated total energy of neutral, cationic and anionic states. The reorganization energy for the hole transport of compounds 1-3, FurylBz-Ph4 and PFDA-Ph4 is close to that of TPD at the same level of calculation. For compounds 2,3 and PFDA-Ph4, the order of Ip is 3 > PFDA-Ph4 > 2. It is indicated that the contribution of aryl groups to Ip follows the sequence: thienyl > furyl > pyrryl. The absorptions of these compounds had been also investegated by time-dependent density functional theory (TD-DFT) methods with 6-31G(d) basis set. The TD-DFT calculations indicate that the absorption maximum of compounds 2 is blue-shifted from PFDA-Ph4, while the absorption maximum of compounds 3 is red-shifted from PFDA-Ph4. Since the electron-withdrawing strength is in the order pyrryl > thienyl > furyl and PFDA-Ph4 has the longer conjugated length between the two amine nitrogen atoms than that of compound 2 and 3. The different bands of emission and reorganization energy can be obtained by changing the substituents and the site of substitution between two triphenylamino units.
Co-reporter:Cuili Zhang;Liming Liu;Yanling Wang
Journal of Molecular Modeling 2008 Volume 14( Issue 11) pp:1053-1064
Publication Date(Web):2008 November
DOI:10.1007/s00894-008-0344-6
The mechanism of the spontaneous initiation of the polymerization of methyl methacrylate (MMA) was investigated theoretically. The six minimum energy paths (MEP) of the possible reactions were calculated using the density functional theory (DFT) in conjunction with the B3LYP functional and 6-31G* basis set. The Diels-Alder initiation mechanism (path (I) and path (II)) with remarkably high energy barriers is not applicable to MMA. Four favorable paths were found (path (III), path (IV), path (V) and path (VI)), which are supporting the Flory mechanism. Path (V) has the lowest active energy. Therefore this path is considered as the main path for the spontaneous polymerization of MMA.
Co-reporter:Han-Lu Wang;Xue-Ye Wang;Ling Wang;Heng-Liang He;Ai-Hong Liu
Chinese Journal of Chemistry 2007 Volume 25(Issue 7) pp:
Publication Date(Web):16 JUL 2007
DOI:10.1002/cjoc.200790174
Quantum-chemical techniques were applied to investigate a series of conjugated polymers: poly(3-octylthien-2,5-ylene-co-pyrid-2,6-ylene) (p1), poly[pyrid-2,6-ylenebis(3-octylthien-2,5-ylene)] (p2) and poly[pyrid-2,5-ylene- bis(3-octylthien-2,5-ylene)] (p3) comprising alternating π-excessive 3-alkylthiophene and π-deficient meta- or para-linked pyridine moieties. Their ground state and excited state structures were optimized with density functional theory B3LYP method, and the optical properties were calculated by the time-dependent density functional theory (TD-DFT) and ZINDO/S methods. Their HOMO-LUMO gaps (ΔH-L), the lowest excitation energies (Eex), ionization potentials (IP) and electron affinities (EA) were obtained by extrapolating those of the polymers to the inverse chain length equal to zero (1/n=0). The calculated results showed that the decrease of pyridylene content increased the HOMO level and decreased the LUMO level while the para-linkage further contributed to it. The IP are in the order: p1>p2>p3 but EA are: p1<p2<p3. In addition, the decrease of the pyridylene content and the para-linked pyridylene in the backbone of the polythiophene resulted in a narrowed energy gap and bathochromic absorption and emission peaks.
Co-reporter:Hengliang Wang;Hanlu Wang;Ling Wang
Journal of Molecular Modeling 2007 Volume 13( Issue 1) pp:147-153
Publication Date(Web):2007 January
DOI:10.1007/s00894-006-0135-x
In the present work, a theoretical study of five bipyrazolic-type organic compounds, 4-{bis[(3,5-dimethyl-1H-pyrazolyl-1-yl)methyl]-amino}phenol (1), N1,N1-bis[(3,5-dimethyl-1H-pyrazol-1-yl)methyl}]-N4,N4-dimethyl-1,4-benzenediamine (2), N,N-bis[(3,5-dimethyl-1H-pyrazol-1-yl)methyl]aniline (3), 4-[bis(3,5-dimethyl pyrazol-1-yl-methyl)-amino]butan-1-ol (4) and ethyl4-[bis(3,5-dimethyl-1H-pyrazol-1-yl-methyl) aminobenzoate] (5), has been performed using density functional theory (DFT) at the B3LYP/6-31G(d) level in order to elucidate the different inhibition efficiencies and reactive sites of these compounds as corrosion inhibitors. The efficiencies of corrosion inhibitors and the global chemical reactivity relate to some parameters, such as EHOMO, ELUMO, gap energy (ΔE) and other parameters, including electronegativity (χ), global hardness (η) and the fraction of electrons transferred from the inhibitor molecule to the metallic atom (ΔN). The calculated results are in agreement with the experimental data on the whole. In addition, the local reactivity has been analyzed through the Fukui function and condensed softness indices.
Co-reporter:Xinliang Yu;Xiaobing Li;Jinwei Gao;Hanlu Wang
Macromolecular Theory and Simulations 2006 Volume 15(Issue 1) pp:94-99
Publication Date(Web):27 DEC 2005
DOI:10.1002/mats.200500057
Summary: A set of new molecular descriptors, RSC, SMC, DHB and MPE, which are obtained directly from polystyrenes monomer structures, are used to predict the Tg values of polystyrenes and generate a QSPR model by multiple linear stepwise regression analysis, with R = 0.959 and s = 15.20 K. Compared with the existing QSPR models, the proposed model requires only four descriptors, which can be obtained by simple calculation and makes it more valuable to predict the Tg values of polymers.
Co-reporter:Xinliang Yu;Hanlu Wang;Xiaobing Li;Jinwei Gao
Journal of Polymer Science Part B: Polymer Physics 2006 Volume 44(Issue 2) pp:409-415
Publication Date(Web):5 DEC 2005
DOI:10.1002/polb.20714
Density functional theory calculations have been carried out for monomers of polymers at the B3LYP/6-31G(d) level. The quantum chemical descriptors of the calculated results, which are the molecular average polarizability, the most positive net atomic charge on hydrogen atoms in a molecule, the energy of the lowest unoccupied molecular orbital, and the molecular dipole moment, have been used to predict the cohesive energy of polymers with the structure (C1H2C2R3R4). A general quantitative structure–property relationship model, with a correlation coefficient of R = 0.986 and a standard error of s = 2636 J/mol, has been built by multiple linear regression analysis. © 2005 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 44: 409–415, 2006
Co-reporter:Chunyang Zhao, Xueye Wang
Journal of Alloys and Compounds (15 May 2017) Volume 704() pp:
Publication Date(Web):15 May 2017
DOI:10.1016/j.jallcom.2017.02.065
•The elastic constants, three modulus, Poisson's ratio and anisotropy index are calculated.•Thermodynamic properties are predicted in wide pressure and temperature ranges.•The calculated results are in good agreement with the experimental data.The density functional theory (DFT) and the quasi-harmonic Debye model are used to investigate the structural, electronic, elastic and thermodynamic properties of Rh3Sc compound under high pressure. Firstly, the calculated lattice parameter of Rh3Sc compound at 0 GPa is in good agreement with the available experimental value. The calculated total density of states (TDOS) at various pressures, 0 GPa, 25 GPa and 50 GPa, show that the stability of Rh3Sc compound increases with increasing pressure. The pressure dependence on the elastic constants Cij, bulk modulus B, shear modulus G, Young's modulus E, B/G ratio, Poisson's ratio (ν) and the anisotropic index (A) were also calculated successfully. Finally, the temperature and pressure variation of the thermal expansion α, heat capacity CV, Debye temperature Θ and entropy S are also calculated in a wide pressure (0–50 GPa) and temperature (0–1500 K) ranges and discussed.