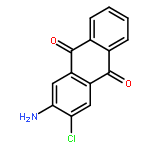
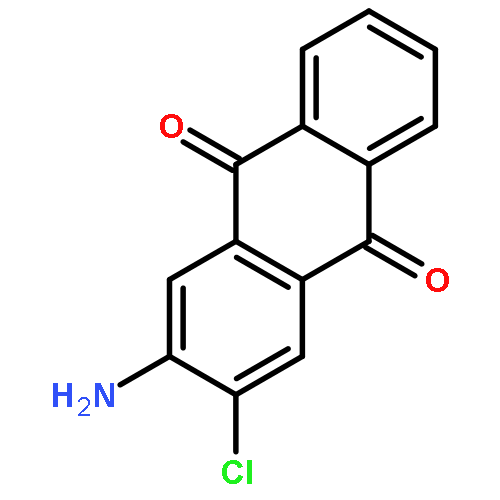
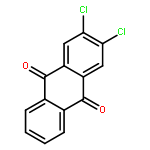
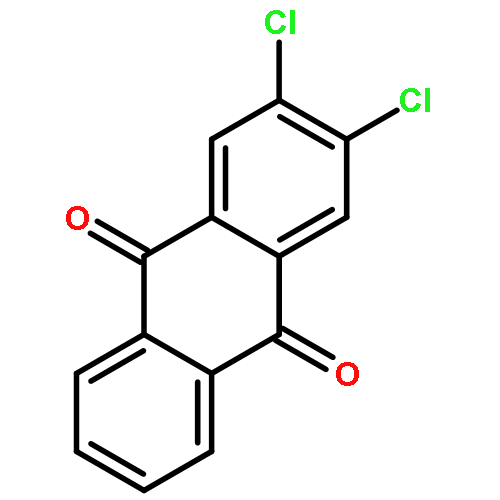
Co-reporter:Jia-Hui Tay, Alonso J. Argüelles, Matthew D. DeMars II, Paul M. Zimmerman, David H. Sherman, and Pavel Nagorny
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8570-8570
Publication Date(Web):June 19, 2017
DOI:10.1021/jacs.7b03198
This work describes the first example of using chiral catalysts to control site-selectivity for the glycosylations of complex polyols such as 6-deoxyerythronolide B and oleandomycin-derived macrolactones. The regiodivergent introduction of sugars at the C3, C5, and C11 positions of macrolactones was achieved by selecting appropriate chiral acids as catalysts or through introduction of stoichiometric boronic acid-based additives. BINOL-based chiral phosphoric acids (CPAs) were used to catalyze highly selective glycosylations at the C5 positions of macrolactones (up to 99:1 rr), whereas the use of SPINOL-based CPAs resulted in selectivity switch and glycosylation of the C3 alcohol (up to 91:9 rr). Additionally, the C11 position of macrolactones was selectively functionalized through traceless protection of the C3/C5 diol with boronic acids prior to glycosylation. Investigation of the reaction mechanism for the CPA-controlled glycosylations revealed the involvement of covalently linked anomeric phosphates rather than oxocarbenium ion pairs as the reactive intermediates.
Co-reporter:Hyungjun Kim, Theodore Goodson III, and Paul M. Zimmerman
The Journal of Physical Chemistry Letters July 20, 2017 Volume 8(Issue 14) pp:3242-3242
Publication Date(Web):June 29, 2017
DOI:10.1021/acs.jpclett.7b01434
Electronic coupling estimates from constrained density functional theory configuration interaction (CDFT-CI) depend critically on choice of density functional. In this Letter, the orbital multielectron self-interaction error (OMSIE), vertical electron affinity (VEA), and vertical ionization potential (VIP) are shown to be the key indicators inherited from the density functional that determine the accuracy of electronic coupling estimates. An error metric η is derived to connect the three properties, based on the linear proportionality between electronic coupling and overlap integral, and the hypothesis that the slope of this line is a function of VEA/VIP, η = (1/Ntestset)Σitestset|−VERef × OMSIE + ΔVE – ΔVE × OMSIE|i. Based on η, BH&HLYP and LRC-ωPBEh are suggested as the best functionals for electron and hole transfer, respectively. Error metric η is therefore a useful predictor of errors in CDFT-CI electronic coupling, showing that the physical correctness of the density functional has a direct effect on the accuracy of the electronic coupling.
Co-reporter:Jacob R. Ludwig, Susan Phan, Christopher C. McAtee, Paul M. Zimmerman, James J. Devery III, and Corinna S. Schindler
Journal of the American Chemical Society August 9, 2017 Volume 139(Issue 31) pp:10832-10832
Publication Date(Web):July 28, 2017
DOI:10.1021/jacs.7b05641
Iron(III)-catalyzed carbonyl-olefin ring-closing metathesis represents a new approach toward the assembly of molecules traditionally generated by olefin–olefin metathesis or olefination. Herein, we report detailed synthetic, spectroscopic, kinetic, and computational studies to determine the mechanistic features imparted by iron(III), substrate, and temperature to the catalytic cycle. These data are consistent with an iron(III)-mediated asynchronous, concerted [2+2]-cycloaddition to form an intermediate oxetane as the turnover-limiting step. Fragmentation of the oxetane via Lewis acid-activation results in the formation of five- and six-membered unsaturated carbocycles.
Co-reporter:M. Ian Childers, Andrew K. Vitek, Lilliana S. Morris, Peter C. B. Widger, Syud M. Ahmed, Paul M. Zimmerman, and Geoffrey W. Coates
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:11048-11048
Publication Date(Web):July 3, 2017
DOI:10.1021/jacs.7b00194
Hydroxy-telechelic poly(propylene oxide) (PPO) is widely used industrially as a midsegment in polyurethane synthesis. These atactic polymers are produced from racemic propylene oxide using chain shuttling agents and double-metal cyanide catalysts. Unlike atactic PPO, isotactic PPO is semicrystalline with a melting temperature of approximately 67 °C. Currently there is no practical route to hydroxy-telechelic isotactic PPO using racemic propylene oxide as the monomer. In this paper, hydroxy-telechelic isotactic PPO is synthesized from racemic propylene oxide with control of molecular weight using enantioselective and isoselective bimetallic catalysts in conjunction with chain shuttling agents. The discovery of an easily accessible bimetallic chromium catalyst is reported for this transformation. Diol, triol, and polymeric chain shuttling agents are used to give hydroxy-telechelic isotactic PPO of varying functionality and structure. Detailed quantum chemical studies are used to reveal the polymerization mechanism and origin of stereoselectivity.
Co-reporter:Amanda L. Dewyer
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 3) pp:501-504
Publication Date(Web):2017/01/18
DOI:10.1039/C6OB02183B
Chemical reaction mechanisms have been frequently studied using computational simulations, but these tools have been primarily effective at examining reaction paths within the scope of chemical intuition. To determine reaction mechanisms that were not already preconceived by chemists, nonstandard simulation tools are required. This perspective introduces new methods developed by the Zimmerman group that are designed to uncover sequences of elementary steps, from first principles and without substantial human guidance. Results from the areas of organo catalysis and transition metal catalysis indicate that new frontiers of knowledge will be gained through continued development and application of reaction discovery simulation techniques.
Co-reporter:Ian M. Pendleton; Mónica H. Pérez-Temprano; Melanie S. Sanford
Journal of the American Chemical Society 2016 Volume 138(Issue 18) pp:6049-6060
Publication Date(Web):April 18, 2016
DOI:10.1021/jacs.6b02714
This report describes a combined experimental and computational investigation of the mechanism of C(sp3)–N bond-forming reductive elimination from sulfonamide-ligated PdIV complexes. After an initial experimental assessment of reactivity, we used ZStruct, a computational combinatorial reaction finding method, to analyze a large number of multistep mechanisms for this process. This study reveals two facile isomerization pathways connecting the experimentally observed PdIV isomers, along with two competing SN2 pathways for C(sp3)–N coupling. One of these pathways involves an unanticipated oxygen–nitrogen exchange of the sulfonamide ligand prior to an inner-sphere SN2-type reductive elimination. The calculated ΔG⧧ values for isomerization and reductive elimination with a series of sulfonamide derivatives are in good agreement with experimental data. Furthermore, the simulations predict relative reaction rates with different sulfonamides, which is successful only after considering competition between the proposed operating mechanisms. Overall, this work shows that the combination of experimental studies and new computational tools can provide fundamental mechanistic insights into complex organometallic reaction pathways.
Co-reporter:Paul M. Zimmerman and Peter Smereka
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:1883-1891
Publication Date(Web):February 25, 2016
DOI:10.1021/acs.jctc.5b01168
The choice of coordinate system strongly affects the convergence properties of vibrational structure computations. Two methods for efficient generation of improved vibrational coordinates are presented and justified by analysis of a model anharmonic two-mode Hessian and numerical computations on polyatomic molecules. To produce optimal coordinates, metrics which quantify off-diagonal couplings over a grid of Hessian matrices are minimized through unitary rotations of the vibrational basis. The first proposed metric minimizes the total squared off-diagonal coupling, and the second minimizes the total squared change in off-diagonal coupling. In this procedure certain anharmonic modes tend to localize, for example X–H stretches. The proposed methods do not rely on prior fitting of the potential energy, vibrational structure computations, or localization metrics, so they are unique from previous vibrational coordinate generation algorithms and are generally applicable to polyatomic molecules. Fitting the potential to the approximate n-mode representation in the optimized bases for all-trans polyenes shows that off-diagonal anharmonic couplings are substantially reduced by the new choices of coordinate system. Convergence of vibrational energies is examined in detail for ethylene, and it is shown that coupling-optimized modes converge in vibrational configuration interaction computations to within 1 cm–1 using only 3-mode couplings, where normal modes require 4-mode couplings for convergence. Comparison of the vibrational configuration interaction convergence with respect to excitation level for the two proposed metrics shows that minimization of the total off-diagonal coupling is most effective for low-cost vibrational structure computations.
Co-reporter:Maxwell W. Li, Ian M. Pendleton, Alex J. Nett, and Paul M. Zimmerman
The Journal of Physical Chemistry A 2016 Volume 120(Issue 8) pp:1135-1144
Publication Date(Web):February 4, 2016
DOI:10.1021/acs.jpca.5b11156
This study employs computational reaction finding tools to probe the unique biphilic reactivity between ammonia-borane (AB) and CO2. The results show that sequential reactions involving multiple equivalents of AB and CO2 can lead to the formation of stable nonplanar B,C,N,O-heterocycles (Cy-BCN). Cy-BCN is shown to emerge through boron–oxygen bond formation, hydroboration, dative bond formation, and single- or double-hydrogen transfers. The most kinetically facile reactions (computed at the coupled cluster singles and doubles with perturbative triples (CCSD(T)) level of theory) result from polarized nitrogen–boron double bonds whereas thermodynamic stability results from formation of covalent boron–oxygen bonds. An important structure, HCOOBHNH2 (DHFAB), contains both of these features and is the key intermediate involved in generation of Cy-BCN. Crucially, it is shown that favorable boron–oxygen bond formation results in production of Cy-BCN species that are more stable than polyaminoboranes. These types of reaction intermediates could serve as building blocks in the formation of B,N-codoped graphene oxide (BCN).
Co-reporter:Yu Zhao, Alex J. Nett, Anne J. McNeil, and Paul M. Zimmerman
Macromolecules 2016 Volume 49(Issue 20) pp:7632-7641
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.macromol.6b01648
The living, chain-growth polymerization of (hetero)aryl monomers by carbene-ligated palladium precatalysts gives conjugated polymers with narrow dispersities as well as specified molecular weights and sequences. Despite this success, the mechanism for Pd precatalyst initiation and subsequent polymer growth is unknown. A quantum chemical study is presented herein to provide insight into thiophene homopolymerization initiated by (IPr)PdCl2(3-chloropyridine) (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) using novel reaction discovery tools for generating and evaluating mechanistic hypotheses. This study reveals the role for the “throw-away” 3-chloropyridine ligand during initiation via a stepwise transmetalation pathway that involves pyridine dissociation, followed by pyridine reassociation to liberate the Grignard byproduct (MgCl2) and stabilize the resulting Pd intermediate. In contrast, 3-chloropyridine association to Pd during propagation hinders catalyst turnover by producing a low-energy, off-cycle intermediate. Throughout these studies, explicit solvent molecules (THF) coordinated to the Grignard reagent are shown to be needed for accurate modeling. Finally, the energetic span model (ESM) reveals that the turnover-limiting step for initiation is transmetalation with pyridine dissociation/reassociation, while the turnover-limiting step for propagation is also transmetalation that is inhibited by pyridine coordination.
Co-reporter:Mitchell L. Smith, Amanda K. Leone, Paul M. Zimmerman, and Anne J. McNeil
ACS Macro Letters 2016 Volume 5(Issue 12) pp:
Publication Date(Web):December 9, 2016
DOI:10.1021/acsmacrolett.6b00886
Polymerizing electron-deficient arenes in a controlled, chain-growth fashion remains a significant challenge despite a decade of research on catalyst-transfer polycondensation. The prevailing hypothesis is that the chain-growth mechanism stalls at a strongly associated metal–polymer π-complex, preventing catalyst turnover. To evaluate this hypothesis, we performed mechanistic studies using thiazole derivatives and identified approaches to improve their chain-growth polymerization. These studies revealed a surprisingly high barrier for chain-walking toward the reactive C–X bond. In addition, a competitive pathway involving chain-transfer to monomer was identified. This pathway is facilitated by ancillary ligand dissociation and N-coordination to the incoming monomer. We found that this chain-transfer pathway can be attenuated by using a rigid ancillary ligand, leading to an improved polymerization. Combined, these studies provide mechanistic insight into the challenges associated with electron-deficient monomers as well as ways to improve their living, chain-growth polymerization. Our mechanistic studies also revealed an unexpected radical anion-mediated oligomerization in the absence of catalyst, as well as a surprising oxidative addition into the thiazole C–S bond in a model system.
Co-reporter:Hyungjun Kim, Theodore Goodson III, and Paul M. Zimmerman
The Journal of Physical Chemistry C 2016 Volume 120(Issue 39) pp:22235-22247
Publication Date(Web):September 13, 2016
DOI:10.1021/acs.jpcc.6b07558
Co-reporter:Joshua A. KammeraadPaul M. Zimmerman
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 24) pp:5074-5079
Publication Date(Web):November 23, 2016
DOI:10.1021/acs.jpclett.6b02501
The difference gradient and derivative coupling vectors span the branching planes of conical intersections between electronic states. While gradients are commonly available in many electronic structure methods, the derivative coupling vectors are not always implemented and ready for use in characterizing conical intersections. This Letter shows how the derivative coupling vectors can be computed to high accuracy (direction and magnitude) using energy and gradient information. The new method is based on the combination of a linear-coupling two-state Hamiltonian and a finite-difference Davidson approach for computing the branching plane. Benchmark cases are provided showing these vectors can be efficiently computed near conical intersections.
Co-reporter:Alex J. Nett; Wanxiang Zhao; Paul M. Zimmerman;John Montgomery
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7636-7639
Publication Date(Web):June 9, 2015
DOI:10.1021/jacs.5b04548
An inhibitory role of 1,5-cyclooctadiene (COD) in nickel-catalyzed C–H functionalization processes was identified and studied. The bound COD participates in C–H activation by capturing the hydride, leading to a stable off-cycle π-allyl complex that greatly diminished overall catalytic efficiency. Computational studies elucidated the origin of the effect and enabled identification of a 1,5-hexadiene-derived pre-catalyst that avoids the off-cycle intermediate and provides catalytic efficiencies that are superior to those of catalysts derived from Ni(COD)2.
Co-reporter:Alan D. Chien
The Journal of Physical Chemistry C 2015 Volume 119(Issue 51) pp:28258-28268
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.jpcc.5b07786
Co-reporter:Dr. Zhankui Sun;Grace A. Winschel;Dr. Paul M. Zimmerman;Dr. Pavel Nagorny
Angewandte Chemie International Edition 2014 Volume 53( Issue 42) pp:11194-11198
Publication Date(Web):
DOI:10.1002/anie.201405128
Abstract
An enantioselective intramolecular chiral phosphoric acid-catalyzed cyclization of unsaturated acetals has been utilized for the synthesis of functionalized chiral piperidines. The chiral enol ether products of these cyclizations undergo subsequent in situ enantioenrichment through acetalization of the minor enantiomer. A new computational reaction exploration method was utilized to elucidate the mechanism and stereoselectivity of this transformation. Rather than confirming the originally postulated cyclization proceeding directly through a vinyl oxocarbenium ion, simulations identified an alternative two-step mechanism involving the formation of a mixed chiral phosphate acetal, which undergoes a concerted, asynchronous SN2′-like displacement to yield the product with stereoselectivity in agreement with experimental observations.
Co-reporter:Dr. Zhankui Sun;Grace A. Winschel;Dr. Paul M. Zimmerman;Dr. Pavel Nagorny
Angewandte Chemie 2014 Volume 126( Issue 42) pp:11376-11380
Publication Date(Web):
DOI:10.1002/ange.201405128
Abstract
An enantioselective intramolecular chiral phosphoric acid-catalyzed cyclization of unsaturated acetals has been utilized for the synthesis of functionalized chiral piperidines. The chiral enol ether products of these cyclizations undergo subsequent in situ enantioenrichment through acetalization of the minor enantiomer. A new computational reaction exploration method was utilized to elucidate the mechanism and stereoselectivity of this transformation. Rather than confirming the originally postulated cyclization proceeding directly through a vinyl oxocarbenium ion, simulations identified an alternative two-step mechanism involving the formation of a mixed chiral phosphate acetal, which undergoes a concerted, asynchronous SN2′-like displacement to yield the product with stereoselectivity in agreement with experimental observations.
Co-reporter:Paul M. Zimmerman, Charles B. Musgrave, and Martin Head-Gordon
Accounts of Chemical Research 2013 Volume 46(Issue 6) pp:1339
Publication Date(Web):February 21, 2013
DOI:10.1021/ar3001734
Singlet fission occurs when a single exciton splits into multiple electron-hole pairs, and could dramatically increase the efficiency of organic solar cells by converting high energy photons into multiple charge carriers. Scientists might exploit singlet fission to its full potential by first understanding the underlying mechanism of this quantum mechanical process. The pursuit of this fundamental mechanism has recently benefited from the development and application of new correlated wave function methods. These methods—called restricted active space spin flip—can capture the most important electron interactions in molecular materials, such as acene crystals, at low computational cost. It is unrealistic to use previous wave function methods due to the excessive computational cost involved in simulating realistic molecular structures at a meaningful level of electron correlation.In this Account, we describe how we use these techniques to compute single exciton and multiple exciton excited states in tetracene and pentacene crystals in order to understand how a single exciton generated from photon absorption undergoes fission to generate two triplets. Our studies indicate that an adiabatic charge transfer intermediate is unlikely to contribute significantly to the fission process because it lies too high in energy. Instead, we propose a new mechanism that involves the direct coupling of an optically allowed single exciton to an optically dark multiexciton. This coupling is facilitated by intermolecular motion of two acene monomers that drives nonadiabatic population transfer between the two states. This transfer occurs in the limit of near degeneracies between adiabatic states where the Born–Oppenheimer approximation of fixed nuclei is no longer valid. Existing theories for singlet fission have not considered this type of coupling between states and, therefore, cannot describe this mechanism.The direct mechanism through intermolecular motion describes many experimentally observed characteristics of these materials, such as the ultrafast time scale of photobleaching and triplet generation during singlet fission in pentacene. We believe this newly discovered mechanism provides fundamental insight to guide the creation of new solar materials that exhibit high efficiencies through multiple charge generation.
Co-reporter:Paul Zimmerman
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 7) pp:3043-3050
Publication Date(Web):June 11, 2013
DOI:10.1021/ct400319w
The growing string method (GSM) is highly useful for locating reaction paths connecting two molecular intermediates. GSM has often been used in a two-step procedure to locate exact transition states (TS), where GSM creates a quality initial structure for a local TS search. This procedure and others like it, however, do not always converge to the desired transition state because the local search is sensitive to the quality of the initial guess. This article describes an integrated technique for simultaneous reaction path and exact transition state search. This is achieved by implementing an eigenvector following optimization algorithm in internal coordinates with Hessian update techniques. After partial convergence of the string, an exact saddle point search begins under the constraint that the maximized eigenmode of the TS node Hessian has significant overlap with the string tangent near the TS. Subsequent optimization maintains connectivity of the string to the TS as well as locks in the TS direction, all but eliminating the possibility that the local search leads to the wrong TS. To verify the robustness of this approach, reaction paths and TSs are found for a benchmark set of more than 100 elementary reactions.
Co-reporter:Yaroslav Ya. Khomutnyk; Alonso J. Argüelles; Grace A. Winschel; Zhankui Sun; Paul M. Zimmerman;Pavel Nagorny
Journal of the American Chemical Society () pp:
Publication Date(Web):December 7, 2015
DOI:10.1021/jacs.5b12528
Mechanistic and computational studies were conducted to elucidate the mechanism and the origins of enantiocontrol for asymmetric chiral phosphoric acid-catalyzed spiroketalization reactions. These studies were designed to differentiate between the SN1-like, SN2-like, and covalent phosphate intermediate-based mechanisms. The chiral phosphoric acid-catalyzed spiroketalization of deuterium-labeled cyclic enol ethers revealed a highly diastereoselective syn-selective protonation/nucleophile addition, thus ruling out long-lived oxocarbenium intermediates. Hammett analysis of the reaction kinetics revealed positive charge accumulation in the transition state (ρ = −2.9). A new computational reaction exploration method along with dynamics simulations supported an asynchronous concerted mechanism with a relatively short-lived polar transition state (average lifetime = 519 ± 240 fs), which is consistent with the observed inverse secondary kinetic isotope effect of 0.85. On the basis of these studies, a transition state model explaining the observed stereochemical outcome has been proposed. This model predicts the enantioselective formation of the observed enantiomer of the product with 92% ee, which matches the experimentally observed value.
Co-reporter:Amanda L. Dewyer and Paul M. Zimmerman
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 3) pp:NaN504-504
Publication Date(Web):2016/12/12
DOI:10.1039/C6OB02183B
Chemical reaction mechanisms have been frequently studied using computational simulations, but these tools have been primarily effective at examining reaction paths within the scope of chemical intuition. To determine reaction mechanisms that were not already preconceived by chemists, nonstandard simulation tools are required. This perspective introduces new methods developed by the Zimmerman group that are designed to uncover sequences of elementary steps, from first principles and without substantial human guidance. Results from the areas of organo catalysis and transition metal catalysis indicate that new frontiers of knowledge will be gained through continued development and application of reaction discovery simulation techniques.
.jpg)






























































![DICHLORO-[1,3-BIS(DIISOPROPYLPHENYL)-2-IMIDAZOLIDINYLIDENE]-(3-CHLOROPYRIDYL)PALLADIUM(II)](http://img.cochemist.com/ccimg/927800/927706-57-8.png)
![DICHLORO-[1,3-BIS(DIISOPROPYLPHENYL)-2-IMIDAZOLIDINYLIDENE]-(3-CHLOROPYRIDYL)PALLADIUM(II)](http://img.cochemist.com/ccimg/927800/927706-57-8_b.png)
![CARBAMIC ACID, [[1-(3-BUTENYL)CYCLOHEXYL]METHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/887200/887131-50-2.png)
![CARBAMIC ACID, [[1-(3-BUTENYL)CYCLOHEXYL]METHYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/887200/887131-50-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bS)-](/data/chemimg/108900/878111-17-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bS)-](/data/chemimg/108900/878111-17-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)




![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1_b.png)







![SPIRO[ISOBENZOFURAN-1(3H),2'-[2H]PYRAN], 3',4',5',6'-TETRAHYDRO-](http://img.cochemist.com/ccimg/139700/139697-95-3.png)
![SPIRO[ISOBENZOFURAN-1(3H),2'-[2H]PYRAN], 3',4',5',6'-TETRAHYDRO-](http://img.cochemist.com/ccimg/139700/139697-95-3_b.png)
![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1.png)
![Benzene, [(1E)-4-bromo-1-methyl-1-butenyl]-](http://img.cochemist.com/ccimg/129000/128902-50-1_b.png)



![9,10-Anthracenedione,1,4-bis[[2-(4-hydroxyphenyl)ethyl]amino]-](http://img.cochemist.com/ccimg/97000/96969-83-4.png)
![9,10-Anthracenedione,1,4-bis[[2-(4-hydroxyphenyl)ethyl]amino]-](http://img.cochemist.com/ccimg/97000/96969-83-4_b.png)

















![1-amino-9,10-dihydro-4-[[4-[(methylamino)methyl]phenyl]amino]-9,10-dioxoanthracene-2-sulphonic acid](http://img.cochemist.com/ccimg/64200/64135-01-9.png)
![1-amino-9,10-dihydro-4-[[4-[(methylamino)methyl]phenyl]amino]-9,10-dioxoanthracene-2-sulphonic acid](http://img.cochemist.com/ccimg/64200/64135-01-9_b.png)







![[1,1'-Biphenyl]-4-propanoicacid, b-oxo-, ethyl ester](http://img.cochemist.com/ccimg/57500/57477-98-2.png)
![[1,1'-Biphenyl]-4-propanoicacid, b-oxo-, ethyl ester](http://img.cochemist.com/ccimg/57500/57477-98-2_b.png)















![1-hydroxy-5-[(1-methylethyl)amino]anthracene-9,10-dione](http://img.cochemist.com/ccimg/34000/33902-49-7.png)
![1-hydroxy-5-[(1-methylethyl)amino]anthracene-9,10-dione](http://img.cochemist.com/ccimg/34000/33902-49-7_b.png)













![9,10-Anthracenedione, 1,5-bis[(2-phenylethyl)amino]-](http://img.cochemist.com/ccimg/15900/15830-98-5.png)
![9,10-Anthracenedione, 1,5-bis[(2-phenylethyl)amino]-](http://img.cochemist.com/ccimg/15900/15830-98-5_b.png)





![2-Anthracenesulfonicacid, 4-[[4-(acetylamino)phenyl]amino]-1-amino-9,10-dihydro-9,10-dioxo-](http://img.cochemist.com/ccimg/6300/6247-34-3.png)
![2-Anthracenesulfonicacid, 4-[[4-(acetylamino)phenyl]amino]-1-amino-9,10-dihydro-9,10-dioxo-](http://img.cochemist.com/ccimg/6300/6247-34-3_b.png)





![9,10-ANTHRACENEDIONE, 1-[(3-METHOXYPROPYL)AMINO]-](http://img.cochemist.com/ccimg/5000/4946-81-0.png)
![9,10-ANTHRACENEDIONE, 1-[(3-METHOXYPROPYL)AMINO]-](http://img.cochemist.com/ccimg/5000/4946-81-0_b.png)




![5-METHYL-2-[[4-(4-METHYL-2-SULFOANILINO)-9,10-DIOXOANTHRACEN-1-YL]AMINO]BENZENESULFONIC ACID](http://img.cochemist.com/ccimg/3500/3443-90-1.png)
![5-METHYL-2-[[4-(4-METHYL-2-SULFOANILINO)-9,10-DIOXOANTHRACEN-1-YL]AMINO]BENZENESULFONIC ACID](http://img.cochemist.com/ccimg/3500/3443-90-1_b.png)









![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)