Co-reporter:Chao-Yun Cai, Li Rao, Yong Rao, Jin-Xuan Guo, Zhi-Zun Xiao, Jing-Yu Cao, Zhi-Shu Huang, Bo Wang
European Journal of Medicinal Chemistry 2017 Volume 130(Volume 130) pp:
Publication Date(Web):21 April 2017
DOI:10.1016/j.ejmech.2017.02.007
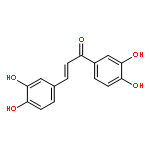
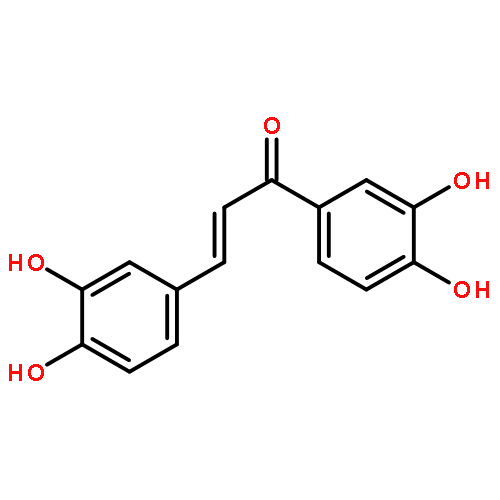
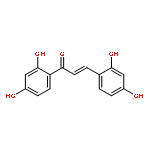
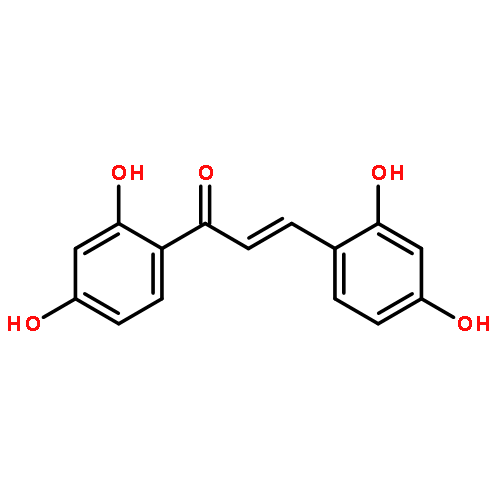
•Twenty-six chalcone and bis-chalcone derivatives were synthesized and four of them are first reported.•Some compounds exhibited excellent inhibitory activities against α-glucosidase, the best IC50 value reaches to 1.0 μM.•Valuable structure-activity relationship was obtained.•The selected compounds (2c, 2g, 2j, 2l) were showed as non-competitive inhibitors.•The compound 2g had a better effect than the positive control 1-deoxynojirimycin on reducing the glucose level in HepG-2 cells and could be a potential drug candidates for treating diabetes.Two series of compounds (chalcones and bis-chalcones) were designed, synthesized, and evaluated as α-glucosidase inhibitors (AGIs) with 1-deoxynojirimycin as positive control in vitro. Most of the compounds with two or four hydroxyl groups showed better inhibitory activities than 1-deoxynojirimycin towards α-glucosidase with noncompetitive mechanism. Moreover, most of the hydroxy bis-chalcones exhibit good α-glucosidase inhibitory activities in enzyme test. Inspiringly, bis-chalcones 2g (at 1 μM concentration) has stronger effect than 1-deoxynojirimycin on reducing the glucose level in HepG-2 cells (human liver cancer cell line).Two series of chalcones and bis-chalcones were synthesized and exhibited α-glucosidase inhibition via a non-competitive mechanism. Moreover, the compound 2g could significantly reduce the glucose level in HepG-2 cells.Download high-res image (126KB)Download full-size image
Co-reporter:Gai-Li Li, Chao-Yun Cai, Jia-Yun He, Li Rao, Lin Ma, Yan Liu, Bo Wang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 7) pp:1431-1438
Publication Date(Web):1 April 2016
DOI:10.1016/j.bmc.2016.01.022
Considerable interest has been attracted in xanthone and its derivatives because of their important biological activities. In this paper, a series of novel 3-arylacyloxyxanthone derivatives 2a–p were synthesized and evaluated for their biological activities toward α-glucosidase. In comparison to the parent 1,3-dihydroxylxanthone 1a, 3-arylacyloxy derivatives 2a–p with additional aromatic ester groups at 3-position show up to 13.7-fold higher inhibitory activities. In particular, the IC50 values of compounds 2i, 2m, 2p reach 13.3, 10.6, 11.6 μM, respectively. These results suggest that addition of aromatic moieties by esterification at the 3-OH of the parent 1,3-dihydroxylxanthone is an efficient way to increase the inhibition against α-glucosidase. Different from previous multi-hydroxylxanthones, these 3-arylacyloxyxanthone derivatives show efficient inhibitory activities may due to the π-stacking or hydrophobic effects of the additional aromatic moieties rather than the H-bonding donor interaction of 3-OH. Structure–activity relationship analysis shows that the substituents on the additional aromatic ring also influence the inhibition. All the oxygen or nitrogen-containing groups, like hydroxyl, methoxy, methaminyl, and alkylsilyloxy, can enhance the inhibitory activities. In addition, the kinetics of enzyme inhibition measured by using Lineweaver–Burk plots shows that selected compounds 2i, 2m and 2p are non-competitive inhibitors. Docking simulations further support our structure–activity relationship analysis that additional aromatic moieties enhance inhibitory activities via hydrophobic effects. The new developed 3-arylacyloxyxanthone derivatives probably bind with α-glucosidase in an allosteric site different from traditional multi-hydroxylxanthones.
Co-reporter:Yan Liu, Lin Ma, Wen-Hua Chen, Hwangseo Park, Zhuofeng Ke, and Bo Wang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 43) pp:13464-13471
Publication Date(Web):October 1, 2013
DOI:10.1021/jp4067235
Newly emerged xanthone derivatives have attracted considerable interests as a novel class of potent α-glucosidase inhibitors. To provide insights into the inhibitory and binding mechanisms of xanthone-based inhibitors toward α-glucosidase, we carried out experimental and theoretical studies on two typical xanthone derivatives, i.e., 1,3,7-trihydroxyxanthone and 1,3-dihydroxybenzoxanthone. The results indicate that these two xanthone derivatives belong to noncompetitive inhibitors and induce a loss in the α-helix content of the secondary structure of α-glucosidase. Docking simulation revealed the existence of multiple binding modes, in which polyhydroxyl groups and expanded aromatic rings acted as two key pharmacophores to form H-bonding and π–π stacking interactions with α-glucosidase. The fact that 1,3,7-tridroxyxanthone and 1,3-dihydroxybenzoxanthone exhibited significant synergetic inhibition to α-glucosidase strongly suggests that both xanthone derivatives simultaneously bind to the distinct noncompetitive sites of yeast’s α-glucosidase. On the basis of the plausible binding clues, synergetic inhibition can be developed to be a promising strategy to achieve enhanced inhibitory activities.
Co-reporter:Gai-Li Li, Jia-Yun He, Aiqin Zhang, Yiqian Wan, Bo Wang, Wen-Hua Chen
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 9) pp:4050-4055
Publication Date(Web):September 2011
DOI:10.1016/j.ejmech.2011.06.003
A series of novel xanthone derivatives 6–16 having non-coplanar and flexible structures were synthesized as potent α-glucosidase inhibitors. Biological evaluation indicated that compounds 6–12 bearing one or two naphthol moieties exhibited up to 30-fold enhanced activities compared with their corresponding parent compounds 2–5, whereas compounds 13–16 bearing one dihydroxylnaphthalenyl group showed decreased activities compared with their corresponding analogs 6–9 having one naphthol group. Among them, compounds 7–8, 10–12 and 15 were more active than 1-deoxynojirimycin, a well-known inhibitor for α-glucosidase. The structure–activity correlations suggested that inhibiting of α-glucosidase was a result of multiple interactions with the enzyme, including π-stacking, hydrophobic effect and conformational flexibility due to the structural non-coplanarity. In addition, compounds 4, 8 and 15 showed non-competitive inhibition.Highlights► Non-coplanar and flexible xanthone derivatives were synthesized in 25–50%. ► The inhibitory activities against yeast’s α-glucosidase were up to 30-fold enhanced. ► π-Stacking, hydrophobic effect and conformational flexibility were found to be predominant factors for modulating the activity. ► Compounds 4, 8 and 15 were found to act as non-competitive inhibitors.
Co-reporter:Quan-Guan Su;Yan Liu;Yu-Chen Cai;Yue-Li Sun
Investigational New Drugs 2011 Volume 29( Issue 6) pp:1230-1240
Publication Date(Web):2011 December
DOI:10.1007/s10637-010-9468-5
To explore the potential anti-tumour activities of xanthone derivatives, 26 hydroxylxanthones and benzoxanthones and their structurally modified analogues were examined for potential cytotoxic activities against eight human cancer cell lines. Most of the xanthone derivatives exhibited a higher degree of cytotoxicity on HepG2 cells than on the other seven cancer cell lines. Compound 24 (1,3,7-Trihydroxy-12H-benzo[b] xanthen-12-one) showed the highest degree of cytotoxicity of the tested compounds against HepG2 cells and demonstrated good tumour specificity by exhibiting a much higher degree of cytotoxicity against HepG2 cells than against normal liver cells (L02). Several valuable structure-activity relationships were derived from the cytotoxicity data. In addition, we found that compound 24 could downregulate the expression of the Mcl-1 protein, induce changes in the mitochondrial membrane potential and induce apoptosis in HepG2 cells via the mitochondrial pathway. Compound 24 was also shown to inhibit topoisomerase (topo) II activity and downregulate the levels of both topo II mRNA and protein in HepG2 cells. The present results suggest that due to its potent cytotoxicity and good tumour selectivity, compound 24 may be exploited as a potential lead compound in the development of a new anti-tumour agent with specific activity against liver cancer.

![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7.png)
![Rhodium, tetrakis[m-[(aS)-1,3-dihydro-1,3-dioxo-a-tricyclo[3.3.1.13,7]dec-1-yl-2H-isoindole-2-acetato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/909400/909389-99-7_b.png)
![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6.png)
![Rhodium, tetrakis[m-[1-[(4-dodecylphenyl)sulfonyl]-L-prolinato-kO2:kO2']]di-, (Rh-Rh)](http://img.cochemist.com/ccimg/179200/179162-34-6_b.png)




![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/194200/194156-28-0.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/194200/194156-28-0_b.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7_b.png)


![TETRAKIS{1-[(4-DODECYLPHENYL)SULFONYL]-(2S)-PROLINATE} DIRHODIUM](http://img.cochemist.com/ccimg/179200/179162-32-4.png)
![TETRAKIS{1-[(4-DODECYLPHENYL)SULFONYL]-(2S)-PROLINATE} DIRHODIUM](http://img.cochemist.com/ccimg/179200/179162-32-4_b.png)











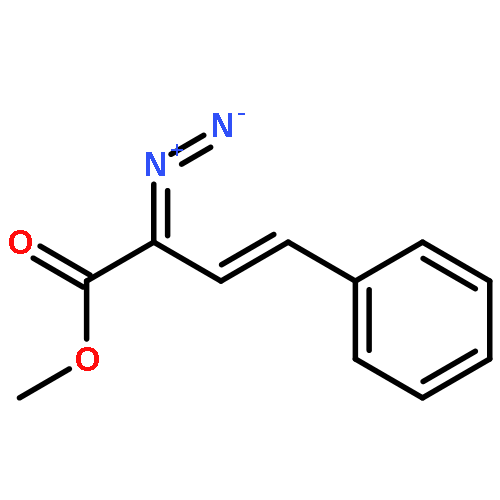




![Ethanone, 1-[5-methoxy-2-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/92000/91971-19-6.png)
![Ethanone, 1-[5-methoxy-2-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/92000/91971-19-6_b.png)
![Benzoic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/83500/83405-98-5.png)
![Benzoic acid, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/83500/83405-98-5_b.png)
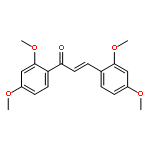



![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3_b.png)
















![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5.png)
![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5_b.png)

