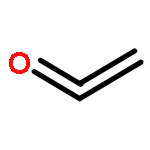
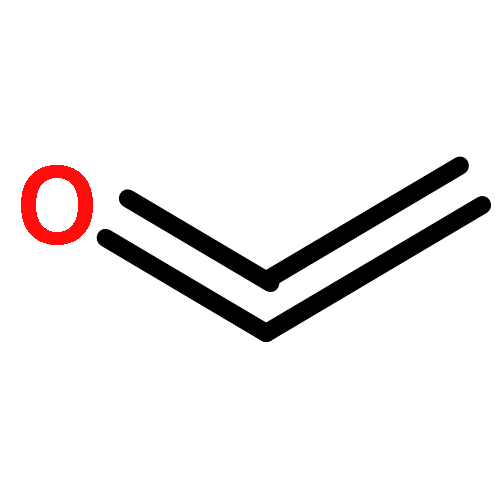
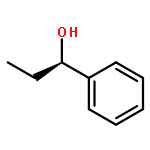
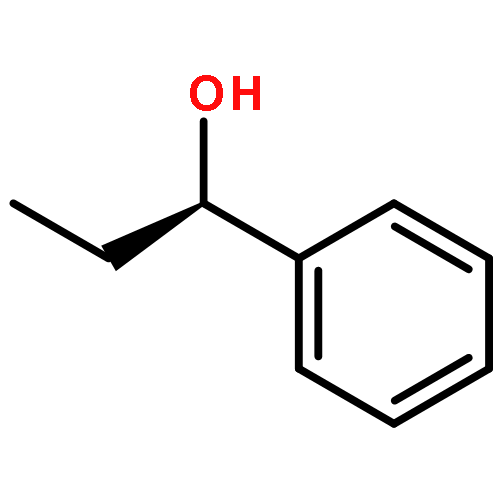
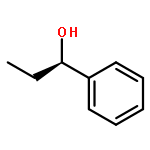
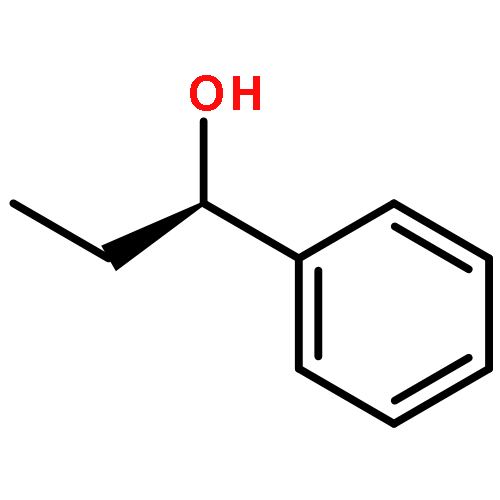
Co-reporter:Hao Xu;Yue Li;Xuchen Shang;Zhenhua Zhu
Structural Chemistry 2017 Volume 28( Issue 6) pp:1959-1968
Publication Date(Web):03 July 2017
DOI:10.1007/s11224-017-0990-3
In this paper, generation mechanism of α,β-unsaturated carbon γ-amino esters catalyzed by triphenylphosphine (PPh3) have been investigated using density functional theory (DFT). Two possible mechanisms (Mechanism A and Mechanism B) are proposed, in which there are three possible reaction pathways (Path A1/A2/A3 in Mechanism A and Path B1/B2/B3 in Mechanism B) except for the generation of the intermediate M1. The calculated results demonstrate that the energy favorable pathways (Path A2 and Path B2) include three process: the first step is an intermolecular proton transfer accompanied by a nucleophilic attack, the second step is an intramolecular proton transfer process, and the last step is the dissociation of PPh3 and the product generation. Furthermore, the reaction pathway associated with the E-isomer is the most favorable pathway and leads to the main product (E-P), which is in good agreement with the experimental results. This work might be helpful for understanding the significant roles of catalyst PPh3 and thus provide valuable insights on the rational design of potential catalysts for this kind of reactions.
Co-reporter:Yue Li;Fangfang Li;Xue Li;Ziyuan Zhou;Chunmei Liu
Structural Chemistry 2016 Volume 27( Issue 4) pp:1165-1173
Publication Date(Web):2016 August
DOI:10.1007/s11224-016-0740-y
In this work, three possible reaction pathways (Path 1, Path 2 and Path 3) for the generation process of cyclic dipeptide from amino acid have been investigated in detail using density functional theory. Path 1 and Path 2 are the intramolecular reaction processes, while Path 3 involves the intermolecular reaction process that assisted with water molecule. Our calculated results indicate that Path 3 is more energy favorable than Path 1 and Path 2. There are four steps in Path 3 proceed from the amino acid to cyclic dipeptide. The first step is two adjacent amino acids to form precursor of dipeptide, the second step is the removal of water molecule of precursor of dipeptide for the formation of the linear dipeptide, the third step is generation of precursor of cyclic dipeptide associated with other hydrogen atom transfer, and the last step is another dehydration process to generate the final product of cyclic dipeptide. Moreover, the obtained results indicate that the generation mechanisms of different cyclic dipeptides are similar, and the energy barrier of the rate-determined step influenced somewhat by the hydrophilic or hydrophobic group linked to the Cα atom. Additionally, the potential energy profiles suggest that the generation reactions of the studied nine cyclic dipeptides are exothermic processes. The detailed mechanisms should be helpful for people to understanding the title reaction at the molecular level, and the proposed novel intermolecular process might provide valuable insights on rational improve reaction condition for this type of reaction.
Co-reporter:Chunmei Liu;Bin Zhang;Mingsheng Tang
Structural Chemistry 2016 Volume 27( Issue 5) pp:1449-1464
Publication Date(Web):2016 October
DOI:10.1007/s11224-016-0764-3
We describe the development of the AMBER force field parameters for 46 nucleases involving most kinds of copper nucleases with high DNA affinities and specificities by MINA approach that could evaluate accurate force constants for batch bonds/angles on the basis of energies of three adjacent lengths/angles geometries. The molecular mechanics (MM) and molecular dynamic simulations on adducts of the 21 representative copper-based nucleases with DNA are in excellent agreement with those of experimental results. Furthermore, to validate the evaluated parameters, the studied structures performed frequency analysis together with normal mode calculations in quantum mechanics and MM calculations. The force field parameters evaluated in this work provide an extension of AMBER force field, and the results of molecular dynamics simulations of adduct of copper nuclease and duplex DNA illustrate the potential utility of these parameters.
Co-reporter:Chunmei Liu, Yanyan Zhu, Mingsheng Tang
Journal of Molecular Graphics and Modelling 2016 Volume 64() pp:11-29
Publication Date(Web):March 2016
DOI:10.1016/j.jmgm.2015.12.003
•Molecular docking predicts groove binding and electrostatic interaction well for the studied copper nucleases.•The DNA binding affinity of copper nucleases influenced by the ligand size, length, functional groups, chelate ring size bound to copper center.•Intercalation modes could be reproduced by “distorted DNA” formed by molecular dynamics simulations.•MM-PBSA approach validated the DNA binding affinity of copper nuclease.In the present work, molecular simulations were performed for the purpose of predicting the binding modes of four types of copper nucleases (a total 33 compounds) with DNA. Our docking results accurately predicted the groove binding and electrostatic interaction for some copper nucleases with B-DNA. The intercalation modes were also reproduced by “gap DNA”. The obtained results demonstrated that the ligand size, length, functional groups and chelate ring size bound to the copper center could influence the binding affinities of copper nucleases. The binding affinities obtained from the docking calculations herein also replicated results found using MM-PBSA approach. The predicted DNA binding modes of copper nucleases with DNA will ultimately help us to better understand the interaction of copper compounds with DNA.
Co-reporter:Mengmeng Zhang, Donghui Wei, Yang Wang, Suiji Li, Jiefei Liu, Yanyan Zhu and Mingsheng Tang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 33) pp:6374-6383
Publication Date(Web):14 May 2014
DOI:10.1039/C4OB00606B
In this paper, two possible mechanisms (mechanisms A and B) on the stereoselective [2 + 2] cycloaddition of aryl(alkyl)ketenes and electron-deficient benzaldehydes catalyzed by N-heterocyclic carbenes (NHCs) have been investigated using density functional theory (DFT). Our calculated results indicate that the favorable mechanism (mechanism A) includes three processes: the first step is the nucleophilic attack on the arylalkylketene by the NHC catalyst to form an intermediate, the second step is the [2 + 2] cycloaddition of the intermediate and benzaldehyde for the formation of a β-lactone, and the last step is the dissociation of the NHC catalyst and the β-lactone. Notably, the [2 + 2] cycloaddition, in which two chiral centers associated with four configurations (SS, RR, SR and RS) are formed, is demonstrated to be both the rate- and stereoselectivity-determining step. Moreover, the reaction pathway associated with the SR configuration is the most favorable pathway and leads to the main product, which is in good agreement with the experimental results. Furthermore, the analysis of global and local reactivity indexes has been performed to explain the role of the NHC catalyst in the [2 + 2] cycloaddition reaction. Therefore, this study will be of great use for the rational design of more efficient catalysts for this kind of cycloaddition.
Co-reporter:Yunxia Li, Yanyan Zhu, Wenjing Zhang, Donghui Wei, Yingying Ran, Qilin Zhao and Mingsheng Tang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 37) pp:20001-20008
Publication Date(Web):31 Jul 2014
DOI:10.1039/C4CP02186J
Reaction mechanisms of the N-heterocyclic carbene (NHC)-catalyzed dimerization of methyl methacrylate were studied using density functional theory (DFT) at the M05-2X/6-31G(d,p) level of theory. Four possible reaction channels (A, B, C, and D) have been investigated in this work. Particularly, we proposed a novel reaction pathway, where the proton transfers are assisted by a different molecule. The calculated results indicate that the channels B and D are more energetically favourable channels. The obtained results suggest that the E-isomer product is the main product, which is in agreement with the experimental results. Further calculations and analyses of global and local reactivity indices reveal the role of the NHC catalysts in the title reaction. The mechanistic insights gained are valuable for not only rational design of more efficient NHC catalysts but also for understanding the similar reaction mechanism.
Co-reporter:Yanyan Zhu;Hongge Zhao;Chunmei Liu;Donghui Wei;Xue Li
Structural Chemistry 2014 Volume 25( Issue 3) pp:699-705
Publication Date(Web):2014 June
DOI:10.1007/s11224-013-0333-y
The inclusion complexes of two modified cyclic decapeptides with 1-phenyl-1-propanol (PP) enantiomers were first studied using the density functional theory B3LYP method. Our calculated results indicated that modified cyclic decapeptide (CM-CDP and DA-CDP) could form stable inclusion complexes. Significantly, based on the structural characteristics and hydrogen bond analyses, we found that the primary driving force of inclusion complex formation is a cooperative work of hydrogen bonds, steric effect, and electronic interactions, which facilitates the enhancement of binding affinity of the PP enantiomers with CM-CDP and DA-CDP. The current study shows that modified cyclic decapeptide is a desirable host molecule for chiral and molecular recognition.
Co-reporter:Zhenyu Li, Donghui Wei, Yang Wang, Yanyan Zhu, and Mingsheng Tang
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:3069-3078
Publication Date(Web):March 16, 2014
DOI:10.1021/jo500194d
The possible reaction mechanisms of stereoselective [4 + 2] cycloaddition of enals and chalcones catalyzed by N-heterocyclic carbene (NHC) have been investigated using density functional theory (DFT). The calculated results indicate that the most favorable reaction channel occurs through five steps. The first step is the nucleophilic attack on the enal by NHC. Then, there are two consecutive acid (AcOH)-assisted proton-transfer steps. Subsequently, the fourth step is the [4 + 2] cycloaddition process associated with the formation of two chiral centers, followed by dissociation of NHC and product. Our computational results demonstrate that the [4 + 2] cycloaddition is the rate-determining and stereoselectivity-determining step. The energy barrier for the SS configurational channel (17.62 kcal/mol) is the lowest one, indicating the SS configurational product should be the main product, which is in agreement with experiment. Moreover, the role of NHC catalyst in the [4 + 2] cycloaddition of enal and chalcone was explored by the analysis of global reactivity indexes. This work should be helpful for realizing the significant roles of catalyst NHC and the additive AcOH and thus provide valuable insights on the rational design of potential catalyst for this kind of reactions.
Co-reporter:Chunmei Liu, Yanyan Zhu, Peipei Chen, and Mingsheng Tang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 5) pp:1197-1209
Publication Date(Web):January 14, 2013
DOI:10.1021/jp306998f
In the present study, molecular dynamic simulations have been performed to investigate the DNA binding affinities and cleavage activities of a new class of mononuclear copper (p-Cu(BPA) and m-Cu(BPA)) and dinuclear copper–platinum (p-Cu(BPA)-Pt and m-Cu(BPA)-Pt) metallonucleases. The simulated results reveal that the two mononuclear nucleases are noncovalent minor groove DNA binders and the two dinuclear ones tend to be bound to DNA in the major groove by a covalent bond between the platinum center and N7 of the guanine base, which is in agreement with the experimental results. The simulated results show that the binding affinities of the four studied nucleases with DNA are in the order of p-Cu(BPA) < m-Cu(BPA) < p-Cu(BPA)-Pt < m-Cu(BPA)-Pt; the binding affinities are dominated by intermolecular binding modes of nucleases with DNA and the intermolecular hydrogen bonds. The distance probability distributions indicate that the hydrogen atoms of DNA sugar could be abstracted by the four nucleases. Specifically, the dinuclear nucleases abstract hydrogen atoms from the deoxyribose sugar linking to G18 base while mononuclear nuclease abstracts hydrogen atoms from the deoxyribose sugars linking to C15 and C16 bases, suggesting that the dinuclear nucleases improve the sequence-selective cleavage of DNA compared with the mononuclear one. Moreover, the differences in calculated DNA conformational dynamics and groove parameters demonstrate that the extent of DNA conformational distortions induced by dinuclear nucleases is greater than that induced by mononuclear nucleases. This investigation provides detailed information showing that dinuclear nucleases have superior DNA binding affinities and nuclease activities as compared with their mononuclear counterparts.
Co-reporter:Wenjing Zhang, Yanyan Zhu, Donghui Wei, Yunxia Li, and Mingsheng Tang
The Journal of Organic Chemistry 2012 Volume 77(Issue 23) pp:10729-10737
Publication Date(Web):November 15, 2012
DOI:10.1021/jo302044n
Density functional theory (DFT) calculations have been performed to provide the first detailed computational study on the mechanism and enantioselectivity for the [4 + 2] cycloaddition reaction of ketenes with N-benzoyldiazenes catalyzed by N-heterocyclic carbenes (NHCs). Two possible mechanisms have been studied: first is the “ketene-first” mechanism (mechanism A), and second is the novel “diazene-first” mechanism (mechanism B). The calculated results reveal that mechanism B is more favorable than mechanism A because it is not only of lower energy barrier but also more consistent with the provided general experimental procedure (Huang, X.-L.; He, L.; Shao, P.-L.; Ye, S. Angew. Chem., Int. Ed.2009, 48, 192–195). The enantioselectivity-determining step is demonstrated to present during the first process of cycloaddition, and the main product configuration is verified to agree with the experimental ee values very well. This study should be of some worth on forecasting how different substituent groups of catalysts and/or reactants affect the enantioselectivity of products. The obtained novel mechanistic insights should be valuable for not only rational design of more efficient NHC catalysts but also understanding the general reaction mechanism of [4 + 2] cycloaddition of ketenes.
Co-reporter:Hongge Zhao;Mingqiong Tong;Juan He
Journal of Molecular Modeling 2012 Volume 18( Issue 3) pp:851-858
Publication Date(Web):2012 March
DOI:10.1007/s00894-011-1119-z
Cyclic peptides are exciting novel hosts for chiral and molecular recognition. In this work, the inclusion complexes of cyclic decapeptide (CDP) with the 1-phenyl-1-propanol enantiomers (E-PP) are firstly studied using the density functional theory (DFT) B3LYP method. Our calculated results indicated that S(-)-1-phenyl-1-propanol (S-PP) could form a more stable inclusion complex with CDP than that of R(+)-1-phenyl-1-propanol (R-PP). The obvious differences in binding energy and thermodynamics data suggest that the cyclic decapeptide could differentiate the two enantiomers. Furthermore, molecular dynamics simulation results have supported the conclusions obtained by DFT. The current investigation shows that cyclic peptide is a desirable host molecule for chiral and molecular recognition.
Co-reporter:Donghui Wei, Yanyan Zhu, Cong Zhang, Dongzhen Sun, Wenjing Zhang, Mingsheng Tang
Journal of Molecular Catalysis A: Chemical 2011 Volume 334(1–2) pp:108-115
Publication Date(Web):4 January 2011
DOI:10.1016/j.molcata.2010.11.004
Recently, N-heterocyclic carbenes (NHCs) have been found to be efficient catalysts for the formal [2 + 2] cycloaddition of aryl(alkyl)ketenes and diazenedicarboxylates to give aza-β-lactams in good enantioselectivity (up to 91% ee) [X.-L. Huang, X.-Y. Chen, S. Ye, J. Org. Chem. 74 (2009) 7585–7587]. However, it is still ambiguous which step is the enantioselectivity-determining step and what the role of NHC catalysts is in this reaction. In this paper, we have suggested a possible mechanism of the title reaction and then theoretically investigated it in detail using density functional theory (DFT). Fully optimized geometries of reactants, products, transition states and intermediates were obtained at the B3LYP/[6-31G (d, p)/LANL2DZ] level of theory, and the results revealed that this reaction had three steps. Our calculated results indicate that the [2 + 2] cycloaddition step is the enantioselectivity-determining step. Moreover, the frontier molecular orbital (FMO) analysis has been carried out to explain why the NHC catalysts can make the [2 + 2] cycloaddition easier to occur, which is mainly due to that the energy gap of FMOs become narrower under the NHC-catalysis condition. Noteworthy, the results of global reactivity indexes analysis are consistent with those of the FMO analysis. Further calculations show that the solvent effect of dichloromethane has no great influence on enantioselectivity of this reaction.Graphical abstractWhy can R1 react with R2 to generate P(R) with a good enantioselectivity under the NHC-catalyzed condition? In this paper, we have investigated the mechanisms of the title reaction using DFT method.Research highlights▶ This study provides a model for predicting the enantioselectivity of the product, which should be helpful in designing other enantioselective catalyst.
Co-reporter:Dongzhen Sun, Yanyan Zhu, Donghui Wei, Cong Zhang, Wenjing Zhang, Mingsheng Tang
Chemical Physics Letters 2010 Volume 495(1–3) pp:33-39
Publication Date(Web):29 July 2010
DOI:10.1016/j.cplett.2010.06.039
Abstract
The multicomponent reaction mechanisms of prop-2-en-1-amine and ethyl propiolate with alloxan were studied using density functional theory. The reaction mechanisms were found to consist of two stages. First, the prop-2-en-1-amine reacts with ethyl propiolate to form an β-aminoacrylate through two competitive channels (channels a and b). Second, a nucleophilic addition of β-aminoacrylate to alloxan occurs via four possible channels (channels b1, b2, b3 and b4). The calculated results revealed that the most energetically favorable path is channel b4 and suggested that the water molecule plays as a proton transfer intermediate in the reaction. Our calculations demonstrated that the reaction occurs easily at room temperature, which agrees well with the experiment.
Co-reporter:Cong Zhang, Yanyan Zhu, Donghui Wei, Dongzhen Sun, Wenjing Zhang and Mingsheng Tang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 8) pp:2913-2919
Publication Date(Web):February 8, 2010
DOI:10.1021/jp910173d
Reaction mechanisms of the 6-benzyl-6-azabicyclo[2.2.1]hept-2-ene with benzoyl isocyanate have been investigated using density functional theory (DFT) at the B3LYP/6-31G(d,p) level of theory. The reaction proceeding along six competitive channels includes two categories. That is, two channels are formally [3,3]-sigmatropic rearrangements and four channels are [4+2] cycloadditions. For urea, the formally [3,3]-sigmatropic rearrangement channel and the [4+2] cycloaddition channels are competitive since they have similar barriers. However, the [4+2] cycloaddition channels are energetically favorable pathways to lead to isourea, with the highest barrier of 12.77 kcal/mol. These polar Diels−Alder (P-DA) reactions are controlled by the charge transfer (CT) at the transition states. Moreover, the main products of this reaction include urea and isourea. Furthermore, difference of two new bond lengths at transition states indicate that the [4+2] cycloadditions in this reaction are asynchronous processes, which is in good agreement with the experiment.
Co-reporter:Donghui Wei, Wenjing Zhang, Yanyan Zhu, Mingsheng Tang
Journal of Molecular Catalysis A: Chemical 2010 326(1–2) pp: 41-47
Publication Date(Web):
DOI:10.1016/j.molcata.2010.04.005
Co-reporter:Mengmeng Zhang, Donghui Wei, Yang Wang, Suiji Li, Jiefei Liu, Yanyan Zhu and Mingsheng Tang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 33) pp:NaN6383-6383
Publication Date(Web):2014/05/14
DOI:10.1039/C4OB00606B
In this paper, two possible mechanisms (mechanisms A and B) on the stereoselective [2 + 2] cycloaddition of aryl(alkyl)ketenes and electron-deficient benzaldehydes catalyzed by N-heterocyclic carbenes (NHCs) have been investigated using density functional theory (DFT). Our calculated results indicate that the favorable mechanism (mechanism A) includes three processes: the first step is the nucleophilic attack on the arylalkylketene by the NHC catalyst to form an intermediate, the second step is the [2 + 2] cycloaddition of the intermediate and benzaldehyde for the formation of a β-lactone, and the last step is the dissociation of the NHC catalyst and the β-lactone. Notably, the [2 + 2] cycloaddition, in which two chiral centers associated with four configurations (SS, RR, SR and RS) are formed, is demonstrated to be both the rate- and stereoselectivity-determining step. Moreover, the reaction pathway associated with the SR configuration is the most favorable pathway and leads to the main product, which is in good agreement with the experimental results. Furthermore, the analysis of global and local reactivity indexes has been performed to explain the role of the NHC catalyst in the [2 + 2] cycloaddition reaction. Therefore, this study will be of great use for the rational design of more efficient catalysts for this kind of cycloaddition.
Co-reporter:Yunxia Li, Yanyan Zhu, Wenjing Zhang, Donghui Wei, Yingying Ran, Qilin Zhao and Mingsheng Tang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 37) pp:NaN20008-20008
Publication Date(Web):2014/07/31
DOI:10.1039/C4CP02186J
Reaction mechanisms of the N-heterocyclic carbene (NHC)-catalyzed dimerization of methyl methacrylate were studied using density functional theory (DFT) at the M05-2X/6-31G(d,p) level of theory. Four possible reaction channels (A, B, C, and D) have been investigated in this work. Particularly, we proposed a novel reaction pathway, where the proton transfers are assisted by a different molecule. The calculated results indicate that the channels B and D are more energetically favourable channels. The obtained results suggest that the E-isomer product is the main product, which is in agreement with the experimental results. Further calculations and analyses of global and local reactivity indices reveal the role of the NHC catalysts in the title reaction. The mechanistic insights gained are valuable for not only rational design of more efficient NHC catalysts but also for understanding the similar reaction mechanism.

![(5aR,10bS)-5a,10b-Dihydro-2-phenyl-4H,6H-indeno[2,1-b][1,2,4]triazolo[4,3-d][1,4]oxazinium Tetrafluoroborate](/data/chemimg/925706-36-1.png)
![(5aR,10bS)-5a,10b-Dihydro-2-phenyl-4H,6H-indeno[2,1-b][1,2,4]triazolo[4,3-d][1,4]oxazinium Tetrafluoroborate](/data/chemimg/925706-36-1_b.png)
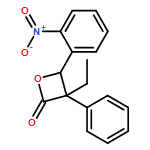
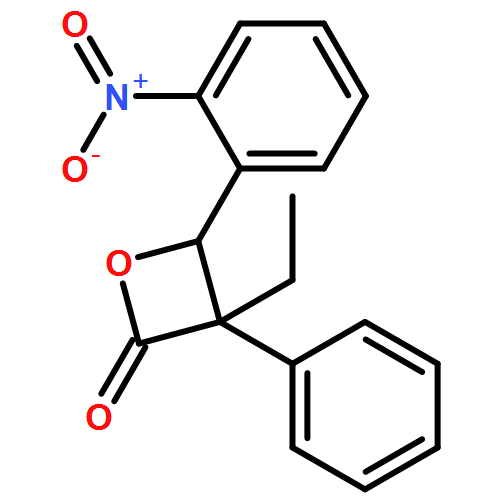


![7-methoxy-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/1206900/1206805-72-2.png)
![7-methoxy-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/1206900/1206805-72-2_b.png)
![7-fluoro-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/1177500/1177455-73-0.png)
![7-fluoro-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/1177500/1177455-73-0_b.png)







![[1,1':2',1''-Terphenyl]-4,4',4'',5'-tetracarboxylic acid (9CI)](/data/chemimg/3723700/623904-16-5.png)
![[1,1':2',1''-Terphenyl]-4,4',4'',5'-tetracarboxylic acid (9CI)](/data/chemimg/3723700/623904-16-5_b.png)


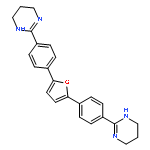
![6-(4,5-dihydro-1H-imidazol-2-yl)-2-{5-[4-(4,5-dihydro-1H-imidazol-2-yl)phenyl]furan-2-yl}-1H-benzimidazole](http://img.cochemist.com/ccimg/216400/216308-23-5.png)
![6-(4,5-dihydro-1H-imidazol-2-yl)-2-{5-[4-(4,5-dihydro-1H-imidazol-2-yl)phenyl]furan-2-yl}-1H-benzimidazole](http://img.cochemist.com/ccimg/216400/216308-23-5_b.png)
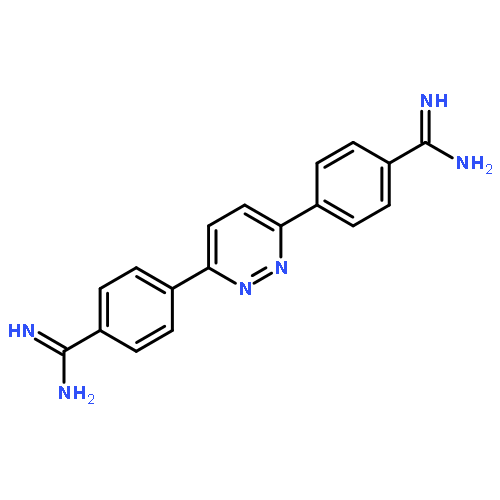
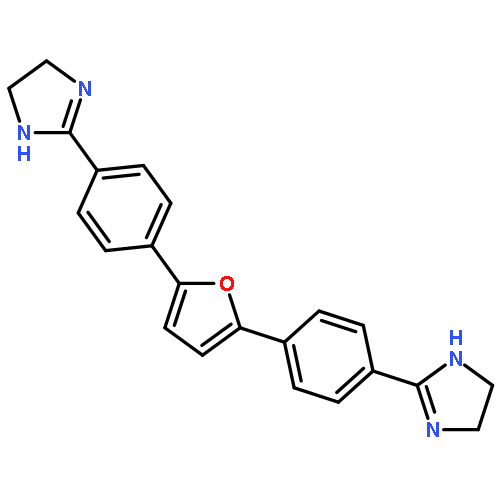
![2-[5-(4-carbamimidoylphenyl)furan-2-yl]-1H-benzimidazole-6-carboximidamide](http://img.cochemist.com/ccimg/216400/216308-19-9.png)
![2-[5-(4-carbamimidoylphenyl)furan-2-yl]-1H-benzimidazole-6-carboximidamide](http://img.cochemist.com/ccimg/216400/216308-19-9_b.png)

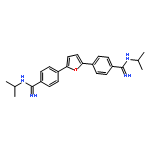
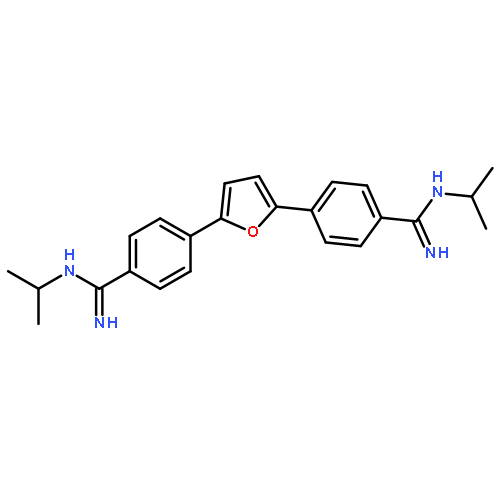






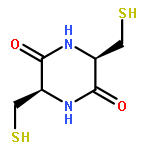
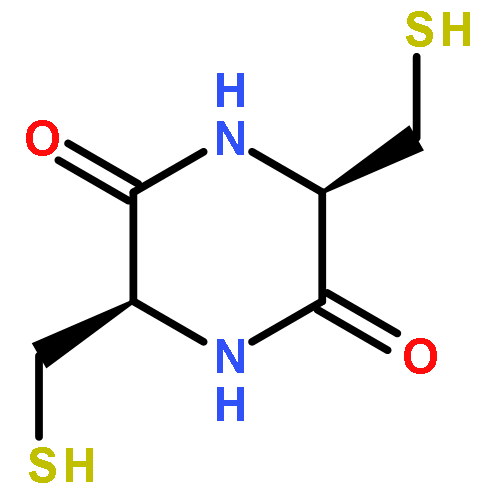


![2,5-Piperazinedione, 3,6-bis[2-(methylthio)ethyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/69800/69744-12-3.png)
![2,5-Piperazinedione, 3,6-bis[2-(methylthio)ethyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/69800/69744-12-3_b.png)

![7-nitro-9H-pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/66900/66889-47-2.png)
![7-nitro-9H-pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/66900/66889-47-2_b.png)
![2,5-Piperazinedione,3,6-bis[(1R)-1-hydroxyethyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/54700/54617-73-1.png)
![2,5-Piperazinedione,3,6-bis[(1R)-1-hydroxyethyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/54700/54617-73-1_b.png)
![2,5-Piperazinedione, 3,6-bis[(1S)-1-methylpropyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/35800/35712-77-7.png)
![2,5-Piperazinedione, 3,6-bis[(1S)-1-methylpropyl]-, (3S,6S)-](http://img.cochemist.com/ccimg/35800/35712-77-7_b.png)
![5-methyl-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/35600/35524-55-1.png)
![5-methyl-9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/35600/35524-55-1_b.png)





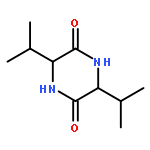
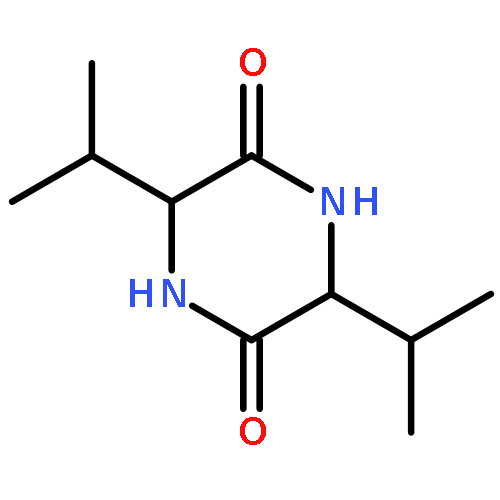
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-phenyl-](http://img.cochemist.com/ccimg/19200/19164-61-5.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-ol, 7-phenyl-](http://img.cochemist.com/ccimg/19200/19164-61-5_b.png)

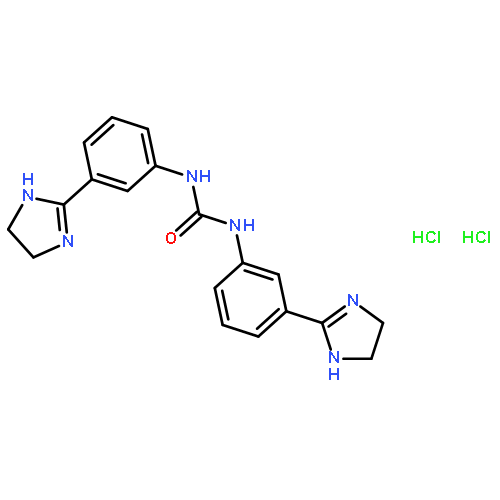




![9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/600/525-24-6.png)
![9H-Pyrrolo[1,2-a]indol-9-one](http://img.cochemist.com/ccimg/600/525-24-6_b.png)