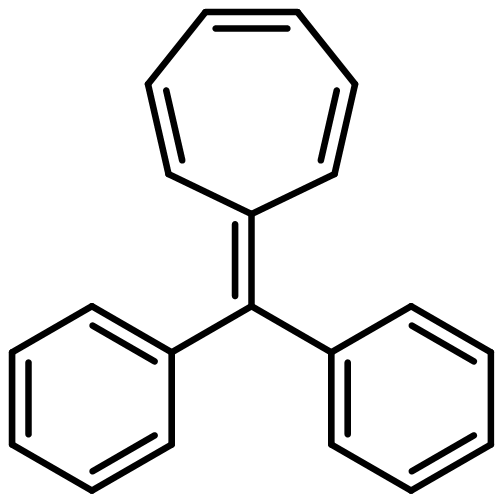
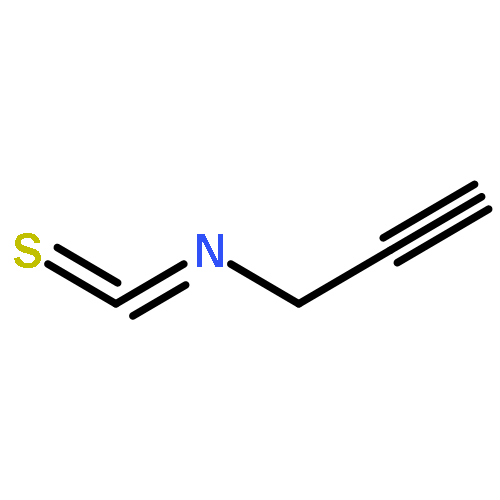
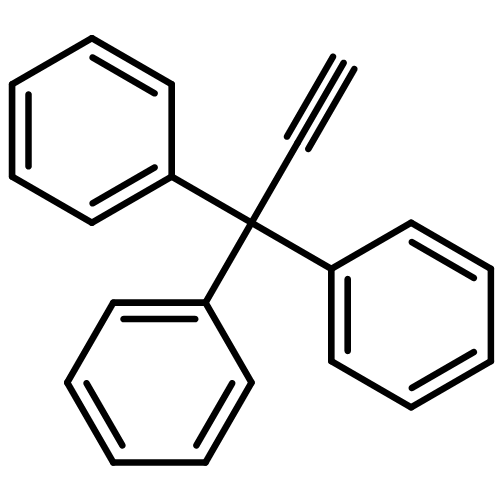
As a rule, acetylides and sulfonyl azides do not undergo electrophilic azide transfer because 1,2,3-triazoles are usually formed. We show now that treatment of tritylethyne with butyllithium followed by exposure to 2,4,6-triisopropylbenzenesulfonyl azide leads to products that are easily explained through the generation of short-lived tritylethynyl azide and its secondary product cyanotritylcarbene. Furthermore, it is demonstrated that tritylcarbenes generally do not produce triphenylethenes exclusively, as was stated in the literature. Instead, these carbenes always yielded also (diphenylmethylidene)cycloheptatrienes (heptafulvenes) as side products. This result is supported by static DFT, coupled cluster, and ab initio molecular dynamics calculations. From these investigations, the fused bicyclobutane intermediate was found to be essential for heptafulvene formation. Although the bicyclobutane is also capable of rearranging to the triphenylethene product, only the heptafulvene pathway is reasonable from the energetics. The ethene is formed straight from cyanotritylcarbene.
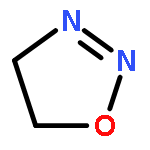
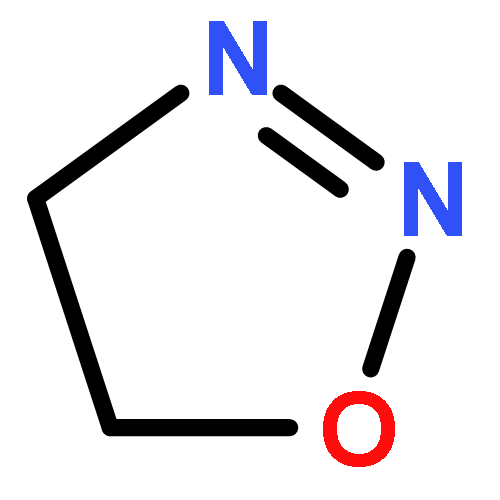
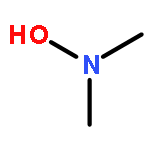
4,5-Dihydro-1,2,3-oxadiazoles are postulated to be key intermediates in the industrial synthesis of ketones from alkenes, in the alkylation of DNA in vivo, and in the decomposition of N-nitrosoureas; they are also a subject of great interest for theoretical chemists. In the presented report, the formation of 4,5-dihydro-1,2,3-oxadiazole and the subsequent decay into secondary products have been studied by NMR monitoring analysis. The elusive properties evading characterization have now been confirmed by 1H, 13C, and 15N NMR spectroscopy, and relevant 2D experiments at very low temperatures. Our experiments with suitably substituted N-nitrosoureas using thallium(I) alkoxides as bases under apolar conditions answer important questions on the existence and the secondary products of 4,5-dihydro-1,2,3-oxadiazole.
Decomposition of the extremely explosive and unstable parent compound of 1-azidoalkynes, HCCN3 (azidoacetylene), was studied in the gas phase, solid argon matrix, and solutions. In the gas phase, this azide decomposes quickly at room temperature with a half-life time (t1/2) of 20 min at an initial pressure (p0) of 0.8 mbar. The decay (p0 = 1.0 mbar) is significantly increased in an atmosphere of O2 with t1/2 of 3 min, in which HC(O)CN was identified as the trapping product of the cyanocarbene intermediate HCCN. Trapping products of this carbene by solvent molecules (CH2Cl2 and CHCl3) were also found during decomposition of the azide in solution, whereas the reaction with a solution of bromine to form dibromoacetonitrile is interpreted as taking place by nucleophilic attack of the alkyne itself. The intermediary formation of triplet HCCN by flash vacuum pyrolysis and photolysis (255 nm) of the azide in the gas phase and in solid argon matrices, respectively, was confirmed by IR spectroscopy and mutual photo-interconversion of HCCN with isomeric cyclo-C(H)CN (azirinylidene) and HCNC by selective irradiations at 16 K.
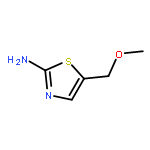
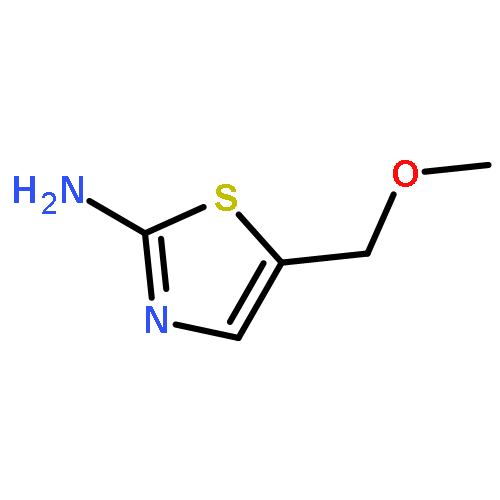
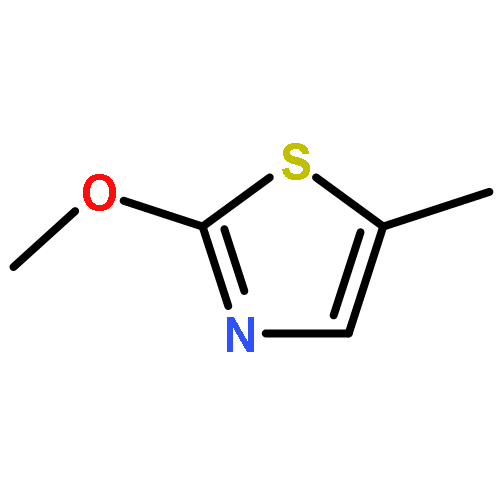
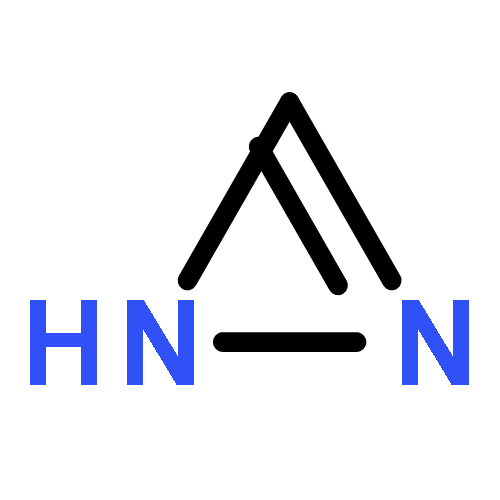
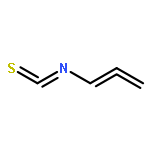
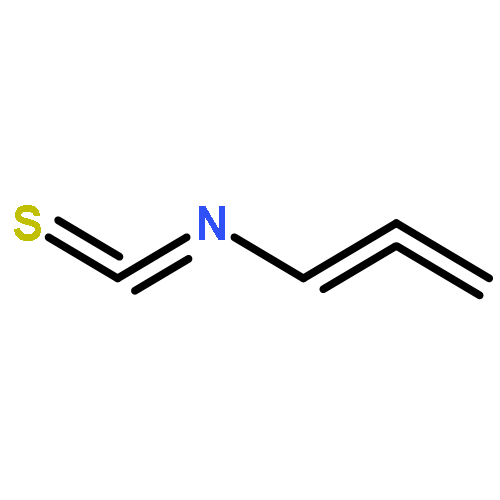
Treatment of allenyl isothiocyanate with a variety of nucleophiles leads to 5-methylthiazoles with a functional group at the 2-position. The same pattern of reactivity is also seen with N-aminophthalimide. In the presence of azide salt, hydrazoic acid, or N,N-disubstituted hydroxylamines, however, allenyl isothiocyanate is converted into bifunctionalized thiazoles. We explain the formation of these products by nucleophilic addition at the isothiocyanato moiety followed by ring closure and an N–N or N–O cleavage reaction to generate short-lived 2-imino-5-methylidenethiazole or 5-methylidenethiazol-2-one. Such intermediates are trapped by addition reactions to give the final heterocyclic compounds. In the case of N,N-disubstituted hydroxylamines, the primary addition products with allenyl isothiocyanate can be detected as unstable intermediates by IR and NMR spectroscopy.
Substituted 1-azidocyclopentenes and 1-azidocyclohexenes were photolyzed to generate 2,3-bridged 2H-azirines. In the case of bridgehead azirines with a six-membered carbocycle, detection by NMR spectroscopic analysis was possible, whereas even kinetically stabilized bridgehead azirines with a five-membered ring could not be characterized by low-temperature NMR spectroscopic analysis. Thus, a recent report on the latter heterocycles was corrected. Depending on the substitution pattern, irradiation of 1-azidocyclopentenes either led to products that can be explained on the basis of short-lived 2,3-bridged 2H-azirines, or gave secondary products generated from triplet nitrenes. The diverse photoreactivity of 2,3-bridged 2H-azirines was also studied by quantum chemical methods (DFT, CCSD(T), CASSCF(6,6)) with respect to the singlet and triplet energy surfaces. The ring-opening processes leading to the corresponding vinyl nitrenes were identified as key steps for the observed reactivity.
1,4-Diazidobuta-1,3-dienes (Z,Z)-10, 17, and 21 were photolyzed and thermolyzed to yield the pyridazines 13, 20, and 23, respectively. To explain these aromatic final products, the generation of highly strained bi-2H-azirin-2-yls 12, 19, and 22 and their valence isomerization were postulated. In the case of meso- and rac-22, nearly quantitative formation from diazide 21, isolation as stable solids, and complete characterization were possible. On the thermolysis of 22, aromatization to 23 was only a side reaction, whereas equilibration of meso- and rac-22 and fragmentation, which led to alkyne 24 and acetonitrile, dominated. Prolonged irradiation of 22 gave mainly the pyrimidine 25. The change of the configuration at C-2 of the 2H-azirine unit was observed not only in the case of bi-2H-azirin-2-yls 22 but also for simple spirocyclic 2H-azirines 29 at a relatively low temperature (75 °C). The fragmentation of rac-22 to give alkyne 24 and two molecules of acetonitrile was also studied by high-level quantum chemical calculations. For a related model system 30 (methyl instead of phenyl groups), two transition states TS-30–31 of comparable energy with multiconfigurational electronic states could be localized on the energy hypersurface for this one-step conversion. The symmetrical transition state complies with the definition of a coarctate mechanism.
The first procedures to prepare 4-bromo-4-methylpentanal and 4-azido-4-methylpentanal are reported. The latter compound and also the parent 4-azidobutanal do not lead to 4,5-dihydro-1,2,3,4-oxatriazoles by intramolecular 1,3-dipolar cycloaddition, although it was claimed to be so in the literature. The NMR spectroscopic data of such heterocycles reported previously do not agree with those of similar substances and are incompatible with 13C NMR spectroscopic chemical shifts calculated by quantum chemical methods in the presented work. These calculations show furthermore that the intramolecular cycloaddition of 4-azidobutanals to give the title compounds is strongly endothermic and thus most probably not possible.
A nitrogen-rich polymer was formed from the reaction of hexamethylene diisocyanate and N-[1-(2-hydroxyethyl)-1H-tetrazol-5-yl]-N-methylhydrazine (3) monomers. Compound 3 was synthesized by a nucleophilic substitution of the methylated sulfur atom of 4-[2-(acetoxy)ethyl]-2-methylthiosemicarbazide (10) with sodium azide and final deprotection of the formed N-[1-2-(acetoxyethyl)-1H-tetrazol-5-yl]-N-methylhydrazine (13). Moreover, the isomer 4-(2-azidoethyl)-2-methylsemicarbazide (18) to 3 was synthesized. Compounds 3, 10, 13 and 4-[2-(trimethylsilyloxy)ethyl]-2-methylthiosemicarbazide (15) were characterized by using vibrational spectroscopy (IR, Raman), mass spectrometry and multinuclear NMR spectroscopy. The crystal structures of 3, 10 and 13 were determined by using single-crystal X-ray diffraction. The molecular weights of the polymers were determined by GPC. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
With a nitrogen content of 93.3 %, “perazidomethane” (CN12) is highly explosive but nevertheless isolable. The title compound, which is accessible from commercially available trichloroacetonitrile in one step, undergoes simple dissociation and trapping reactions as well as more-complex transformations (for example, in the presence of norbornene; see scheme).
Mit einem Stickstoffgehalt von 93.3 % ist „Perazidomethan“, CN12, hochexplosiv, aber dennoch isolierbar. Die in einer Stufe aus käuflichem Trichloracetonitril zugängliche Titelverbindung geht sowohl einfache Dissoziations- und Abfangreaktionen als auch komplexere Umwandlungen ein (z. B. in Gegenwart von Norbornen; siehe Schema).
New and relatively stable methylene-2H-azirines 1 have been prepared by photolysis of allenyl azides or from 2-halo-2H-azirines by elimination of HX (X = halogen). The reaction of these methylene-2H-azirines with nucleophiles led to the highly stereo- and regio-selective formation of novel 1-aminovinyl derivatives with good to excellent yields. The trapping reactions of the less stable methylene-2H-azirines gave rise to similar results. Moreover, we were able to prove that the previous report on 2-(phenylsulfonyl)acrylonitrile (9) was based on incorrect data. For this reason, the latter compound can be supposed to be firstly described in this paper. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Trotz extremer Ringspannung und Reaktivität lassen sich die NMR-spektroskopisch beobachtbaren und aus den Aziden 1 leicht zugänglichen Heterocyclen 2 in neuartigen Additions- und Cycloadditionsreaktionen stereoselektiv umsetzen.
3,4-Diazidocyclobutenes 16 were prepared from the corresponding dihalides. Some of these diazides, such as parent compound 16 d and phenyl-substituted derivatives 16 c,f, underwent spontaneous stereoselective electrocyclic ring opening below room temperature, whereas the tetraalkyl derivatives of 16 had to be heated to force the same reaction. In most cases, the resulting 1,4-diazidobuta-1,3-dienes 8 were isolated to study their photochemical transformation into bi-2H-azirin-2-yls 9 via intermediate mono-azirines 17. Except for starting materials with a low number of substituents such as 9 d and 9 f, title compounds 9 underwent a thermal valence isomerization which led exclusively to pyridazines 18 at surprisingly low temperatures. Based on quantum-chemical calculations for the parent bi-2H-azirinyl-2-yl 9 d at the UB3LYP/6-31+G(d) and MR-MP2/TZV(2df,2p) levels, the valence isomerization process is best explained by simultaneous homolytic cleavage of both CN single bonds of 9 to generate energetically favorable N,N′ diradicals 26, which cyclize to 18. The theoretical studies indicate also that one stereoisomer of 9, namely, the rac compound, should undergo valence isomerization more easily than the other, which is in conformity with different rates of these rearrangement reactions found experimentally. For the tetramethyl-bi-2H-azirin-2-yls 9 g, which are better models for the experimentally studied compounds, simultaneous homolytic cleavage of both CN single bonds is also predicted by the calculations, although the intermediate diradicals 26 g are significantly higher in energy than those of the parent system 9 d.
Take the strain: Despite extreme ring strain and reactivity, the heterocycles 2, which are easily obtained from azides 1 and detectable by NMR spectroscopy, can be converted stereoselectively by novel addition and cycloaddition reactions.
Reagensfrei und unter sehr milden Bedingungen liefern die isolierbaren Vorstufen 3 die hochgespannten Cycloalkine 4 (Cycloheptin, Cyclohexin, Norbornin), die durch Cycloadditionen abgefangen werden können.
Cyclic ketene N,X-acetals 1 are electron-rich dipolarophiles that undergo 1,3-dipolar cycloaddition reactions with organic azides 2 ranging from alkyl to strongly electron-deficient azides, e.g., picryl azide (2L; R1=2,4,6-(NO2)3C6H2) and sulfonyl azides 2M–O (R1=XSO2; cf. Scheme 1). Reactions of the latter with the most-nucleophilic ketene N,N-acetals 1A provided the first examples for two-step HOMO(dipolarophile)–LUMO(1,3-dipole)-controlled 1,3-dipolar cycloadditions via intermediate zwitterions 3. To set the stage for an exploration of the frontier between concerted and two-step 1,3-dipolar cycloadditions of this type, we first describe the scope and limitations of concerted cycloadditions of 2 to 1 and delineate a number of zwitterions 3. Alkyl azides 2A–C add exclusively to ketene N,N-acetals that are derived from 1H-tetrazole (see 1A) and 1H-imidazole (see 1B,C), while almost all aryl azides yield cycloadducts 4 with the ketene N,X-acetals (X=NR, O, S) employed, except for the case of extreme steric hindrance of the 1,3-dipole (see 2E; R1=2,4,6-(tBu)3C6H2). The most electron-deficient paradigm, 2L, affords zwitterions 16D,E in the reactions with 1A, while ketene N,O- and N,S-acetals furnish products of unstable intermediate cycloadducts. By tuning the electronic and steric demands of aryl azides to those of ketene N,N-acetals 1A, we discovered new borderlines between concerted and two-step 1,3-dipolar cycloadditions that involve similar pairs of dipoles and dipolarophiles: 4-Nitrophenyl azide (2G) and the 2,2-dimethylpropylidene dipolarophile 1A (R, R=H, tBu) gave a cycloadduct 13 H, while 2-nitrophenyl azide (2 H) and the same dipolarophile afforded a zwitterion 16A. Isopropylidene dipolarophile 1A (R=Me) reacted with both 2G and 2 H to afford cycloadducts 13G,J) but furnished a zwitterion 16B with 2,4-dinitrophenyl azide (2I). Likewise, 1A (R=Me) reacted with the isomeric encumbered nitrophenyl azides 2J and 2K to yield a cycloadduct 13L and a zwitterion 16C, respectively. These examples suggest that, in principle, a host of such borderlines exist which can be crossed by means of small structural variations of the reactants. Eventually, we use 15N-NMR spectroscopy for the first time to characterize spirocyclic cycloadducts 10–14 and 17 (Table 6), and zwitterions 16 (Table 7).
1-Azaspiro[2.4]hepta-1,4,6-trienes 3 a–c have been prepared by photolysis or thermolysis of 6-azidofulvenes 5 a–c, which were accessible by nucleophilic substitution reactions of the precursors 4 a,b or by nucleophilic addition of hydrazoic acid to ethenylidenecyclopentadiene (6 c). The UV photoelectron spectrum of 2-methyl-1-azaspiro[2.4]hepta-1,4,6-triene (3 c) has been recorded and analyzed by making use of density functional theory (DFT) B3LYP calculations. Substantial homoconjugative interactions have been determined. The lone-pair orbital n(N) of the 2H-azirine nitrogen atom interacts with the π1 orbital of the cyclopentadiene ring. The energies of these orbitals are lowered or increased by 0.95 or 0.91 eV with respect to the two parent compounds cyclopentadiene (7) and 3-methyl-2H-azirine (9), respectively. In addition, in compound 3 c the π(CN) orbital of the three-membered ring interacts with a σ orbital of the cyclopentadiene unit and is destabilized by 0.47 eV by this effect.
Propargyl isothiocyanates 3 and buta-2,3-dienyl isothiocyanates 20 were prepared conventionally from amines and thiophosgene, or by a new, one-pot procedure using nucleophilic substitution to generate azides, which in turn served as the starting materials for a Staudinger reaction, followed by treatment of the resulting iminophosphoranes or iminophosphates with CS2. Equilibration, through a [3,3] sigmatropic rearrangement, of 3 and the allenyl thiocyanates 4 was established by flash vacuum pyrolysis or by thermolysis in solution. Even the reversible isomerization of the parent compounds 3f and 4f favors the allenyl thiocyanate. In the case of 20, an irreversible rearrangement reaction gave high yields of 2-thiocyanatobuta-1,3-dienes 21. A sequence of two [3,3] migration steps transformed 1,4-diisothiocyanatobut-2-ynes 3m and 3n into 2,3-dithiocyanatobuta-1,3-dienes 21m and 22, respectively. These reactions demonstrate that the [3,3] sigmatropic rearrangement of mustard oils, to afford high yields of thiocyanates bearing the thermodynamically less stable functional group, is possible if the conversion gives rise to a more stable carbon skeleton. The equilibration of 1-thiocyanatopent-2-en-4-ynes 12 and 1-thiocyanatopenta-1,2,4-trienes 14 could be explained by tandem [3,3]−[3,3] sigmatropic rearrangements through short lived 3-isothiocyanatopent-1-en-4-ynes 13. Thiocyanato-substituted vinylallenes 4k and 14 tended to electrocyclize to give cyclobutenes 7 and 15, respectively. Thiocyanates 21a, 21b, 21m and 14a, which exhibit a buta-1,3-diene structure, could be used in Diels−Alder reactions to afford the cycloadducts 25a, 25b, 26a, 26m, 28m, 29m, and 30.
A reinvestigation of the reaction between 2,3-diphenyl-2H-azirine (1) and phenyldiazomethane (2) has shown that a literature report has to be corrected since no vinyl azide 4 but rather the allylic compound 3-azido-1,2,3-triphenyl-1-propene (3) is produced. This stable substance, which can also be prepared by substitution reactions of allylic bromide (E)-10 or from alcohol (E)-11, may be separated into its geometrical isomers (E)-3 and (Z)-3, although these equilibrate through rapid [3,3] sigmatropic migration of the azido group. Attempts to synthesize 4 by dehydration of azido alcohols 7 using methanesulfonyl chloride and sulfur dioxide or by elimination of hydrogen chloride from azides 15 led only to 3 and 2-benzyl-2,3-diphenyl-2H-azirine (14). This heterocycle, which can also be prepared by Neber rearrangement, has been found to be the thermal and photochemical decomposition product of the unstable vinyl azides 4. However, dehydrations of 7 using thionyl chloride at low temperature have led to the first isolation of 1-azido-1,2,3-triphenyl-1-propenes (4). Starting with 3 and various other allylic azides, rearrangement reactions involving sigmatropic shift of the azido group or photochemical cis-trans isomerization have been investigated, as have base-catalyzed (prototropic) rearrangements to give vinyl azides.
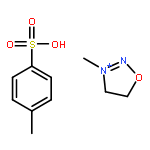
Just by the introduction of isomerizable groups and successive [3,3] or [2,3] rearrangements, the diols 1 can be transformed into the new synthetic building blocks 2. The conversion often proceeds with high stereoselectivity or even stereospecificity, and in some cases in a one-pot reaction. X, Y=NHCOCCl3, N3, P(O)Ph2, 4-SO2C6H4Me, S(O)Ar, SCOR.
Nur durch Einführung umlagerungsfähiger Gruppen und nachfolgende [3,3]- oder [2,3]-Isomerisierungen bilden sich aus den Diolen 1 die neuartigen Synthesebausteine 2. Die Umsetzungen verlaufen häufig stereoselektiv oder sogar stereospezifisch, und einige gelingen im Eintopfverfahren. X, Y = NHCOCCl3, N3, P(O)Ph2, 4-SO2C6H4Me, S(O)Ar, SCOR.
The present article summarizes the synthesis of allenes, which bear a functional group with at least two heteroatoms, by [2,3] or [3,3] sigmatropic rearrangements of appropriate propargyl precursors. Recently, this method has been extended to prepare new types of functionalized allenes such as isocyanates, isothiocyanates, isoselenocyanates, azides, thiocarbonates, and azo compounds. The title compounds are very reactive as shown by rapid intra- and intermolecular consecutive reactions. This reactivity can be used to synthesize heterocycles and doubly functionalized 1,3-butadienes.