Co-reporter:Zhi Lin, Jia Ji, Shuaixiang Zhou, Fang Zhang, Jiequn Wu, Yinlong Guo, and Wen Liu
Journal of the American Chemical Society September 6, 2017 Volume 139(Issue 35) pp:12105-12105
Publication Date(Web):August 18, 2017
DOI:10.1021/jacs.7b05337
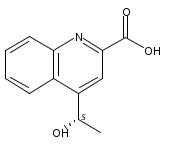
Thiostrepton (TSR), an archetypal member of the family of ribosomally synthesized and post-translationally modified thiopeptide antibiotics, possesses a biologically important quinaldic acid (QA) moiety within the side-ring system of its characteristic thiopeptide framework. QA is derived from an independent l-Trp residue; however, its associated transformation process remains poorly understood. We here report that during the formation of QA, the key expansion of an indole to a quinoline relies on the activities of the pyridoxal-5′-phosphate-dependent protein TsrA and the flavoprotein TsrE. These proteins act in tandem to process the precursor 2-methyl- l-Trp through reversible transamination and selective oxygenation, thereby initiating a highly reactive rearrangement in which selective C2–N1 bond cleavage via hydrolysis for indole ring-opening is closely coupled with C2′–N1 bond formation via condensation for recyclization and ring expansion in the production of a quinoline ketone intermediate. This indole ring-expansion mechanism is unusual, and represents a new strategy found in nature for l-Trp-based functionalization.
Co-reporter:Zhi Lin, Qingli He, Wen Liu
Current Opinion in Biotechnology 2017 Volume 48(Volume 48) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.copbio.2017.06.008
•The specialization of a common thiopeptide framework leads to individual members.•Bio-inspired engineering of thiopeptides expands molecular diversity and utility.•Thiopeptide antibiotics feature a highly evolvable ‘template’-biosynthetic logic.•Future thiopeptide engineering relies on the further appreciation of this logic.Thiopeptide antibiotics, which are a class of sulfur-rich and highly modified peptide natural products, exhibit a wide variety of important biological properties. These antibiotics are ribosomally synthesized and arise from post-translational modifications, exemplifying a process through which nature develops the structural complexity from Ser/Thr and Cys-rich precursor peptides. Following a brief review of the knowledge gained from nature in terms of the formation of a common thiopeptide scaffold and its specialization to individual members, we highlight the significance of bio-inspired engineering, which has greatly expanded the molecular diversity and utility of thiopeptide antibiotics regarding the search for clinically useful agents, investigation into new mechanisms of action and access to typically ‘inaccessible’ biosynthetic processes over the past two years.Download high-res image (194KB)Download full-size image
Co-reporter:Qingfei Zheng;Hui Fang
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 16) pp:3376-3390
Publication Date(Web):2017/04/18
DOI:10.1039/C7OB00466D
Thiopeptide antibiotics are a class of typical ribosomally synthesized and post-translationally modified peptides (RiPPs) with complex chemical structures that are difficult to construct via chemical synthesis. To date, more than 100 thiopeptides have been discovered, and most of these compounds exhibit remarkable biological activities, such as antibacterial, antitumor and immunosuppressive activities. Therefore, studies of the biosynthesis of thiopeptides can contribute to the development of new drug leads and facilitate the understanding of the complex post-translational modifications (PTMs) of peptides and/or proteins. Since the biosynthetic gene clusters of thiopeptides were first discovered in 2009, several research studies regarding the biochemistry and enzymology of thiopeptide biosyntheses have been reported, indicating that their characteristic framework is constructed via a cascade of common PTMs and that additional specific PTMs diversify the molecules. In this review, we primarily summarize recent advances in understanding the biosynthesis of thiopeptide antibiotics and propose some potential applications based on our insights into the biosynthetic logic and machinery.
Co-reporter:Qingfei Zheng;Zhuhua Wu;Peng Sun;Dandan Chen;Zhenhua Tian
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 1) pp:88-91
Publication Date(Web):2016/12/20
DOI:10.1039/C6OB02567F
We herein report the isolation and characterization of a key linear intermediate in the biosynthetic pathway of pyrroindomycins, the potent spirotetramate natural products produced by Streptomyces rugosporus. This polyene intermediate bears a γ-hydroxymethyl group that is exocyclic to the tetramate moiety, indicating that a serine residue serves as the three-carbon unit for tetramate formation and chain-elongation termination. The further conversion involves an acetylation–elimination of the exocyclic γ-hydroxymethyl group to generate a γ-methylene group, which is indispensable for intramolecular [4 + 2] cross-bridging to construct the characteristic pentacyclic core. The findings presented in this study provide new insights into the biosynthesis of pyrroindomycins, and thus suggest a common paradigm for both spirotetramates and spirotetronates in processing the exocyclic γ-hydroxymethyl group of the five-membered heterocycle.
Co-reporter:Xiaojun Li;Qingfei Zheng;Jun Yin;Shuanhu Gao
Chemical Communications 2017 vol. 53(Issue 34) pp:4695-4697
Publication Date(Web):2017/04/25
DOI:10.1039/C7CC01929G
We here report that the biosynthesis of equisetin, a fungal tetramate natural product with potent anti-infectious activity, relies on Fsa2, a Diels–Alderase that constructs the trans-decalin system of the molecule in a stereo-selective manner. This finding led to the development of a concise chemo-enzymatic route toward the synthesis of equisetin, which involves facile preparation of a linear polyene precursor via 7-steps and Fsa2 activity for equisetin maturation through an intramolecular Diels–Alder reaction, thus exemplifying the significance of the combination of chemical and biological methods to achieve structurally complex cyclic natural products and their derivatives.
Co-reporter:Ming Chen;Yipeng Zhang;Yanan Du;Qunfei Zhao;Qinglin Zhang;Jiequn Wu
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 26) pp:5472-5475
Publication Date(Web):2017/07/05
DOI:10.1039/C7OB01284E
In this study, we characterized CaeB6 as a selective hydroxylase and CaeG1 as an O-methyltransferase in the biosynthesis of the 2,2′-bipyridine natural products caerulomycins (CAEs). The C3-hydroxylation activity of CaeB6 competes with the C4-O-methylation activity of CaeG1 and thereby branches the CAE pathway from a common C4-O-demethylated 2,2′-bipyridine intermediate. CaeG1-catalyzed C4-O-methylation leads to a main route that produces the major product CAE-A in Actinoalloteichus cyanogriseus NRRL B-2194. In contrast, CaeB6-catalyzed C3-hydroxylation results in a shunt route in which CaeG1 causes C4-O-methylation and subsequent C3-O-methylation to produce a series of minor CAE products. These findings provide new insights into the biosynthetic pathway of CAEs and a synthetic biology strategy for the selective functionalization of the 2,2′-bipyridine core.
Co-reporter:Ming Chen, Bo Pang, Ya-nan Du, Yi-peng Zhang, Wen Liu
Synthetic and Systems Biotechnology 2017 Volume 2, Issue 2(Issue 2) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.synbio.2017.07.002
2,2′-Bipyridine (2,2′-BiPy) is an attractive core structure present in a number of biologically active natural products, including the structurally related antibiotics caerulomycins (CAEs) and collismycins (COLs). Their biosynthetic pathways share a similar key 2,2′-BiPy-l-leucine intermediate, which is desulfurated or sulfurated at C5, arises from a polyketide synthase/nonribosomal peptide synthetase hybrid assembly line. Focusing on the common off-line modification steps, we here report that the removal of the “auxiliary” l-leucine residue relies on the metallo-dependent amidohydrolase activity of CaeD or ColD. This activity leads to the production of similar 2,2′-BiPy carboxylate products that then receive an oxime functionality that is characteristic for both CAEs and COLs. Unlike many metallo-dependent amidohydrolase superfamily proteins that have been previously reported, these proteins (particularly CaeD) exhibited a strong zinc ion-binding capacity that was proven by site-specific mutagenesis studies to be essential to proteolytic activity. The kinetics of the conversions that respectively involve CaeD and ColD were analyzed, showing the differences in the efficiency and substrate specificity of these two proteins. These findings would generate interest in the metallo-dependent amidohydrolase superfamily proteins that are involved in the biosynthesis of bioactive natural products.
Co-reporter:Min Wang; Qunfei Zhao; Qinglin Zhang
Journal of the American Chemical Society 2016 Volume 138(Issue 20) pp:6348-6351
Publication Date(Web):May 12, 2016
DOI:10.1021/jacs.6b01751
Pyridoxal-5′-phosphate (PLP)-dependent proteins constitute one of the largest and most important families of enzymes in living organisms. These proteins participate in numerous biochemical processes, many of which have not been characterized, and transform substrates containing an amino group through various reactions that share aldimine as a common intermediate. Herein, we report that the PLP-dependent enzymes CcbF and LmbF, which are highly related in phylogenesis, process cysteine S-conjugated intermediates in different ways and associate with individual downstream enzyme(s) toward distinct S-functionalization of the lincosamide antibiotics celesticetin and lincomycin A. CcbF catalyzes an unusual conversion that involves decarboxylation-coupled oxidative deamination of the cysteinyl group during the formation of a two-carbon alcohol linker, whereas LmbF is responsible for β-elimination, followed by S-methylation to produce a methylmercapto group. The two tailoring routes are variable and exchangeable with each other, allowing for in vitro combinatorial biosynthesis of a number of hybrid lincosamide antibiotics, including the natural product Bu-2545. These findings demonstrate the wide diversity of PLP chemistry in enzymatic catalysis and its promising applicability in creation of new molecules.
Co-reporter:Bo Pang, Min Wang and Wen Liu
Natural Product Reports 2016 vol. 33(Issue 2) pp:162-173
Publication Date(Web):25 Nov 2015
DOI:10.1039/C5NP00095E
Covering: 2009 to 2015
Modular polyketide synthases (PKSs) and non-ribosomal peptide synthetases (NRPSs) are multifunctional megaenzymes that serve as templates to program the assembly of short carboxylic acids and amino acids in a primarily co-linear manner. The variation, combination, permutation and evolution of their functional units (e.g., modules, domains and proteins) along with their association with external enzymes have resulted in the generation of numerous versions of templates, the roles of which have not been fully recognized in the structural diversification of polyketides, non-ribosomal peptides and their hybrids present in nature. In this Highlight, we focus on the assembly-line enzymology and associated chemistry by providing examples of some newly characterized cyclization reactions that occur on and off the assembly lines during and after chain elongation for the purpose of elucidating the template effects of PKSs and NRPSs. A fundamental understanding of the underlying biosynthetic logic would facilitate the elucidation of chemical information contained within the PKS or NRPS templates and benefit the development of strategies for genome mining, biosynthesis-inspired chemical synthesis and combinatorial biosynthesis.
Co-reporter:Qingfei Zheng, Zhenhua Tian, Wen Liu
Current Opinion in Chemical Biology 2016 Volume 31() pp:95-102
Publication Date(Web):April 2016
DOI:10.1016/j.cbpa.2016.01.020
•[4+2] Cycloaddition and spiroketalization are different cyclization reactions.•‘[4+2] Cycloadditionases’ and ‘spiroketalases’ have been identified in nature.•There are several similarities between the two types of enzymes.•They both allow for stereochemical control and/or rate enhancement in reactions.•Each type of enzymes feature a convergent evolution from variable protein folds.Diels-Alder-like [4+2] cycloaddition and ketalization of dihydroxy ketones are cyclization reactions with different mechanisms that produce characteristic cyclohexene and spiroketal units, respectively. Here, we review newly identified, naturally occurring ‘[4+2] cycloadditionases’ and ‘spiroketalases’ and reveal several similarities between the two types of enzymes. During catalysis, these enzymes control product stereochemistry or/and enhance the transformation rate. They exhibit convergent evolution of [4+2] cycloaddition or spiroketalization activity, which is likely dependent on interactions of variable protein folds with specialized chemical structures. An understanding of these similarities is expected to allow for establishment of the underlying principles for the application and catalyst design of associated enzymatic reactions in organic chemistry and synthetic biology.
Co-reporter:Qingfei Zheng, Shoufeng Wang, Rijing Liao, and Wen Liu
ACS Chemical Biology 2016 Volume 11(Issue 10) pp:2673
Publication Date(Web):August 25, 2016
DOI:10.1021/acschembio.6b00419
Side-ring-modified thiostrepton (TSR) derivatives that vary in their quinaldic acid (QA) substitution possess more potent biological activities and better pharmaceutical properties than the parent compound. In this work, we sought to introduce fluorine onto C-7′ or C-8′ of the TSR QA moiety via precursor-directed mutational biosynthesis to obtain new TSR variants. Unexpectedly, instead of the target product, the exogenous chemical feeding of 7-F-QA into the ΔtsrT mutant strain resulted in a unique TSR analog with an incomplete side-ring structure and an unoxidized QA moiety (1). Accordingly, two cytochrome P450 genes, tsrP and tsrR, were in-frame deleted to elucidate the candidate responsible for the monooxidation of the QA moiety in TSR. The unfluorinated analog of compound 1 that was thus isolated from ΔtsrP (2) and the abolishment of TSR production in ΔtsrR revealed not only the biosynthetic logic of the TSR side-ring but also the essential checkpoint in TSR maturation before macro-ring closure.
Co-reporter:Zhi Lin;Dandan Chen
Science China Chemistry 2016 Volume 59( Issue 9) pp:1175-1187
Publication Date(Web):2016 September
DOI:10.1007/s11426-016-0062-x
Natural products are often secondary metabolites in living organisms with a wide variety of biological activities. The diversification of their structures, aiming to the search for biologically active small molecules by expanding chemical and functional spaces, is a major area of current interest in synthetic chemistry. However, developing synthetic accessibility and efficiency often faces challenges associated with structural complexity. Synthetic biology has recently emerged and is promising to accomplish complex molecules; by contrast, the application to structural diversification of natural products relies on the understanding, development and utilization of compatible biosynthetic machinery. Here, we review the strategies primarily concerning the artificial evolution of microbial natural products whose biosynthesis features template enzymology, including ribosomally synthesized and post-translationally modified peptides as well as the assembly line-resultant polyketides, non-ribosomal peptides and hybrids. The establishment of these approaches largely facilitates the expansion of the molecular diversity and utility through bioengineering at different stages/levels of biosynthetic pathways.
Co-reporter:Qingfei Zheng;Shoufeng Wang;Panpan Duan;Rijing Liao;Dandan Chen
PNAS 2016 Volume 113 (Issue 50 ) pp:14318-14323
Publication Date(Web):2016-12-13
DOI:10.1073/pnas.1612607113
Thiostrepton (TSR), an archetypal bimacrocyclic thiopeptide antibiotic that arises from complex posttranslational modifications
of a genetically encoded precursor peptide, possesses a quinaldic acid (QA) moiety within the side-ring system of a thiopeptide-characteristic
framework. Focusing on selective engineering of the QA moiety, i.e., by fluorination or methylation, we have recently designed
and biosynthesized biologically more active TSR analogs. Using these analogs as chemical probes, we uncovered an unusual indirect
mechanism of TSR-type thiopeptides, which are able to act against intracellular pathogens through host autophagy induction
in addition to direct targeting of bacterial ribosome. Herein, we report the accumulation of 6′-fluoro-7′, 8′-epoxy-TSR, a
key intermediate in the preparation of the analog 6′-fluoro-TSR. This unexpected finding led to unveiling of the TSR maturation
process, which involves an unusual dual activity of TsrI, an α/β-hydrolase fold protein, for cascade C-N bond cleavage and
formation during side-ring system construction. These two functions of TsrI rely on the same catalytic triad, Ser72-His200-Asp191,
which first mediates endopeptidyl hydrolysis that occurs selectively between the residues Met-1 and Ile1 for removal of the
leader peptide and then triggers epoxide ring opening for closure of the QA-containing side-ring system in a regio- and stereo-specific
manner. The former reaction likely requires the formation of an acyl-Ser72 enzyme intermediate; in contrast, the latter is
independent of Ser72. Consequently, C-6′ fluorination of QA lowers the reactivity of the epoxide intermediate and, thereby,
allows the dissection of the TsrI-associated enzymatic process that proceeds rapidly and typically is difficult to be realized
during TSR biosynthesis.
Co-reporter:Qingfei Zheng, Qinglan Wang, Shoufeng Wang, Jiequn Wu, Qian Gao, Wen Liu
Chemistry & Biology 2015 Volume 22(Issue 8) pp:1002-1007
Publication Date(Web):20 August 2015
DOI:10.1016/j.chembiol.2015.06.019
•Thiostreptons are active against the intracellular pathogen Mycobacterium marinum•Thiostreptons induce host autophagy by promoting ER stress•The activity of autophagy induction is sensitive to variation of the quinaldic unit•Thiostreptons have a dual action on both the pathogens and the infected host cellsThiostrepton (TSR) is an archetypal thiopeptide antibiotic possessing a quinaldic acid (QA) moiety in the side ring system. According to the mechanism of TSR previously known to target bacterial ribosome, we recently designed and biosynthesized several TSR derivatives that varied in QA substitution. Utilizing these thiopeptide antibiotics to treat the intracellular pathogen Mycobacterium marinum, we herein report a novel mode of action of TSRs, which induce ER stress-mediated autophagy to enhance host cell defense. This intracellular response, which is sensitive to the modification of the QA group, serves as an indirect but unignorable mechanism for eliminating intracellular pathogens. TSRs are thus the only type of antibiotics, to our knowledge, with the dual action on both the parasitic bacteria and the infected host cells. The newly observed mechanism of TSRs may inspire the future change in the treatment of intracellular pathogens, by taking host response into account.Figure optionsDownload full-size imageDownload high-quality image (155 K)Download as PowerPoint slide
Co-reporter:Heng Guo, Jiang Wang, Yeming Li, Yi Yu, Qingfei Zheng, Jiequn Wu and Wen Liu
Chemical Science 2014 vol. 5(Issue 1) pp:240-246
Publication Date(Web):26 Sep 2013
DOI:10.1039/C3SC52015C
The ribosomal origin of thiopeptide antibiotics, a class of sulfur-rich and highly modified poly(thi)azolyl natural products, has recently been uncovered and features complex post-translational modifications (PTMs) of a precursor peptide. Based on molecular engineering and production improvement, we report insight into the biosynthesis of two bicyclic thiopeptide compounds, thiostrepton and nosiheptide. The PTMs of thiostrepton tolerate variations in the first two amino acids of the core peptide part of the precursor peptide: (1) the mutation of Ile1 to Val had no apparent effect on molecular maturation, suggesting that attachment of the quinaldic moiety at position 1 is not residue-dependent for the construction of the side ring system; and (2) the change of Ala2 to Ser led exclusively to the production of an analog that bears a corresponding dehydroamino acid residue, indicating that dehydration at position 2 is site-selective or that the oxazoline formed by cyclodehydration is inaccessible for maturation. For nosiheptide biosynthesis in particular, we provide the first structural evidence that construction of the specific side ring system precedes formation of the common central heterocycle domain and therefore propose that formation of a characteristic thiopeptide framework interweaves both common and specific PTMs that are interdependent. The above efforts benefited from the development of a uniform approach to examine the effectiveness for trans expression of gene encoding precursor peptides and associated PTM capacity. This approach is independent of knowledge regarding organism-specific regulatory mechanisms and potentially applicable to other systems that produce ribosomally synthesized peptide natural products.
Co-reporter:Qingfei Zheng, Shoufeng Wang, Wen Liu
Tetrahedron 2014 70(42) pp: 7686-7690
Publication Date(Web):
DOI:10.1016/j.tet.2014.06.076
Co-reporter:Dr. Peng Sun;Qunfei Zhao;Hua Zhang;Dr. Jiequn Wu; Dr. Wen Liu
ChemBioChem 2014 Volume 15( Issue 5) pp:660-664
Publication Date(Web):
DOI:10.1002/cbic.201300616
Abstract
Natural avermectins (AVEs) share a 6,6-spiroketal moiety with an exclusive R configuration at the C21 spirocyclic junction. Herein, we report the characterization of nine AVE-like spiroketals of two types (C21 S and R) in a mutant strain that lacks spirocyclase activity. Comparative analysis of their structures facilitated evaluation of the effect of stereochemistry on endogenous biotransformations and biological activities of the spiroketals.
Co-reporter:Peng Sun ; Qunfei Zhao ; Futao Yu ; Hua Zhang ; Zhuhua Wu ; Yinyan Wang ; Yan Wang ; Qinglin Zhang
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1540-1548
Publication Date(Web):January 7, 2013
DOI:10.1021/ja311339u
Avermectins (AVEs), which are widely used for the treatment of agricultural parasitic diseases, belong to a family of 6,6-spiroketal moiety-containing, macrolide natural products. AVE biosynthesis is known to employ a type I polyketide synthase (PKS) system to assemble the molecular skeleton for further functionalization. It remains unknown how and when spiroketal formation proceeds, particularly regarding the role of AveC, a unique protein in the pathway that shares no sequence homology to any enzyme of known function. Here, we report the unprecedented, dual function of AveC by correlating its activity with spiroketal formation and modification during the AVE biosynthetic process. The findings in this study were supported by characterizing extremely unstable intermediates, products and their spontaneous derivative products from the simplified chemical profile and by comparative analysis of in vitro biotransformations and in vivo complementations mediated by AveC and MeiC (the counterpart in biosynthesizing the naturally occurring, AVE-like meilingmycins). AveC catalyzes the stereospecific spiroketalization of a dihydroxy-ketone polyketide intermediate and the optional dehydration to determine the regiospecific saturation characteristics of spiroketal diversity. These reactions take place between the closures of the hexene ring and 16-membered macrolide and the formation of the hexahydrobenzofuran unit. MeiC can replace the spirocyclase activity of AveC, but it lacks the independent dehydratase activity. Elucidation of the generality and specificity of AveC-type proteins allows for the rationalization of previously published results that were not completely understood, suggesting that enzyme-mediated spiroketal formation was initially underestimated, but is, in fact, widespread in nature for the control of stereoselectivity.
Co-reporter:Qi Zhang and Wen Liu
Natural Product Reports 2013 vol. 30(Issue 2) pp:218-226
Publication Date(Web):19 Dec 2012
DOI:10.1039/C2NP20107K
Covering: 2009 to 2012
Thiopeptide antibiotics, a growing class of highly modified polythiazolyl peptides, have long been known for their remarkable bioactivities and unusual modes of actions. Uncovering of the ribosomal origin of thiopeptides now sets the stage for appreciating the initially undervalued modifications of the precursor peptides to afford the astonishing structure complexity. In this Highlight, we discuss the advances during the past four years in understanding the generality and specificity of thiopeptide biosynthesis, and on this basis, in expanding the structural diversity by pathway engineering.
Co-reporter:Qi Zhang, Bo Pang, Wei Ding, and Wen Liu
ACS Catalysis 2013 Volume 3(Issue 7) pp:1439
Publication Date(Web):May 21, 2013
DOI:10.1021/cs400211x
Polyketides comprise a large and highly diverse group of natural products produced by polyketide synthases (PKSs), and many of these compounds display remarkable biological activities. Although PKSs share a common mechanism in the assembly of polyketides from short carboxylic acid precursors, different types of PKSs have been classified according to their structures and modes of action. This review discusses a growing group of bacterial PKSs that are structurally type I but act in an iterative manner to produce aromatic polyketides. We summarize the genetic and biochemical features of these enzymes and compare them with other types of PKSs with an emphasis on the evolutionary relationship. We also discuss the different mechanisms for polyketide off-loading and the diverse post-PKS modifications on the resulting aromatic rings. Insights into bacterial iterative type I PKSs for aromatic polyketide formation and the relevant tailoring enzymes may guide rational bioengineering efforts to produce novel natural products.Keywords: aromatic polyketides; iterative type I PKSs (iPKSs); polyketide natural products; polyketide synthases (PKSs); post-iPKS modifications
Co-reporter:Lei Shao, Junsheng Chen, Chunxia Wang, Ji-an Li, Yumin Tang, Daijie Chen, Wen Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1438-1441
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.064
Gentamicin is an aminoglycoside antibiotic obtained from cultures of Micromonospora as the important anti-infective agents. Gentamicin which lacks 3′-hydroxyl group can avoid the attack from the modification enzymes of antibiotic-resistant bacteria in clinic. Consequently, C-3′ dehydroxylation is the key step in gentamicins biosynthesis. We suppose that there are some enzymes responsible for converting intermediate JI-20A to 3′,4′-bisdehydroxylated final product gentamicin C1a, while phosphorylation of 3′-OH is possibly the first step for C-3′ dehydroxylation. The gentamicin biosynthetic gene gntI, encoding an aminoglycoside phosphotransferase, was cloned from Micromonospora echinospora ATCC15835 and overexpressed in Escherichia coli. The resulting phosphotransferase was purified, and the kinetic parameters for Kanamycin A, Kanamycin B, Neomycin B and Amikacin were determined. Elucidation of NMR data of phosphorylated kanamycin B has unambiguously demonstrated a regiospecific phosphorylation of 3′-hydroxyl of the 6-aminohexose ring. The results described here partly confirm that the 3′-dehydroxylation step is preceded by a 3′ phosphorylation step. It is predicted that GntI belongs to a new aminoglycoside phosphotransferase group involved with aminoglycoside antibiotics biosynthesis pathway.
Co-reporter:Yan Yan;Dr. Jing Chen;Lihan Zhang;Qingfei Zheng;Ying Han;Hua Zhang;Daozhong Zhang;Dr. Takayoshi Awakawa;Dr. Ikuro Abe;Dr. Wen Liu
Angewandte Chemie International Edition 2013 Volume 52( Issue 47) pp:12308-12312
Publication Date(Web):
DOI:10.1002/anie.201305569
Co-reporter:Yan Yan;Dr. Jing Chen;Lihan Zhang;Qingfei Zheng;Ying Han;Hua Zhang;Daozhong Zhang;Dr. Takayoshi Awakawa;Dr. Ikuro Abe;Dr. Wen Liu
Angewandte Chemie 2013 Volume 125( Issue 47) pp:12534-12538
Publication Date(Web):
DOI:10.1002/ange.201305569
Co-reporter:Qiongqiong Wu ; Zhuhua Wu ; Xudong Qu
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17342-17345
Publication Date(Web):October 12, 2012
DOI:10.1021/ja304829g
The natural products pyrroindomycins (PYRs), active against various drug-resistant pathogens, possess a characteristic, cyclohexene ring spiro-linked tetramate moiety. In this study, investigation into PYR biosynthesis revealed two new proteins, both of which, phylogenetically distinct from but functionally substitutable to each other in vivo, individually catalyze a Dieckmann cyclization in vitro for converting an N-acetoacetyl-l-alanyl thioester into a tetramate. Their counterparts are commonly present in the biosynthetic pathways of spiro and polyether tetronates, supporting a uniform paradigm for tetronate/tetramate formation, which features an enzymatic way to generate the C–X (X = O or N) bond first and the C–C bond next in building of the 5-membered heterocycle.
Co-reporter:Xudong Qu ; Bo Pang ; Zhicong Zhang ; Ming Chen ; Zhuhua Wu ; Qunfei Zhao ; Qinglin Zhang ; Yinyan Wang ; Yun Liu
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9038-9041
Publication Date(Web):May 17, 2012
DOI:10.1021/ja3016457
Caerulomycins (CAEs) and collismycins (COLs), which mainly differ in sulfur decoration, are two groups of structurally similar natural products containing a 2,2′-bipyridine (2,2′-BP) core, derivatives of which have been widely used in chemistry. The biosynthetic pathways of CAEs and COLs remain elusive. In this work, cloning of the CAE biosynthetic gene cluster allowed us to mine a highly conserved gene cluster encoding COL biosynthesis in a Streptomyces strain that was previously unknown as a 2,2′-BP producer. In vitro and in vivo investigations into the biosynthesis revealed that CAEs and COLs share a common paradigm featuring an atypical hybrid polyketide synthase/nonribosomal peptide synthetase system that programs the 2,2′-BP formation. This likely involves an unusual intramolecular cyclization/rearrangement sequence, and a difference in processing of the sulfhydryl group derived from the same precursor cysteine drives the biosynthetic route toward CAEs or COLs.
Co-reporter:Yan Yan, Lihan Zhang, Takuya Ito, Xudong Qu, Yoshinori Asakawa, Takayoshi Awakawa, Ikuro Abe, and Wen Liu
Organic Letters 2012 Volume 14(Issue 16) pp:4142-4145
Publication Date(Web):August 3, 2012
DOI:10.1021/ol301785x
We herein report comparative analysis of two versions of the biosynthetic gene clusters of antimycins, a natural product family possessing up to 44 distinct entities. The biosynthetic pathway of antimycins is amenable to the high structural variation of the substrates, supported by successes in heterologous expression of the ant cluster and in fluorine incorporation. The latter facilitated the investigation of the structure–activity relationship into the usually invariable 3-formamidosalicylic acid moiety of the molecules.
Co-reporter:Lian Duan, Shoufeng Wang, Rijing Liao, Wen Liu
Chemistry & Biology 2012 Volume 19(Issue 4) pp:443-448
Publication Date(Web):20 April 2012
DOI:10.1016/j.chembiol.2012.02.008
Thiostrepton (TSR), often referred as to a parent compound in the thiopeptide family, is a bimacrocyclic member that features a quinaldic acid (QA) moiety-containing side ring appended to the characteristic core system. QA biosynthesis requires an unusual ring-expanding conversion, showing a methyl transfer onto and a rearrangement of the indole part of L-tryptophan to give a quinoline ketone. Herein, we report that the process involves the activities of the radical methyltransferase TsrT, aminotransferase TsrA, dehydrogenase TsrE, and cyclase TsrD. TsrU, a stereospecific oxidoreductase, catalyzes the further conversion of the ketone into an enantiomerically pure S-alcohol. Elucidation of this chemistry, which is common in the biosynthesis of a number of thiopeptides sharing a QA side ring system, facilitates analog generation, as shown by the achievement of region-specific fluorination of thiostrepton with the improved antibacterial activity.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (177 K)Download as PowerPoint slideHighlights► Quinoline ketone formation involves a ring-expanding conversion of L-tryptophan ► Further stereospecific reduction produces an enantiomerically pure S-alcohol ► Quinaldic acid-forming chemistry is common in thiopeptide biosynthesis ► Region-specific fluorination of thiostrepton improved the antibacterial activity
Co-reporter:Rijing Liao
Journal of the American Chemical Society 2011 Volume 133(Issue 9) pp:2852-2855
Publication Date(Web):February 16, 2011
DOI:10.1021/ja1111173
Thiopeptides are a class of clinically interesting and highly modified peptide antibiotics. Their biosyntheses share a common paradigm for characteristic core formation but differ in tailoring to afford individual members. Herein we report an unusual deesterification−amidation process in thiostrepton maturation to furnish the terminal amide moiety. TsrB, serving as a carboxylesterase, catalyzes the hydrolysis of the methyl ester intermediate to provide the carboxylate intermediate, which can be converted to the amide product by an amidotransferase, TsrC. These findings revealed a C-terminal methylation of the precursor peptide, which is cryptic in thiostrepton biosynthesis but potentially common in the formation of its homologous series of thiopeptides that vary in the C-terminal form as methyl ester, carboxylate, or amide.
Co-reporter:Xudong Qu, Nan Jiang, Fei Xu, Lei Shao, Gongli Tang, Barrie Wilkinson and Wen Liu
Molecular BioSystems 2011 vol. 7(Issue 3) pp:852-861
Publication Date(Web):15 Dec 2010
DOI:10.1039/C0MB00234H
Sanglifehrin A (SFA), a potent cyclophilin inhibitor produced by Streptomyces flaveolus DSM 9954, bears a unique [5.5] spirolactam moiety conjugated with a 22-membered, highly functionalized macrolide through a linear carbon chain. SFA displays a diverse range of biological activities and offers significant therapeutic potential. However, the structural complexity of SFA poses a tremendous challenge for new analogue development via chemical synthesis. Based on a rational prediction of its biosynthetic origin, herein we report the cloning, sequencing and characterization of the gene cluster responsible for SFA biosynthesis. Analysis of the 92776 bp contiguous DNA region reveals a mixed polyketide synthase (PKS)/non-ribosomal peptide synthetase (NRPS) pathway which includes a variety of unique features for unusual PKS and NRPS building block formation. Our findings suggest that SFA biosynthesis requires a crotonyl-CoA reductase/carboxylase (CCR) for generation of the putative unusual PKS starter unit (2R)-2-ethylmalonamyl-CoA, an iterative type I PKS for the putative atypical extender unit (2S)-2-(2-oxo-butyl)malonyl-CoA and a phenylalanine hydroxylase for the NRPS extender unit (2S)-m-tyrosine. A spontaneous ketalization of significant note, may trigger spirolactam formation in a stereo-selective manner. This study provides a framework for the application of combinatorial biosynthesis methods in order to expand the structural diversity of SFA.
Co-reporter:Dr. Xudong Qu;Dr. Chun Lei ;Dr. Wen Liu
Angewandte Chemie 2011 Volume 123( Issue 41) pp:9825-9828
Publication Date(Web):
DOI:10.1002/ange.201103085
Co-reporter:Dr. Xudong Qu;Dr. Chun Lei ;Dr. Wen Liu
Angewandte Chemie International Edition 2011 Volume 50( Issue 41) pp:9651-9654
Publication Date(Web):
DOI:10.1002/anie.201103085
Co-reporter:Yi Yu ; Heng Guo ; Qi Zhang ; Lian Duan ; Ying Ding ; Rijing Liao ; Chun Lei ; Ben Shen
Journal of the American Chemical Society 2010 Volume 132(Issue 46) pp:16324-16326
Publication Date(Web):November 3, 2010
DOI:10.1021/ja106571g
The carboxyl-terminal amide group has been often found in many bioactive peptide natural products, including nosiheptide belonging to the over 80 entity-containing thiopeptide family. Upon functional characterization of a novel protein NosA in nosiheptide biosynthesis, herein we report an unusual C-terminal amide forming strategy in general for maturating certain amide-terminated thiopeptides by processing their precursor peptides featuring a serine extension. NosA acts on an intermediate bearing a bis-dehydroalanine tail and catalyzes an enamide dealkylation to remove the acrylate unit originating from the extended serine residue.
Co-reporter:Wei Ding, Chun Lei, Qingli He, Qinglin Zhang, Yurong Bi, Wen Liu
Chemistry & Biology 2010 Volume 17(Issue 5) pp:495-503
Publication Date(Web):28 May 2010
DOI:10.1016/j.chembiol.2010.04.009
The enzymes 6-methylsalicylic acid (6-MSA) synthases (6-MSASs) are involved in the building of an aryl moiety in many bioactive secondary metabolites produced by fungi and bacteria. Using the bacterial 6-MSAS ChlB1 in the biosynthesis of spirotetronate antibiotic chlorothricin (CHL) as a model, functional analysis of its dehydratase (DH) and ketoreductase (KR) domains by site-specific mutagenesis revealed that selective ketoreduction is not essential for polyketide chain extension. Promiscuity of the ketoacylsynthase domain in β functionality recognition allows for engineering ChlB1 to an orsellinic acid (OSA) synthase (OSAS) by inactivating KR at the active site. The engineered ChlB1 is compatible with the enzymes for late-stage tailoring in CHL biosynthesis, featuring specific protein recognitions that facilitate variable aryl group incorporation. The resulting spirotetronates, which bear an OSA-derived aryl group, exhibited antibacterial activities comparable to those of the parent products.Highlights► The 6-MSA synthase ChlB1 can be engineered to generate an OSA synthase ► ChlB1-controlled ketoreduction is not essential for polyketide chain extension ► The engineered ChlB1 is compatible with the chlorothricin biosynthetic machinery ► The specific protein recognitions facilitate spirotetronate generation
Co-reporter:Ying Ding, Yi Yu, Haixue Pan, Heng Guo, Yeming Li and Wen Liu
Molecular BioSystems 2010 vol. 6(Issue 7) pp:1180-1185
Publication Date(Web):17 May 2010
DOI:10.1039/C005121G
Characterization of the biosynthetic gene cluster of glycosylated antibiotic nocathiacin I (NOC-I) here adds new insights to thiopeptide biosynthesis, showing the NOC-specific tailoring and unusual sugar formation. NOC-I biosynthesis shares the paradigm for forming a common thiopeptide core and the generality for converting to an e series member, as that of the parent compound nosiheptide (NOS). This may permit the production of NOC-I in the genetically amenable, NOS-producing strain by building NOC-specific genes for pathway engineering.
Co-reporter:Wei Ding, Wei Deng, Mancheng Tang, Qi Zhang, Gongli Tang, Yurong Bi and Wen Liu
Molecular BioSystems 2010 vol. 6(Issue 6) pp:1071-1081
Publication Date(Web):16 Mar 2010
DOI:10.1039/B926358F
Azinomycin B is a potent antitumor antibiotic that features a set of unusual, densely assembled functionalities. Among them, the 3-methoxy-5-methylnaphthoic acid (NPA) moiety provides an important noncovalent association with DNA, and may, therefore, contribute to the specificity of DNA alkylation for biological activity exhibition. We have previously cloned and sequenced the azinomycin B biosynthetic gene cluster, and proposed that four enzymes: AziB, AziB1, AziB2, and AziA1, are involved in the naphthoate moiety formation and incorporation. In this study, we report in vivo and/or in vitro characterizations of the P450 hydroxylase AziB1, the O-methyltransferase AziB2, and the substrate specificity of the non-ribosomal peptide synthetase (NRPS) AziA1, providing insights into the timing of individual steps in the late-stage modification of 5-methyl-NPA synthesized by the iterative type I polyketide synthase AziB. AziB1 catalyzes a regiospecific hydroxylation at the C3 position of the free naphthoic acid 5-methyl-NPA to produce 3-hydroxy-5-methyl-NPA, and the resulting hydroxyl group is subsequently O-methylated by AziB2 to furnish the methoxy functionality. The di-domain NRPS AziA1 specifically incorporates 3-methoxy-5-methyl-NPA via an unusual A domain to initiate the backbone formation of azinomycin B. AziA1 activates several analogues of the natural starter unit, suggesting a potential for production by metabolic engineering of new azinomycin analogues differing in their NPA moieties.
Co-reporter:Rijing Liao, Lian Duan, Chun Lei, Haixue Pan, Ying Ding, Qi Zhang, Daijie Chen, Ben Shen, Yi Yu, Wen Liu
Chemistry & Biology 2009 Volume 16(Issue 2) pp:141-147
Publication Date(Web):27 February 2009
DOI:10.1016/j.chembiol.2009.01.007
Thiopeptides, with potent activity against various drug-resistant pathogens, contain a characteristic macrocyclic core consisting of multiple thiazoles, dehydroamino acids, and a 6-membered nitrogen heterocycle. Their biosynthetic pathways remain elusive, in spite of great efforts by in vivo feeding experiments. Here, cloning, sequencing, and characterization of the thiostrepton and siomycin A gene clusters unveiled a biosynthetic paradigm for the thiopeptide specific core formation, featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. The paradigm generality for thiopeptide biosynthesis was supported by genome mining and ultimate confirmation of the thiocillin I production in Bacillus cereus ATCC 14579, a strain that was previously unknown as a thiopeptide producer. These findings set the stage to accelerate the discovery of thiopeptides by prediction at the genetic level and to generate structural diversity by applying combinatorial biosynthesis methods.
Co-reporter:Yi Yu, Lian Duan, Qi Zhang, Rijing Liao, Ying Ding, Haixue Pan, Evelyn Wendt-Pienkowski, Gongli Tang, Ben Shen and Wen Liu
ACS Chemical Biology 2009 Volume 4(Issue 10) pp:855
Publication Date(Web):August 13, 2009
DOI:10.1021/cb900133x
Nosiheptide (NOS), belonging to the e series of thiopeptide antibiotics that exhibit potent activity against various bacterial pathogens, bears a unique indole side ring system and regiospecific hydroxyl groups on the characteristic macrocyclic core. Here, cloning, sequencing, and characterization of the nos gene cluster from Streptomyces actuosus ATCC 25421 as a model for this series of thiopeptides has unveiled new insights into their biosynthesis. Bioinformatics-based sequence analysis and in vivo investigation into the gene functions show that NOS biosynthesis shares a common strategy with recently characterized b or c series thiopeptides for forming the characteristic macrocyclic core, which features a ribosomally synthesized precursor peptide with conserved posttranslational modifications. However, it apparently proceeds via a different route for tailoring the thiopeptide framework, allowing the final product to exhibit the distinct structural characteristics of e series thiopeptides, such as the indole side ring system. Chemical complementation supports the notion that the S-adenosylmethionine-dependent protein NosL may play a central role in converting tryptophan to the key 3-methylindole moiety by an unusual carbon side chain rearrangement, most likely via a radical-initiated mechanism. Characterization of the indole side ring-opened analogue of NOS from the nosN mutant strain is consistent with the proposed methyltransferase activity of its encoded protein, shedding light into the timing of the individual steps for indole side ring biosynthesis. These results also suggest the feasibility of engineering novel thiopeptides for drug discovery by manipulating the NOS biosynthetic machinery.
Co-reporter:Qing-Li He Dr.;Xin-Ying Jia Dr.;Man-Cheng Tang;Zhen-Hua Tian Dr.;Gong-Li Tang
ChemBioChem 2009 Volume 10( Issue 5) pp:813-819
Publication Date(Web):
DOI:10.1002/cbic.200800714
Co-reporter:Qunfei Zhao, Qingli He, Wei Ding, Mancheng Tang, Qianjin Kang, Yi Yu, Wei Deng, Qi Zhang, Jie Fang, Gongli Tang, Wen Liu
Chemistry & Biology 2008 Volume 15(Issue 7) pp:693-705
Publication Date(Web):21 July 2008
DOI:10.1016/j.chembiol.2008.05.021
Azinomycin B is a complex natural product containing densely assembled functionalities with potent antitumor activity. Cloning and sequence analysis of the azi gene cluster revealed an iterative type I polyketide synthase (PKS) gene, five nonribosomal peptide synthetases (NRPSs) genes and numerous genes encoding the biosynthesis of unusual building blocks and tailoring steps for azinomycin B production. Characterization of AziB as a 5-methyl-naphthoic acid (NPA) synthase showed a distinct selective reduction pattern in aromatic polyketide biosynthesis governed by bacterial iterative type I PKSs. Heterologous expression established the PKS-post modification route from 5-methyl-NPA to reach the first building block 3-methoxy-5-methyl-NPA. This proposed azinomycin B biosynthetic pathway sets the stage to investigate the enzymatic mechanisms for building structurally unique and pharmaceutically important groups, including the unprecedented azabicyclic ring system and highly active epoxide moiety.
Co-reporter:Xin-Ying Jia, Zhen-Hua Tian, Lei Shao, Xu-Dong Qu, Qun-Fei Zhao, Jian Tang, Gong-Li Tang, Wen Liu
Chemistry & Biology 2006 Volume 13(Issue 6) pp:575-585
Publication Date(Web):June 2006
DOI:10.1016/j.chembiol.2006.03.008
The biosynthetic gene cluster for chlorothricin (CHL) was localized to a 122 kb contiguous DNA from Streptomyces antibioticus DSM 40725, and its involvement in CHL biosynthesis was confirmed by gene inactivation and complementation. Bioinformatic analysis of the sequenced 111.989 kb DNA region revealed 42 open reading frames, 35 of which were defined to constitute the CHL gene cluster. An assembly model for CHL biosynthesis from D-olivose, 2-methoxy-5-chloro-6-methylsalicyclic acid, and chlorothricolide building blocks was proposed. This work represents cloning of a gene cluster for spirotetronate antibiotic biosynthesis and sets the stage to investigate the unusual macrolide biosynthesis including tandem Diels-Alder cyclizations, Baeyer-Villiger oxidation, and incorporation of an enoylpyruvate unit.
Co-reporter:Wen Liu, Siliang Zhang, Yun Chen, Wei Deng, Jiequn Wu
Journal of Biotechnology (October 2008) Volume 136(Supplement) pp:
Publication Date(Web):1 October 2008
DOI:10.1016/j.jbiotec.2008.07.035
Co-reporter:Peng Sun; Qunfei Zhao; Zhuhua Wu; Wen Zhang
Journal of Natural Products () pp:
Publication Date(Web):January 22, 2015
DOI:10.1021/np500468f
Three new 1,19-seco-avermectin (AVE) analogues were isolated from the ΔaveCDE mutant Streptomyces avermectinius strain. Their structures were elucidated by detailed spectroscopic analysis. This is the first report of 1,19-seco-AVE analogues. In an in vitro assay these compounds displayed cytotoxicity against Saos-2, MG-63, and B16 cell lines.
Co-reporter:Ming Chen, Yipeng Zhang, Yanan Du, Qunfei Zhao, Qinglin Zhang, Jiequn Wu and Wen Liu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 26) pp:NaN5475-5475
Publication Date(Web):2017/06/16
DOI:10.1039/C7OB01284E
In this study, we characterized CaeB6 as a selective hydroxylase and CaeG1 as an O-methyltransferase in the biosynthesis of the 2,2′-bipyridine natural products caerulomycins (CAEs). The C3-hydroxylation activity of CaeB6 competes with the C4-O-methylation activity of CaeG1 and thereby branches the CAE pathway from a common C4-O-demethylated 2,2′-bipyridine intermediate. CaeG1-catalyzed C4-O-methylation leads to a main route that produces the major product CAE-A in Actinoalloteichus cyanogriseus NRRL B-2194. In contrast, CaeB6-catalyzed C3-hydroxylation results in a shunt route in which CaeG1 causes C4-O-methylation and subsequent C3-O-methylation to produce a series of minor CAE products. These findings provide new insights into the biosynthetic pathway of CAEs and a synthetic biology strategy for the selective functionalization of the 2,2′-bipyridine core.
Co-reporter:Shoufeng Wang, Qingfei Zheng, Jianfeng Wang, Zhixiong Zhao, Qingye Li, Yunsong Yu, Renxiao Wang and Wen Liu
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 2) pp:NaN109-109
Publication Date(Web):2014/12/05
DOI:10.1039/C4QO00288A
Thiostrepton is a potent archetypal thiopeptide antibiotic. According to its mechanism known to target bacterial ribosome, we show here a rational design upon modeling of this molecule into the ribosome complex and an effective biosynthesis of new thiopeptide antibiotics through regioselective modifications. The resulting derivatives exhibit a series of anticipated and unanticipated pharmaceutical advantages, including improvement in activity against a number of drug-resistant pathogens and in water solubility that has largely affected the clinical use of thiostrepton.
Co-reporter:Heng Guo, Jiang Wang, Yeming Li, Yi Yu, Qingfei Zheng, Jiequn Wu and Wen Liu
Chemical Science (2010-Present) 2014 - vol. 5(Issue 1) pp:NaN246-246
Publication Date(Web):2013/09/26
DOI:10.1039/C3SC52015C
The ribosomal origin of thiopeptide antibiotics, a class of sulfur-rich and highly modified poly(thi)azolyl natural products, has recently been uncovered and features complex post-translational modifications (PTMs) of a precursor peptide. Based on molecular engineering and production improvement, we report insight into the biosynthesis of two bicyclic thiopeptide compounds, thiostrepton and nosiheptide. The PTMs of thiostrepton tolerate variations in the first two amino acids of the core peptide part of the precursor peptide: (1) the mutation of Ile1 to Val had no apparent effect on molecular maturation, suggesting that attachment of the quinaldic moiety at position 1 is not residue-dependent for the construction of the side ring system; and (2) the change of Ala2 to Ser led exclusively to the production of an analog that bears a corresponding dehydroamino acid residue, indicating that dehydration at position 2 is site-selective or that the oxazoline formed by cyclodehydration is inaccessible for maturation. For nosiheptide biosynthesis in particular, we provide the first structural evidence that construction of the specific side ring system precedes formation of the common central heterocycle domain and therefore propose that formation of a characteristic thiopeptide framework interweaves both common and specific PTMs that are interdependent. The above efforts benefited from the development of a uniform approach to examine the effectiveness for trans expression of gene encoding precursor peptides and associated PTM capacity. This approach is independent of knowledge regarding organism-specific regulatory mechanisms and potentially applicable to other systems that produce ribosomally synthesized peptide natural products.
Co-reporter:Panpan Duan, Qingfei Zheng, Zhi Lin, Shoufeng Wang, Dandan Chen and Wen Liu
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 10) pp:NaN1258-1258
Publication Date(Web):2016/07/29
DOI:10.1039/C6QO00320F
The quinaldic acid (QA) moiety in the side ring of thiostrepton (TSR), which can be modified regioselectively via precursor-directed mutational biosynthesis, was proven to be biologically relevant but tunable, affecting TSR's outstanding antibacterial activities. In this study, we sought to obtain TSR derivatives with varying amino acid residues connected to the QA moiety. The generation of these TSR derivatives relied on single “base”-based mutagenesis, and six new TSR-type compounds were obtained. Moreover, the simultaneous mutation of Ile1 and Ala2 in the TSR side ring resulted in a naturally occurring compound, siomycin (SIO), together with a new component, SIO-Dha2Ser. The anti-infection assays indicated that all of these new compounds could act as both antimicrobial agents and autophagy inducers, and these two kinds of activities can also be separated via regioselective modifications on the TSR side ring.
Co-reporter:Xiaojun Li, Qingfei Zheng, Jun Yin, Wen Liu and Shuanhu Gao
Chemical Communications 2017 - vol. 53(Issue 34) pp:NaN4697-4697
Publication Date(Web):2017/04/04
DOI:10.1039/C7CC01929G
We here report that the biosynthesis of equisetin, a fungal tetramate natural product with potent anti-infectious activity, relies on Fsa2, a Diels–Alderase that constructs the trans-decalin system of the molecule in a stereo-selective manner. This finding led to the development of a concise chemo-enzymatic route toward the synthesis of equisetin, which involves facile preparation of a linear polyene precursor via 7-steps and Fsa2 activity for equisetin maturation through an intramolecular Diels–Alder reaction, thus exemplifying the significance of the combination of chemical and biological methods to achieve structurally complex cyclic natural products and their derivatives.
Co-reporter:Qingfei Zheng, Hui Fang and Wen Liu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 16) pp:NaN3390-3390
Publication Date(Web):2017/03/22
DOI:10.1039/C7OB00466D
Thiopeptide antibiotics are a class of typical ribosomally synthesized and post-translationally modified peptides (RiPPs) with complex chemical structures that are difficult to construct via chemical synthesis. To date, more than 100 thiopeptides have been discovered, and most of these compounds exhibit remarkable biological activities, such as antibacterial, antitumor and immunosuppressive activities. Therefore, studies of the biosynthesis of thiopeptides can contribute to the development of new drug leads and facilitate the understanding of the complex post-translational modifications (PTMs) of peptides and/or proteins. Since the biosynthetic gene clusters of thiopeptides were first discovered in 2009, several research studies regarding the biochemistry and enzymology of thiopeptide biosyntheses have been reported, indicating that their characteristic framework is constructed via a cascade of common PTMs and that additional specific PTMs diversify the molecules. In this review, we primarily summarize recent advances in understanding the biosynthesis of thiopeptide antibiotics and propose some potential applications based on our insights into the biosynthetic logic and machinery.
Co-reporter:Qingfei Zheng, Zhuhua Wu, Peng Sun, Dandan Chen, Zhenhua Tian and Wen Liu
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 1) pp:NaN91-91
Publication Date(Web):2016/12/06
DOI:10.1039/C6OB02567F
We herein report the isolation and characterization of a key linear intermediate in the biosynthetic pathway of pyrroindomycins, the potent spirotetramate natural products produced by Streptomyces rugosporus. This polyene intermediate bears a γ-hydroxymethyl group that is exocyclic to the tetramate moiety, indicating that a serine residue serves as the three-carbon unit for tetramate formation and chain-elongation termination. The further conversion involves an acetylation–elimination of the exocyclic γ-hydroxymethyl group to generate a γ-methylene group, which is indispensable for intramolecular [4 + 2] cross-bridging to construct the characteristic pentacyclic core. The findings presented in this study provide new insights into the biosynthesis of pyrroindomycins, and thus suggest a common paradigm for both spirotetramates and spirotetronates in processing the exocyclic γ-hydroxymethyl group of the five-membered heterocycle.













![[1,1'-Binaphthalene]-2,2'-diol,3,3'-dibromo-, (1S)-](http://img.cochemist.com/ccimg/119800/119707-74-3.png)
![[1,1'-Binaphthalene]-2,2'-diol,3,3'-dibromo-, (1S)-](http://img.cochemist.com/ccimg/119800/119707-74-3_b.png)
![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-diphenyl-, (1S)-](http://img.cochemist.com/ccimg/102500/102490-05-1.png)
![[1,1'-Binaphthalene]-2,2'-diol, 3,3'-diphenyl-, (1S)-](http://img.cochemist.com/ccimg/102500/102490-05-1_b.png)
![methyl 6,8-dideoxy-7-O-methyl-6-[(1-methylprolyl)amino]-1-thiooctopyranoside](http://img.cochemist.com/ccimg/75100/75007-09-9.png)
![methyl 6,8-dideoxy-7-O-methyl-6-[(1-methylprolyl)amino]-1-thiooctopyranoside](http://img.cochemist.com/ccimg/75100/75007-09-9_b.png)




![2-hydroxyethyl 6,8-dideoxy-7-O-methyl-6-[(1-methylprolyl)amino]-1-thiooctopyranoside](http://img.cochemist.com/ccimg/19300/19246-70-9.png)
![2-hydroxyethyl 6,8-dideoxy-7-O-methyl-6-[(1-methylprolyl)amino]-1-thiooctopyranoside](http://img.cochemist.com/ccimg/19300/19246-70-9_b.png)


![D-erythro-a-D-galacto-Octopyranoside,2-[(2-hydroxybenzoyl)oxy]ethyl6,8-dideoxy-7-O-methyl-6-[[[(2S)-1-methyl-2-pyrrolidinyl]carbonyl]amino]-1-thio-](http://img.cochemist.com/ccimg/2600/2520-21-0.png)
![D-erythro-a-D-galacto-Octopyranoside,2-[(2-hydroxybenzoyl)oxy]ethyl6,8-dideoxy-7-O-methyl-6-[[[(2S)-1-methyl-2-pyrrolidinyl]carbonyl]amino]-1-thio-](http://img.cochemist.com/ccimg/2600/2520-21-0_b.png)





![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)
![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9.png)
![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9_b.png)




![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, 2-methylpropyl ester](http://img.cochemist.com/ccimg/874700/874619-30-4.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, 2-methylpropyl ester](http://img.cochemist.com/ccimg/874700/874619-30-4_b.png)

























![D-myo-Inositol,1-O-[2-[[(2R)-2-(acetylamino)-3-mercapto-1-oxopropyl]amino]-2-deoxy-a-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/192200/192126-76-4.png)
![D-myo-Inositol,1-O-[2-[[(2R)-2-(acetylamino)-3-mercapto-1-oxopropyl]amino]-2-deoxy-a-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/192200/192126-76-4_b.png)














![[(2s,4e,7e)-1-(3-hexyl-4-oxooxetan-2-yl)trideca-4,7-dien-2-yl] (2s)-2-formamido-3-methylpentanoate](http://img.cochemist.com/ccimg/96900/96829-59-3.png)
![[(2s,4e,7e)-1-(3-hexyl-4-oxooxetan-2-yl)trideca-4,7-dien-2-yl] (2s)-2-formamido-3-methylpentanoate](http://img.cochemist.com/ccimg/96900/96829-59-3_b.png)







![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/68000/67909-99-3.png)
![Carbamic acid, [2-(1H-indol-3-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/68000/67909-99-3_b.png)










![18H-16a,19-Metheno-16aH-benzo[e]naphtho[2,1-m][1,4]dioxacyclopentadecin-14-carboxylicacid, 4-[[4-O-[3-O-(3-chloro-6-methoxy-2-methylbenzoyl)-2,6-dideoxy-b-D-arabino-hexopyranosyl]-2,6-dideoxy-b-D-arabino-hexopyranosyl]oxy]-1,2,3,4,4a,6a,7,8,9,10,12a,15,16,21,21a,21b-hexadecahydro-22-hydroxy-15,21a-dimethyl-18,21-dioxo-,(4S,4aS,6aR,11E,12aR,15R,16aS,21aR,21bR)-](http://img.cochemist.com/ccimg/34800/34707-92-1.png)
![18H-16a,19-Metheno-16aH-benzo[e]naphtho[2,1-m][1,4]dioxacyclopentadecin-14-carboxylicacid, 4-[[4-O-[3-O-(3-chloro-6-methoxy-2-methylbenzoyl)-2,6-dideoxy-b-D-arabino-hexopyranosyl]-2,6-dideoxy-b-D-arabino-hexopyranosyl]oxy]-1,2,3,4,4a,6a,7,8,9,10,12a,15,16,21,21a,21b-hexadecahydro-22-hydroxy-15,21a-dimethyl-18,21-dioxo-,(4S,4aS,6aR,11E,12aR,15R,16aS,21aR,21bR)-](http://img.cochemist.com/ccimg/34800/34707-92-1_b.png)
![L-Cysteine, S-acetyl-N-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/3739400/21948-02-7.png)
![L-Cysteine, S-acetyl-N-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/3739400/21948-02-7_b.png)











![Benzo[h]quinoline,2-methyl-](http://img.cochemist.com/ccimg/700/605-88-9.png)
![Benzo[h]quinoline,2-methyl-](http://img.cochemist.com/ccimg/700/605-88-9_b.png)







