Co-reporter:Douglas A. Hansen, Aaron A. Koch, and David H. Sherman
Journal of the American Chemical Society September 27, 2017 Volume 139(Issue 38) pp:13450-13450
Publication Date(Web):August 24, 2017
DOI:10.1021/jacs.7b06432
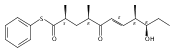
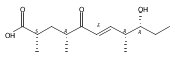
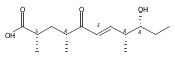
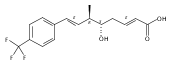
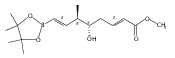
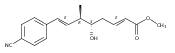
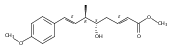
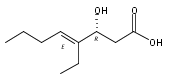
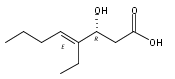
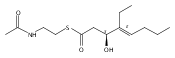
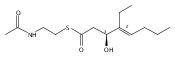
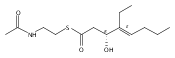
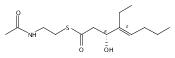
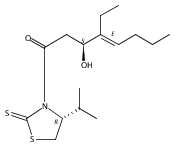
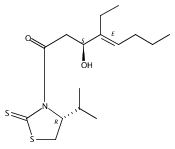
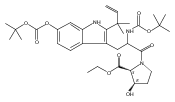
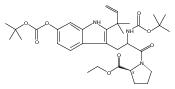
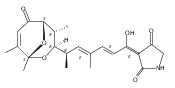
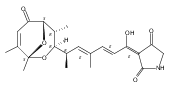
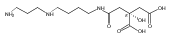
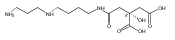
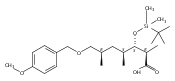
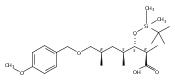
Polyketide biosynthetic pathways have been engineered to generate natural product analogs for over two decades. However, manipulation of modular type I polyketide synthases (PKSs) to make unnatural metabolites commonly results in attenuated yields or entirely inactive pathways, and the mechanistic basis for compromised production is rarely elucidated since rate-limiting or inactive domain(s) remain unidentified. Accordingly, we synthesized and assayed a series of modified pikromycin (Pik) pentaketides that mimic early pathway engineering to probe the substrate tolerance of the PikAIII-TE module in vitro. Truncated pentaketides were processed with varying efficiencies to corresponding macrolactones, while pentaketides with epimerized chiral centers were poorly processed by PikAIII-TE and failed to generate 12-membered ring products. Isolation and identification of extended but prematurely offloaded shunt products suggested that the Pik thioesterase (TE) domain has limited substrate flexibility and functions as a gatekeeper in the processing of unnatural substrates. Synthesis of an analogous hexaketide with an epimerized nucleophilic hydroxyl group allowed for direct evaluation of the substrate stereoselectivity of the excised TE domain. The epimerized hexaketide failed to undergo cyclization and was exclusively hydrolyzed, confirming the TE domain as a key catalytic bottleneck. In an accompanying paper, we engineer the standalone Pik thioesterase to yield a thioesterase (TES148C) and module (PikAIII-TES148C) that display gain-of-function processing of substrates with inverted hydroxyl groups.
Co-reporter:Aaron A. Koch, Douglas A. Hansen, Vikram V. Shende, Lawrence R. Furan, K. N. Houk, Gonzalo Jiménez-Osés, and David H. Sherman
Journal of the American Chemical Society September 27, 2017 Volume 139(Issue 38) pp:13456-13456
Publication Date(Web):August 24, 2017
DOI:10.1021/jacs.7b06436
Macrolactonization of natural product analogs presents a significant challenge to both biosynthetic assembly and synthetic chemistry. In the preceding paper, we identified a thioesterase (TE) domain catalytic bottleneck processing unnatural substrates in the pikromycin (Pik) system, preventing the formation of epimerized macrolactones. Here, we perform molecular dynamics simulations showing the epimerized hexaketide was accommodated within the Pik TE active site; however, intrinsic conformational preferences of the substrate resulted in predominately unproductive conformations, in agreement with the observed hydrolysis. Accordingly, we engineered the stereoselective Pik TE to yield a variant (TES148C) with improved reaction kinetics and gain-of-function processing of an unnatural, epimerized hexaketide. Quantum mechanical comparison of model TES148C and TEWT reaction coordinate diagrams revealed a change in mechanism from a stepwise addition–elimination (TEWT) to a lower energy concerted acyl substitution (TES148C), accounting for the gain-of-function and improved reaction kinetics. Finally, we introduced the S148C mutation into a polyketide synthase module (PikAIII-TE) to impart increased substrate flexibility, enabling the production of diastereomeric macrolactones.
Co-reporter:Jia-Hui Tay, Alonso J. Argüelles, Matthew D. DeMars II, Paul M. Zimmerman, David H. Sherman, and Pavel Nagorny
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8570-8570
Publication Date(Web):June 19, 2017
DOI:10.1021/jacs.7b03198
This work describes the first example of using chiral catalysts to control site-selectivity for the glycosylations of complex polyols such as 6-deoxyerythronolide B and oleandomycin-derived macrolactones. The regiodivergent introduction of sugars at the C3, C5, and C11 positions of macrolactones was achieved by selecting appropriate chiral acids as catalysts or through introduction of stoichiometric boronic acid-based additives. BINOL-based chiral phosphoric acids (CPAs) were used to catalyze highly selective glycosylations at the C5 positions of macrolactones (up to 99:1 rr), whereas the use of SPINOL-based CPAs resulted in selectivity switch and glycosylation of the C3 alcohol (up to 91:9 rr). Additionally, the C11 position of macrolactones was selectively functionalized through traceless protection of the C3/C5 diol with boronic acids prior to glycosylation. Investigation of the reaction mechanism for the CPA-controlled glycosylations revealed the involvement of covalently linked anomeric phosphates rather than oxocarbenium ion pairs as the reactive intermediates.
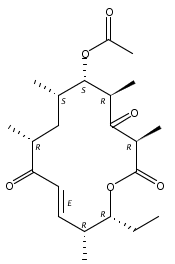
Co-reporter:Andrew N. Lowell, Matthew D. DeMars II, Samuel T. Slocum, Fengan Yu, Krithika Anand, Joseph A. Chemler, Nisha Korakavi, Jennifer K. Priessnitz, Sung Ryeol Park, Aaron A. Koch, Pamela J. Schultz, and David H. Sherman
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7913-7913
Publication Date(Web):May 19, 2017
DOI:10.1021/jacs.7b02875
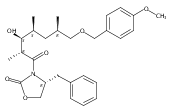
Polyketide synthases (PKSs) represent a powerful catalytic platform capable of effecting multiple carbon–carbon bond forming reactions and oxidation state adjustments. We explored the functionality of two terminal PKS modules that produce the 16-membered tylosin macrocycle, using them as biocatalysts in the chemoenzymatic synthesis of tylactone and its subsequent elaboration to complete the first total synthesis of the juvenimicin, M-4365, and rosamicin classes of macrolide antibiotics via late-stage diversification. Synthetic chemistry was employed to generate the tylactone hexaketide chain elongation intermediate that was accepted by the juvenimicin (Juv) ketosynthase of the penultimate JuvEIV PKS module. The hexaketide is processed through two complete modules (JuvEIV and JuvEV) in vitro, which catalyze elongation and functionalization of two ketide units followed by cyclization of the resulting octaketide into tylactone. After macrolactonization, a combination of in vivo glycosylation, selective in vitro cytochrome P450-mediated oxidation, and chemical oxidation was used to complete the scalable construction of a series of macrolide natural products in as few as 15 linear steps (21 total) with an overall yield of 4.6%.
Co-reporter:Michael M. Gilbert, Matthew D. DeMars II, Song Yang, Jessica M. Grandner, Shoulei Wang, Hengbin Wang, Alison R. H. Narayan, David H. Sherman, K. N. Houk, and John Montgomery
ACS Central Science December 27, 2017 Volume 3(Issue 12) pp:1304-1304
Publication Date(Web):November 15, 2017
DOI:10.1021/acscentsci.7b00450
The diversification of late stage synthetic intermediates provides significant advantages in efficiency in comparison to conventional linear approaches. Despite these advantages, accessing varying ring scaffolds and functional group patterns from a common intermediate poses considerable challenges using existing methods. The combination of regiodivergent nickel-catalyzed C–C couplings and site-selective biocatalytic C–H oxidations using the cytochrome P450 enzyme PikC addresses this problem by enabling a single late-stage linear intermediate to be converted to macrolactones of differing ring size and with diverse patterns of oxidation. The approach is made possible by a novel strategy for site-selective biocatalytic oxidation using a single biocatalyst, with site selectivity being governed by a temporarily installed directing group. Site selectivities of C–H oxidation by this directed approach can overcome positional bias due to C–H bond strength, acidity, inductive influences, steric accessibility, or immediate proximity to the directing group, thus providing complementarity to existing approaches.
Co-reporter:Amy E. Fraley, Marc Garcia-Borràs, Ashootosh Tripathi, Dheeraj Khare, Eduardo V. Mercado-Marin, Hong Tran, Qingyun Dan, Gabrielle P. Webb, Katharine R. Watts, Phillip Crews, Richmond Sarpong, Robert M. Williams, Janet L. Smith, K. N. Houk, and David H. Sherman
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:12060-12060
Publication Date(Web):August 4, 2017
DOI:10.1021/jacs.7b06773
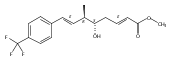
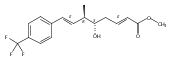
Malbrancheamide is a dichlorinated fungal indole alkaloid isolated from both Malbranchea aurantiaca and Malbranchea graminicola that belongs to a family of natural products containing a characteristic bicyclo[2.2.2]diazaoctane core. The introduction of chlorine atoms on the indole ring of malbrancheamide differentiates it from other members of this family and contributes significantly to its biological activity. In this study, we characterized the two flavin-dependent halogenases involved in the late-stage halogenation of malbrancheamide in two different fungal strains. MalA and MalA′ catalyze the iterative dichlorination and monobromination of the free substrate premalbrancheamide as the final steps in the malbrancheamide biosynthetic pathway. Two unnatural bromo-chloro-malbrancheamide analogues were generated through MalA-mediated chemoenzymatic synthesis. Structural analysis and computational studies of MalA′ in complex with three substrates revealed that the enzyme represents a new class of zinc-binding flavin-dependent halogenases and provides new insights into a potentially unique reaction mechanism.
Co-reporter:Matthew D. DeMars II, Fang Sheng, Sung Ryeol Park, Andrew N. Lowell, Larissa M. Podust, and David H. Sherman
ACS Chemical Biology 2016 Volume 11(Issue 9) pp:2642
Publication Date(Web):July 15, 2016
DOI:10.1021/acschembio.6b00479
Cytochrome P450 monooxygenases (P450s) are some of nature’s most ubiquitous and versatile enzymes for performing oxidative metabolic transformations. Their unmatched ability to selectively functionalize inert C–H bonds has led to their increasing employment in academic and industrial settings for the production of fine and commodity chemicals. Many of the most interesting and potentially biocatalytically useful P450s come from microorganisms, where they catalyze key tailoring reactions in natural product biosynthetic pathways. While most of these enzymes act on structurally complex pathway intermediates with high selectivity, they often exhibit narrow substrate scope, thus limiting their broader application. In the present study, we investigated the reactivity of the P450 MycCI from the mycinamicin biosynthetic pathway toward a variety of macrocyclic compounds and discovered that the enzyme exhibits appreciable activity on several 16-membered ring macrolactones independent of their glycosylation state. These results were corroborated by performing equilibrium substrate binding experiments, steady-state kinetics studies, and X-ray crystallographic analysis of MycCI bound to its native substrate mycinamicin VIII. We also characterized TylHI, a homologous P450 from the tylosin pathway, and showed that its substrate scope is severely restricted compared to MycCI. Thus, the ability of the latter to hydroxylate both macrocyclic aglycones and macrolides sets it apart from related biosynthetic P450s and highlights its potential for developing novel P450 biocatalysts with broad substrate scope and high regioselectivity.
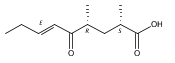
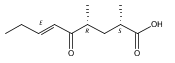
Co-reporter:Xingwang Zhang, Zhong Li, Lei Du, George E. Chlipala, Patricia C. Lopez, Wei Zhang, David H. Sherman, Shengying Li
Tetrahedron Letters 2016 Volume 57(Issue 52) pp:5919-5923
Publication Date(Web):28 December 2016
DOI:10.1016/j.tetlet.2016.11.080
•The first linear tirandamycin (K) was isolated from Streptomyces sp. 307–9 (ΔtamI).•Tirandamycin K provides new insights into tirandamycin biosynthesis.•The bicyclic ketal ring is necessary for the antibiotic activity of tirandamycins.Tirandamycin K (7), the first linear 7,13;9,13-diseco-tirandamycin derivative, was isolated from the tamI (encoding the TamI P450 monooxygenase) disruption mutant strain (ΔtamI) of marine Streptomyces sp. 307–9. Its chemical structure with relative and absolute configurations was elucidated by a combination of extensive spectroscopic analyses and biosynthetic inferences. Structural elucidation of this unusual compound provides new insights into tirandamycin biosynthesis. Moreover, examination of the biological activity of 7 confirms the essential function of the bicyclic ketal ring for antibiotic activities of tirandamycins.
Co-reporter:Ashootosh Tripathi;Si-Sun Choi
Journal of Industrial Microbiology & Biotechnology 2016 Volume 43( Issue 8) pp:1189-1193
Publication Date(Web):2016 August
DOI:10.1007/s10295-016-1790-2
Tautomycetin (TMC) is a linear polyketide metabolite produced by Streptomyces sp. CK4412 that has been reported to possess multiple biological functions including T cell-specific immunosuppressive and anticancer activities that occur through a mechanism of differential inhibition of protein phosphatases such as PP1, PP2A, and SHP2. We previously reported the characterization of the entire TMC biosynthetic gene cluster constituted by multifunctional type I polyketide synthase (PKS) assembly and suggested that the linear form of TMC could be generated via free acid chain termination by a narrow TMC thioesterase (TE) pocket. The modular nature of the assembly presents a unique opportunity to alter or interchange the native biosynthetic domains to produce targeted variants of TMC. Herein, we report swapping of the TMC TE domain sequence with the exact counterpart of the macrocyclic polyketide pikromycin (PIK) TE. PIK TE-swapped Streptomyces sp. CK4412 mutant produced not only TMC, but also a cyclized form of TMC, implying that the bioengineering based in vivo custom construct can be exploited to produce engineered macrolactones with new structural functionality.
Co-reporter:Shasha Li; Andrew N. Lowell; Fengan Yu; Avi Raveh; Sean A. Newmister; Nathan Bair; Jeffrey M. Schaub; Robert M. Williams
Journal of the American Chemical Society 2015 Volume 137(Issue 49) pp:15366-15369
Publication Date(Web):December 2, 2015
DOI:10.1021/jacs.5b10136
Hapalindoles are bioactive indole alkaloids with fascinating polycyclic ring systems whose biosynthetic assembly mechanism has remained unknown since their initial discovery in the 1980s. In this study, we describe the fam gene cluster from the cyanobacterium Fischerella ambigua UTEX 1903 encoding hapalindole and ambiguine biosynthesis along with the characterization of two aromatic prenyltransferases, FamD1 and FamD2, and a previously undescribed cyclase, FamC1. These studies demonstrate that FamD2 and FamC1 act in concert to form the tetracyclic core ring system of the hapalindoles from cis-indole isonitrile and geranyl pyrophosphate through a presumed biosynthetic Cope rearrangement and subsequent 6-exo-trig cyclization/electrophilic aromatic substitution reaction.
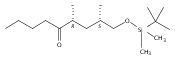
Co-reporter:Douglas A. Hansen; Aaron A. Koch
Journal of the American Chemical Society 2015 Volume 137(Issue 11) pp:3735-3738
Publication Date(Web):March 2, 2015
DOI:10.1021/ja511743n
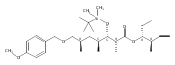
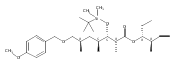
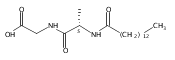
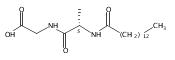
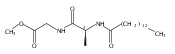
Biochemical characterization of polyketide synthases (PKSs) has relied on synthetic substrates functionalized as electrophilic esters to acylate the enzyme and initiate the catalytic cycle. In these efforts, N-acetylcysteamine thioesters have typically been employed for in vitro studies of full PKS modules as well as excised domains. However, substrate engineering approaches to control the catalytic cycle of a full PKS module harboring multiple domains remain underexplored. This study examines a series of alternatively activated native hexaketide substrates on the catalytic outcome of PikAIV, the sixth and final module of the pikromycin (Pik) pathway. We demonstrate the ability to control product formation with greater than 10:1 selectivity for either full module catalysis, leading to a 14-membered macrolactone, or direct cyclization to a 12-membered ring. This outcome was achieved through modifying the type of hexaketide ester employed, demonstrating the utility of substrate engineering in PKS functional studies and biocatalysis.
Co-reporter:Joseph A. Chemler; Ashootosh Tripathi; Douglas A. Hansen; Mark O’Neil-Johnson; Russell B. Williams; Courtney Starks; Sung Ryeol Park
Journal of the American Chemical Society 2015 Volume 137(Issue 33) pp:10603-10609
Publication Date(Web):July 31, 2015
DOI:10.1021/jacs.5b04842
The structural scaffolds of many complex natural products are produced by multifunctional type I polyketide synthase (PKS) enzymes that operate as biosynthetic assembly lines. The modular nature of these mega-enzymes presents an opportunity to construct custom biocatalysts built in a lego-like fashion by inserting, deleting, or exchanging native or foreign domains to produce targeted variants of natural polyketides. However, previously engineered PKS enzymes are often impaired resulting in limited production compared to native systems. Here, we show a versatile method for generating and identifying functional chimeric PKS enzymes for synthesizing custom macrolactones and macrolides. PKS genes from the pikromycin and erythromycin pathways were hybridized in Saccharomyces cerevisiae to generate hybrid libraries. We used a 96-well plate format for plasmid purification, transformations, sequencing, protein expression, in vitro reactions and analysis of metabolite formation. Active chimeric enzymes were identified with new functionality. Streptomyces venezuelae strains that expressed these PKS chimeras were capable of producing engineered macrolactones. Furthermore, a macrolactone generated from selected PKS chimeras was fully functionalized into a novel macrolide analogue. This method permits the engineering of PKS pathways as modular building blocks for the production of new antibiotic-like molecules.
Co-reporter:Ken Chih-Chien Cheng; Shugeng Cao; Avi Raveh; Ryan MacArthur; Patricia Dranchak; George Chlipala; Matthew T. Okoneski; Rajarshi Guha; Richard T. Eastman△; Jing Yuan△; Pamela J. Schultz; Xin-zhuan Su△; Giselle Tamayo-Castillo; Teatulohi Matainaho; Jon Clardy; David H. Sherman;James Inglese
Journal of Natural Products 2015 Volume 78(Issue 10) pp:2411-2422
Publication Date(Web):October 14, 2015
DOI:10.1021/acs.jnatprod.5b00489
Methods to identify the bioactive diversity within natural product extracts (NPEs) continue to evolve. NPEs constitute complex mixtures of chemical substances varying in structure, composition, and abundance. NPEs can therefore be challenging to evaluate efficiently with high-throughput screening approaches designed to test pure substances. Here we facilitate the rapid identification and prioritization of antimalarial NPEs using a pharmacologically driven, quantitative high-throughput-screening (qHTS) paradigm. In qHTS each NPE is tested across a concentration range from which sigmoidal response, efficacy, and apparent EC50s can be used to rank order NPEs for subsequent organism reculture, extraction, and fractionation. Using an NPE library derived from diverse marine microorganisms we observed potent antimalarial activity from two Streptomyces sp. extracts identified from thousands tested using qHTS. Seven compounds were isolated from two phylogenetically related Streptomyces species: Streptomyces ballenaensis collected from Costa Rica and Streptomyces bangulaensis collected from Papua New Guinea. Among them we identified actinoramides A and B, belonging to the unusually elaborated nonproteinogenic amino-acid-containing tetrapeptide series of natural products. In addition, we characterized a series of new compounds, including an artifact, 25-epi-actinoramide A, and actinoramides D, E, and F, which are closely related biosynthetic congeners of the previously reported metabolites.
Co-reporter:Andrew N. Lowell;Nicholas Santoro;Steven M. Swaney;Thomas J. McQuade;Pamela J. Schultz;Martha J. Larsen
Chemical Biology & Drug Design 2015 Volume 86( Issue 6) pp:1331-1338
Publication Date(Web):
DOI:10.1111/cbdd.12614
Novel antimicrobials that effectively inhibit bacterial growth are essential to fight the growing threat of antibiotic resistance. A promising target is the bacterial ribosome, a 2.5 MDa organelle susceptible to several biorthogonal modes of action used by different classes of antibiotics. To promote the discovery of unique inhibitors, we have miniaturized a coupled transcription/translation assay using E. coli and applied it to screen a natural product library of ~30 000 extracts. We significantly reduced the scale of the assay to 2 μL in a 1536-well plate format and decreased the effective concentration of costly reagents. The improved assay returned 1327 hits (4.6% hit rate) with %CV and Z′ values of 8.5% and 0.74, respectively. This assay represents a significant advance in molecular screening, both in miniaturization and its application to a natural product extract library, and we intend to apply it to a broad array of pathogenic microbes in the search for novel anti-infective agents.
Co-reporter:Alpa Sidhu, Justin R. Miller, Ashootosh Tripathi, Danielle M. Garshott, Amy L. Brownell, Daniel J. Chiego, Carl Arevang, Qinghua Zeng, Leah C. Jackson, Shelby A. Bechler, Michael U. Callaghan, George H. Yoo, Seema Sethi, Ho-Sheng Lin, Joseph H. Callaghan, Giselle Tamayo-Castillo, David H. Sherman, Randal J. Kaufman, and Andrew M. Fribley
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 11) pp:1122
Publication Date(Web):September 8, 2015
DOI:10.1021/acsmedchemlett.5b00133
Oral squamous cell carcinoma (OSCC) is the most common cancer affecting the oral cavity, and US clinics will register about 30,000 new patients in 2015. Current treatment modalities include chemotherapy, surgery, and radiotherapy, which often result in astonishing disfigurement. Cancers of the head and neck display enhanced levels of glucose-regulated proteins and translation initiation factors associated with endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). Previous work demonstrated that chemically enforced UPR could overwhelm these adaptive features and selectively kill malignant cells. The threonyl-tRNA synthetase (ThRS) inhibitor borrelidin and two congeners were discovered in a cell-based chemical genomic screen. Borrelidin increased XBP1 splicing and led to accumulation of phosphorylated eIF2α and UPR-associated genes, prior to death in panel of OSCC cells. Murine embryonic fibroblasts (MEFs) null for GCN2 and PERK were less able to accumulate UPR markers and were resistant to borrelidin. This study demonstrates that UPR induction is a feature of ThRS inhibition and adds to a growing body of literature suggesting ThRS inhibitors might selectively target cancer cells.Keywords: borrelidin; Chop; ER stress; high throughput screen; natural products; oral cancer; oral squamous cell carcinoma; protein folding; UPR; Xbp1, BiP/GRP78
Co-reporter:Ashootosh Tripathi ; Michael M. Schofield ; George E. Chlipala ; Pamela J. Schultz ; Isaiah Yim ; Sean A. Newmister ; Tyler D. Nusca ; Jamie B. Scaglione ; Philip C. Hanna ; Giselle Tamayo-Castillo
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1579-1586
Publication Date(Web):January 8, 2014
DOI:10.1021/ja4115924
Siderophores are high-affinity iron chelators produced by microorganisms and frequently contribute to the virulence of human pathogens. Targeted inhibition of the biosynthesis of siderophores staphyloferrin B of Staphylococcus aureus and petrobactin of Bacillus anthracis hold considerable potential as a single or combined treatment for methicillin-resistant S. aureus (MRSA) and anthrax infection, respectively. The biosynthetic pathways for both siderophores involve a nonribosomal peptide synthetase independent siderophore (NIS) synthetase, including SbnE in staphyloferrin B and AsbA in petrobactin. In this study, we developed a biochemical assay specific for NIS synthetases to screen for inhibitors of SbnE and AsbA against a library of marine microbial-derived natural product extracts (NPEs). Analysis of the NPE derived from Streptomyces tempisquensis led to the isolation of the novel antibiotics baulamycins A (BmcA, 6) and B (BmcB, 7). BmcA and BmcB displayed in vitro activity with IC50 values of 4.8 μM and 19 μM against SbnE and 180 μM and 200 μM against AsbA, respectively. Kinetic analysis showed that the compounds function as reversible competitive enzyme inhibitors. Liquid culture studies with S. aureus, B. anthracis, E. coli, and several other bacterial pathogens demonstrated the capacity of these natural products to penetrate bacterial barriers and inhibit growth of both Gram-positive and Gram-negative species. These studies provide proof-of-concept that natural product inhibitors targeting siderophore virulence factors can provide access to novel broad-spectrum antibiotics, which may serve as important leads for the development of potent anti-infective agents
Co-reporter:Wei Zhang ; Yi Liu ; Jinyong Yan ; Shaona Cao ; Fali Bai ; Ying Yang ; Shaohua Huang ; Lishan Yao ; Yojiro Anzai ; Fumio Kato ; Larissa M. Podust ; David H. Sherman ;Shengying Li
Journal of the American Chemical Society 2014 Volume 136(Issue 9) pp:3640-3646
Publication Date(Web):February 12, 2014
DOI:10.1021/ja4130302
Cytochrome P450 enzymes are capable of catalyzing a great variety of synthetically useful reactions such as selective C–H functionalization. Surrogate redox partners are widely used for reconstitution of P450 activity based on the assumption that the choice of these auxiliary proteins or their mode of action does not affect the type and selectivity of reactions catalyzed by P450s. Herein, we present an exceptional example to challenge this postulate. MycG, a multifunctional biosynthetic P450 monooxygenase responsible for hydroxylation and epoxidation of 16-membered ring macrolide mycinamicins, is shown to catalyze the unnatural N-demethylation(s) of a range of mycinamicin substrates when partnered with the free Rhodococcus reductase domain RhFRED or the engineered Rhodococcus-spinach hybrid reductase RhFRED-Fdx. By contrast, MycG fused with the RhFRED or RhFRED-Fdx reductase domain mediates only physiological oxidations. This finding highlights the larger potential role of variant redox partner protein–protein interactions in modulating the catalytic activity of P450 enzymes.
Co-reporter:Solymar Negretti ; Alison R. H. Narayan ; Karoline C. Chiou ; Petrea M. Kells ; Jessica L. Stachowski ; Douglas A. Hansen ; Larissa M. Podust ; John Montgomery
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4901-4904
Publication Date(Web):March 14, 2014
DOI:10.1021/ja5016052
Highly regioselective remote hydroxylation of a natural product scaffold is demonstrated by exploiting the anchoring mechanism of the biosynthetic P450 monooxygenase PikCD50N-RhFRED. Previous studies have revealed structural and biochemical evidence for the role of a salt bridge between the desosamine N,N-dimethylamino functionality of the natural substrate YC-17 and carboxylate residues within the active site of the enzyme, and selectivity in subsequent C–H bond functionalization. In the present study, a substrate-engineering approach was conducted that involves replacing desosamine with varied synthetic N,N-dimethylamino anchoring groups. We then determined their ability to mediate enzymatic total turnover numbers approaching or exceeding that of the natural sugar, while enabling ready introduction and removal of these amino anchoring groups from the substrate. The data establish that the size, stereochemistry, and rigidity of the anchoring group influence the regioselectivity of enzymatic hydroxylation. The natural anchoring group desosamine affords a 1:1 mixture of regioisomers, while synthetic anchors shift YC-17 analogue C-10/C-12 hydroxylation from 20:1 to 1:4. The work demonstrates the utility of substrate engineering as an orthogonal approach to protein engineering for modulation of regioselective C–H functionalization in biocatalysis.
Co-reporter:Dr. Sean A. Newmister; David H. Sherman
ChemBioChem 2014 Volume 15( Issue 8) pp:1079-1081
Publication Date(Web):
DOI:10.1002/cbic.201402095
Co-reporter:Douglas A. Hansen ; Christopher M. Rath ; Eli B. Eisman ; Alison R. H. Narayan ; Jeffrey D. Kittendorf ; Jonathan D. Mortison ; Yeo Joon Yoon
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11232-11238
Publication Date(Web):June 18, 2013
DOI:10.1021/ja404134f
A biocatalytic platform that employs the final two monomodular type I polyketide synthases of the pikromycin pathway in vitro followed by direct appendage of d-desosamine and final C–H oxidation(s) in vivo was developed and applied toward the synthesis of a suite of 12- and 14-membered ring macrolide natural products. This methodology delivered both compound classes in 13 steps (longest linear sequence) from commercially available (R)-Roche ester in >10% overall yields.
Co-reporter:Dr. Wei Zhang;Dr. Jeffrey L. Fortman;Dr. Jacob C. Carlson;Dr. Jinyong Yan;Yi Liu;Fali Bai;Dr. Wenna Guan;Dr. Junyong Jia; Teatulohi Matainaho; David H. Sherman; Shengying Li
ChemBioChem 2013 Volume 14( Issue 3) pp:301-306
Publication Date(Web):
DOI:10.1002/cbic.201200743
Co-reporter:Joseph A. Chemler ; Tonia J. Buchholz ; Todd W. Geders ; David L. Akey ; Christopher M. Rath ; George E. Chlipala ; Janet L. Smith
Journal of the American Chemical Society 2012 Volume 134(Issue 17) pp:7359-7366
Publication Date(Web):April 5, 2012
DOI:10.1021/ja2112228
Germicidin synthase (Gcs) from Streptomyces coelicolor is a type III polyketide synthase (PKS) with broad substrate flexibility for acyl groups linked through a thioester bond to either coenzyme A (CoA) or acyl carrier protein (ACP). Germicidin synthesis was reconstituted in vitro by coupling Gcs with fatty acid biosynthesis. Since Gcs has broad substrate flexibility, we directly compared the kinetic properties of Gcs with both acyl-ACP and acyl-CoA. The catalytic efficiency of Gcs for acyl-ACP was 10-fold higher than for acyl-CoA, suggesting a strong preference toward carrier protein starter unit transfer. The 2.9 Å germicidin synthase crystal structure revealed canonical type III PKS architecture along with an unusual helical bundle of unknown function that appears to extend the dimerization interface. A pair of arginine residues adjacent to the active site affect catalytic activity but not ACP binding. This investigation provides new and surprising information about the interactions between type III PKSs and ACPs that will facilitate the construction of engineered systems for production of novel polyketides.
Co-reporter:Kyle L. Bolduc, Scott D. Larsen and David H. Sherman
Chemical Communications 2012 vol. 48(Issue 51) pp:6414-6416
Publication Date(Web):02 May 2012
DOI:10.1039/C2CC32417B
A flexible and divergent synthesis of cryptophycin unit A analogues is described. This method relies on iridium-catalysed stereo- and enantioselective crotylation and chemoselective one-pot oxidative olefination to access common intermediate 8. Heck, cross metathesis, and Suzuki-Miyaura reactions are illustrated for the generation of methyl ester unit A analogues 10a–d.
Co-reporter:Shengying Li, Krithika Srinivasan, Hong Tran, Fengan Yu, Jennifer M. Finefield, James D. Sunderhaus, Timothy J. McAfoos, Sachiko Tsukamoto, Robert M. Williams and David H. Sherman
MedChemComm 2012 vol. 3(Issue 8) pp:987-996
Publication Date(Web):16 Apr 2012
DOI:10.1039/C2MD20029E
The biosynthesis of fungal bicyclo[2.2.2]diazaoctane indole alkaloids with a wide spectrum of biological activities have attracted increasing interest. Until recently, the details of these biosynthetic pathways have remained largely unknown due to lack of information on the fungal derived biosynthetic gene clusters. Herein, we report identification of three new fungal gene clusters responsible for biosynthesis of a select group of bicyclo[2.2.2]diazaoctane indole alkaloids including (+)-notoamide, paraherquamide and malbrancheamide by genome mining. In each gene cluster, we identified a non-ribosomal peptide synthetase, a variant number of prenyltransferases, and a series of oxidases responsible for the diverse tailoring modifications of the cyclodipeptide structural core. Based on the comparative analysis of four natural product metabolic systems including (+)/(−)-notoamide, paraherquamide and malbrancheamide, we were able to propose an enzyme for each step in the respective biosynthetic pathways through deep gene annotation and on-going biochemical studies. We proposed that two different types of intramolecular Diels-Alderases operate to generate the monooxopiperazine and dioxopiperazine ring systems for this class of alkaloid natural products.
Co-reporter:Yousong Ding ; Christopher M. Rath ; Kyle L. Bolduc ; Kristina Håkansson
Journal of the American Chemical Society 2011 Volume 133(Issue 37) pp:14492-14495
Publication Date(Web):August 8, 2011
DOI:10.1021/ja204716f
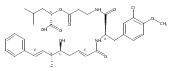
Cryptophycins (Crp) are a group of cyanobacterial depsipeptides with activity against drug-resistant tumors. Although they have been shown to be promising, further efforts are required to return these highly potent compounds to the clinic through a new generation of analogues with improved medicinal properties. Herein, we report a chemosynthetic route relying on the multifunctional enzyme CrpD-M2 that incorporates a 2-hydroxy acid moiety (unit D) into Crp analogues. CrpD-M2 is a unique non-ribosomal peptide synthetase (NRPS) module comprised of condensation–adenylation–ketoreduction–thiolation (C-A-KR-T) domains. We interrogated A-domain 2-keto and 2-hydroxy acid activation and loading, and KR domain activity in the presence of NADPH and NADH. The resulting 2-hydroxy acid was elongated with three synthetic Crp chain elongation intermediate analogues through ester bond formation catalyzed by CrpD-M2 C domain. Finally, the enzyme-bound seco-Crp products were macrolactonized by the Crp thioesterase. Analysis of these sequential steps was enabled through LC-FTICR-MS of enzyme-bound intermediates and products. This novel chemoenzymatic synthesis of Crp involves four sequential catalytic steps leading to the incorporation of a 2-hydroxy acid moiety in the final chain elongation intermediate. The presented work constitutes the first example where a NRPS-embedded KR domain is employed for assembly of a fully elaborated natural product, and serves as a proof-of-principle for chemoenzymatic synthesis of new Crp analogues.
Co-reporter:Shengying Li, Jennifer M. Finefield, James D. Sunderhaus, Timothy J. McAfoos, Robert M. Williams, and David H. Sherman
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:788-791
Publication Date(Web):December 20, 2011
DOI:10.1021/ja2093212
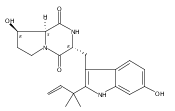
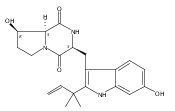
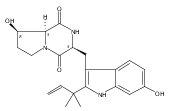
Notoamides produced by Aspergillus spp. bearing the bicyclo[2.2.2]diazaoctane core structure with unusual structural diversity represent a compelling system to understand the biosynthesis of fungal prenylated indole alkaloids. Herein, we report the in vitro characterization of NotB, which catalyzes the indole 2,3-oxidation of notoamide E (13), leading to notoamides C (11) and D (12) through an apparent pinacol-like rearrangement. This unique enzymatic reaction with high substrate specificity, together with the information derived from precursor incorporation experiments using [13C]2–[15N]2 quadruply labeled notoamide S (10), demonstrates 10 as a pivotal branching point in notoamide biosynthesis.
Co-reporter:Shilah A. Bonnett, Christopher M. Rath, Abdur-Rafay Shareef, Joanna R. Joels, Joseph A. Chemler, Kristina Håkansson, Kevin Reynolds, David H. Sherman
Chemistry & Biology 2011 Volume 18(Issue 9) pp:1075-1081
Publication Date(Web):23 September 2011
DOI:10.1016/j.chembiol.2011.07.016
Polyketide natural products generated by type I modular polyketide synthases (PKSs) are vital components in our drug repertoire. To reprogram these biosynthetic assembly lines, we must first understand the steps that occur within the modular “black boxes.” Herein, key steps of acyl-CoA extender unit selection are explored by in vitro biochemical analysis of the PikAIV PKS model system. Two complementary approaches are employed: a fluorescent-probe assay for steady-state kinetic analysis, and Fourier Transform Ion Cyclotron Resonance-mass spectrometry (FTICR-MS) to monitor active-site occupancy. Findings from five enzyme variants and four model substrates have enabled a model to be proposed involving catalysis based upon acyl-CoA substrate loading followed by differential rates of hydrolysis. These efforts suggest a strategy for future pathway engineering efforts using unnatural extender units with slow rates of hydrolytic off-loading from the acyltransferase domain.Highlights► A new model for CoA-extender unit selection in type I PKSs was developed based on differential rates of extender unit AT domain hydrolysis ► FTICR mass spectrometry enabled PKS active-site occupancy to be determined ► A complementary fluorescent reporter assay enabled steady-state kinetic analysis to be performed ► Results of acyl-CoA extender unit substrate specificity studies were applied to generate a new macrolactone from an unnatural ethylmalonyl-CoA extender unit
Co-reporter:Patricia G. Cruz, Douglas S. Auld, Pamela J. Schultz, Scott Lovell, Kevin P. Battaile, Ryan MacArthur, Min Shen, Giselle Tamayo-Castillo, James Inglese, David H. Sherman
Chemistry & Biology 2011 Volume 18(Issue 11) pp:1442-1452
Publication Date(Web):23 November 2011
DOI:10.1016/j.chembiol.2011.08.011
The chemical diversity of nature has tremendous potential for the discovery of molecular probes and medicinal agents. However, sensitivity of HTS assays to interfering components of crude extracts derived from plants, and macro- and microorganisms has curtailed their use in lead discovery. Here, we describe a process for leveraging the concentration-response curves obtained from quantitative HTS to improve the initial selection of “actives” from a library of partially fractionated natural product extracts derived from marine actinomycetes and fungi. By using pharmacological activity, the first-pass CRC paradigm improves the probability that labor-intensive subsequent steps of reculturing, extraction, and bioassay-guided isolation of active component(s) target the most promising strains and growth conditions. We illustrate how this process identified a family of fungal metabolites as potent inhibitors of firefly luciferase, subsequently resolved in molecular detail by X-ray crystallography.Highlights► Quantitative high-throughput screening of natural product extracts ► Retrospective single-concentration HTS analysis across 35 assays ► Strategy for identification, isolation, and structural elucidation of secondary metabolites ► Cocrystal structure of potent natural product inhibitor with firefly luciferase
Co-reporter:Christopher M. Rath, Benjamin Janto, Josh Earl, Azad Ahmed, Fen Z. Hu, Luisa Hiller, Meg Dahlgren, Rachael Kreft, Fengan Yu, Jeremy J. Wolff, Hye Kyong Kweon, Michael A. Christiansen, Kristina Håkansson, Robert M. Williams, Garth D. Ehrlich, and David H. Sherman
ACS Chemical Biology 2011 Volume 6(Issue 11) pp:1244
Publication Date(Web):August 29, 2011
DOI:10.1021/cb200244t
In many macroorganisms, the ultimate source of potent biologically active natural products has remained elusive due to an inability to identify and culture the producing symbiotic microorganisms. As a model system for developing a meta-omic approach to identify and characterize natural product pathways from invertebrate-derived microbial consortia, we chose to investigate the ET-743 (Yondelis) biosynthetic pathway. This molecule is an approved anticancer agent obtained in low abundance (10–4–10–5 % w/w) from the tunicate Ecteinascidia turbinata and is generated in suitable quantities for clinical use by a lengthy semisynthetic process. On the basis of structural similarities to three bacterial secondary metabolites, we hypothesized that ET-743 is the product of a marine bacterial symbiont. Using metagenomic sequencing of total DNA from the tunicate/microbial consortium, we targeted and assembled a 35 kb contig containing 25 genes that comprise the core of the NRPS biosynthetic pathway for this valuable anticancer agent. Rigorous sequence analysis based on codon usage of two large unlinked contigs suggests that Candidatus Endoecteinascidia frumentensis produces the ET-743 metabolite. Subsequent metaproteomic analysis confirmed expression of three key biosynthetic proteins. Moreover, the predicted activity of an enzyme for assembly of the tetrahydroisoquinoline core of ET-743 was verified in vitro. This work provides a foundation for direct production of the drug and new analogues through metabolic engineering. We expect that the interdisciplinary approach described is applicable to diverse host–symbiont systems that generate valuable natural products for drug discovery and development.
Co-reporter:Dr. Liangcai Gu;Eli B. Eisman;Dr. Somnath Dutta;Dr. Titus M. Franzmann;Dr. Stefan Walter;Dr. William H. Gerwick;Dr. Georgios Skiniotis;Dr. David H. Sherman
Angewandte Chemie 2011 Volume 123( Issue 12) pp:2847-2850
Publication Date(Web):
DOI:10.1002/ange.201005280
Co-reporter:Dr. Liangcai Gu;Eli B. Eisman;Dr. Somnath Dutta;Dr. Titus M. Franzmann;Dr. Stefan Walter;Dr. William H. Gerwick;Dr. Georgios Skiniotis;Dr. David H. Sherman
Angewandte Chemie International Edition 2011 Volume 50( Issue 12) pp:2795-2798
Publication Date(Web):
DOI:10.1002/anie.201005280
Co-reporter:Yousong Ding ; Jeffrey R. de Wet ; James Cavalcoli ; Shengying Li ; Thomas J. Greshock ; Kenneth A. Miller ; Jennifer M. Finefield ; James D. Sunderhaus ; Timothy J. McAfoos ; Sachiko Tsukamoto ; Robert M. Williams
Journal of the American Chemical Society 2010 Volume 132(Issue 36) pp:12733-12740
Publication Date(Web):August 19, 2010
DOI:10.1021/ja1049302
Stephacidin and notoamide natural products belong to a group of prenylated indole alkaloids containing a core bicyclo[2.2.2]diazaoctane ring system. These bioactive fungal secondary metabolites have a range of unusual structural and stereochemical features but their biosynthesis has remained uncharacterized. Herein, we report the first biosynthetic gene cluster for this class of fungal alkaloids based on whole genome sequencing of a marine-derived Aspergillus sp. Two central pathway enzymes catalyzing both normal and reverse prenyltransfer reactions were characterized in detail. Our results establish the early steps for creation of the prenylated indole alkaloid structure and suggest a scheme for the biosynthesis of stephacidin and notoamide metabolites. The work provides the first genetic and biochemical insights for understanding the structural diversity of this important family of fungal alkaloids.
Co-reporter:Tonia J. Buchholz, Christopher M. Rath, Nicole B. Lopanik, Noah P. Gardner, Kristina Håkansson, David H. Sherman
Chemistry & Biology 2010 Volume 17(Issue 10) pp:1092-1100
Publication Date(Web):29 October 2010
DOI:10.1016/j.chembiol.2010.08.008
In vitro analysis of natural product biosynthetic gene products isolated from unculturable symbiotic bacteria is necessary to probe the functionalities of these enzymes. Herein, we report the biochemical characterization of BryR, the 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase (HMGS) homolog implicated in β-branching at C13 and C21 of the core ring system from the bryostatin metabolic pathway (Bry). We confirmed the activity of BryR using two complementary methods, radio-SDS PAGE, and Fourier transform ion cyclotron resonance-mass spectrometry (FTICR-MS). The activity of BryR depended on pairing of the native acetoacetyl-BryM3 acceptor acyl carrier protein (ACP) with an appropriate donor acetyl-ACP from a heterologous HMGS cassette. Additionally, the ability of BryR to discriminate between various ACPs was assessed using a surface plasmon resonance (SPR)-based protein-protein binding assay. Our data suggest that specificity for a protein-bound acyl group is a distinguishing feature between HMGS homologs found in PKS or PKS/NRPS biosynthetic pathways and those of primary metabolism. These findings reveal an important example of molecular recognition between protein components that are essential for biosynthetic fidelity in natural product assembly and modification.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (113 K)Download as PowerPoint slideHighlights► Biochemical analysis of BryR verifies its role in PKS β-branching ► Surrogate acetyl-ACP donors of BryR identified by radio-SDS PAGE, FTICR-MS, and SPR ► Specificity for protein-bound acyl groups is an important feature of PKS biosynthesis
Co-reporter:JamieB. Scaglione Dr.;DavidL. Akey Dr.;Rachel Sullivan;JeffreyD. Kittendorf Dr.;ChristopherM. Rath;Eung-Soo Kim Dr.;JanetL. Smith Dr.;DavidH. Sherman Dr.
Angewandte Chemie 2010 Volume 122( Issue 33) pp:5862-5866
Publication Date(Web):
DOI:10.1002/ange.201000032
Co-reporter:JamieB. Scaglione Dr.;DavidL. Akey Dr.;Rachel Sullivan;JeffreyD. Kittendorf Dr.;ChristopherM. Rath;Eung-Soo Kim Dr.;JanetL. Smith Dr.;DavidH. Sherman Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 33) pp:5726-5730
Publication Date(Web):
DOI:10.1002/anie.201000032
Co-reporter:Jonathan D. Mortison and David H. Sherman
The Journal of Organic Chemistry 2010 Volume 75(Issue 21) pp:7041-7051
Publication Date(Web):September 30, 2010
DOI:10.1021/jo101124n
Natural product biosynthetic pathways have evolved enzymes with myriad activities that represent an expansive array of chemical transformations for constructing secondary metabolites. Recently, harnessing the biosynthetic potential of these enzymes through chemoenzymatic synthesis has provided a powerful tool that often rivals the most sophisticated methodologies in modern synthetic chemistry and provides new opportunities for accessing chemical diversity. Herein, we describe our research efforts with enzymes from a broad collection of biosynthetic systems, highlighting recent progress in this exciting field.
Co-reporter:Jacob C. Carlson;J. L. Fortman Dr.;Yojiro Anzai Dr.;Shengying Li;Douglas A. Burr Dr.
ChemBioChem 2010 Volume 11( Issue 4) pp:564-572
Publication Date(Web):
DOI:10.1002/cbic.200900658
Abstract
The structurally intriguing bicyclic ketal moiety of tirandamycin is common to several acyl-tetramic acid antibiotics, and is a key determinant of biological activity. We have identified the tirandamycin biosynthetic gene cluster from the environmental marine isolate Streptomyces sp. 307–9, thus providing the first genetic insight into the biosynthesis of this natural product scaffold. Sequence analysis revealed a hybrid polyketide synthase–nonribosomal peptide synthetase gene cluster with a colinear domain organization, which is entirely consistent with the core structure of the tirandamycins. We also identified genes within the cluster that encode candidate tailoring enzymes for elaboration and modification of the bicyclic ketal system. Disruption of tamI, which encodes a presumed cytochrome P450, led to a mutant strain deficient in production of late stage tirandamycins that instead accumulated tirandamycin C, an intermediate devoid of any post assembly-line oxidative modifications.
Co-reporter:Jonathan D. Mortison ; Jeffrey D. Kittendorf
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15784-15793
Publication Date(Web):October 7, 2009
DOI:10.1021/ja9060596
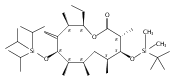
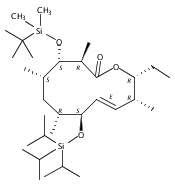
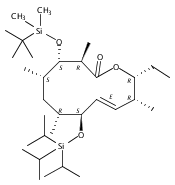
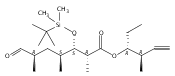
The 6-deoxyerythronolide B synthase (DEBS) and pikromycin (Pik) polyketide synthase (PKS) are unique multifunctional enzyme systems that are responsible for the biosynthesis of the erythromycin and pikromycin 14-membered ring aglycones, respectively. Together, these natural product biosynthetic systems provide excellent platforms to examine the fundamental structural and catalytic elements that govern polyketide assembly, processing, and macrocyclization. In these studies, the native pentaketide intermediate for DEBS was synthesized and employed for in vitro chemoenzymatic synthesis of macrolactone products in engineered monomodules Ery5, Ery5-TE, and Ery6. A comparative analysis was performed with the corresponding Pik module 5 (PikAIII) and module 6 (PikAIV), dissecting key similarities and differences between these highly related PKSs. The data revealed that individual modules in the DEBS and Pik PKSs possess distinctive molecular selectivity profiles and suggest that substrate recognition has evolved unique characteristics in each system.
Co-reporter:Liangcai Gu ; Bo Wang ; Amol Kulkarni ; Jennifer J. Gehret ; Kayla R. Lloyd ; Lena Gerwick ; William H. Gerwick ; Peter Wipf ; Kristina Håkansson ; Janet L. Smith
Journal of the American Chemical Society 2009 Volume 131(Issue 44) pp:16033-16035
Publication Date(Web):October 16, 2009
DOI:10.1021/ja9071578
Biosynthetic innovation in natural product systems is driven by the recruitment of new genes and enzymes into these complex pathways. Here, an unprecedented decarboxylative chain termination mechanism is described for the polyketide synthase of curacin A, an anticancer lead compound isolated from the marine cyanobacterium Lyngbya majuscula. The unusual chain termination module containing adjacent sulfotransferase (ST) and thioesterase (TE) catalytic domains embedded in CurM was biochemically characterized. The TE was proved to catalyze a hydrolytic chain release of the polyketide chain elongation intermediate. Moreover, a selective ST-mediated sulfonation of the (R)-β-hydroxyl group was found to precede TE-mediated hydrolysis, triggering a successive decarboxylative elimination and resulting in the formation of a rare terminal olefin in the final metabolite.
Co-reporter:Tonia J. Buchholz, Todd W. Geders, Frank E. Bartley III, Kevin A. Reynolds, Janet L. Smith and David H. Sherman
ACS Chemical Biology 2009 Volume 4(Issue 1) pp:41
Publication Date(Web):January 16, 2009
DOI:10.1021/cb8002607
Bacterial type I polyketide synthases (PKSs) assemble structurally diverse natural products of significant clinical value from simple metabolic building blocks. The synthesis of these compounds occurs in a processive fashion along a large multiprotein complex. Transfer of the acyl intermediate across interpolypeptide junctions is mediated, at least in large part, by N- and C-terminal docking domains. We report here a comprehensive analysis of the binding affinity and selectivity for the complete set of discrete docking domain pairs in the pikromycin and erythromycin PKS systems. Despite disconnection from their parent module, each cognate pair of docking domains retained exquisite binding selectivity. Further insights were obtained by X-ray crystallographic analysis of the PikAIII/PikAIV docking domain interface. This new information revealed a series of key interacting residues that enabled development of a structural model for the recently proposed H2-T2 class of polypeptides involved in PKS intermodular molecular recognition.
Co-reporter:Jacob C. Carlson, Shengying Li, Douglas A. Burr and David H. Sherman
Journal of Natural Products 2009 Volume 72(Issue 11) pp:2076-2079
Publication Date(Web):November 2, 2009
DOI:10.1021/np9005597
The novel dienoyl tetramic acids tirandamycin C (1) and tirandamycin D (2) with activity against vancomycin-resistant Enterococcus faecalis were isolated from the marine environmental isolate Streptomyces sp. 307-9, which also produces the previously identified compounds tirandamycins A (3) and B (4). Spectroscopic analysis of 1 and 2 indicated structural similarity to 3 and 4, with differences only in the pattern of pendant oxygenation on the bicyclic ketal system. The isolation of these putative biosynthetic intermediates was enabled by their sequestration on an adsorbent resin during early stationary-phase fermentation.
Co-reporter:Jeffrey D. Kittendorf, David H. Sherman
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 6) pp:2137-2146
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmc.2008.10.082
The methymycin/pikromycin (Pik) macrolide pathway represents a robust metabolic system for analysis of modular polyketide biosynthesis. The enzymes that comprise this biosynthetic pathway display unprecedented substrate flexibility, combining to produce six structurally diverse macrolide antibiotics in Streptomyces venezuelae. Thus, it is appealing to consider that the pikromycin biosynthetic enzymes could be leveraged for high-throughput production of novel macrolide antibiotics. Accordingly, efforts over the past decade have focused on the detailed investigation of the six-module polyketide synthase, desosamine sugar assembly and glycosyl transfer, and the cytochrome P450 monooxygenase that is responsible for hydroxylation. This review summarizes the advances in understanding of pikromycin biosynthesis that have been gained during the course of these investigations.
Co-reporter:Liangcai Gu,
Bo Wang,
Amol Kulkarni,
Todd W. Geders,
Rashel V. Grindberg,
Lena Gerwick,
Kristina Håkansson,
Peter Wipf,
Janet L. Smith,
William H. Gerwick
&
David H. Sherman
Nature 2009 459(7247) pp:731
Publication Date(Web):2009-06-04
DOI:10.1038/nature07870
Co-evolution of enzymes for metabolic diversification is not well understood, especially at the biochemical level. Here, the biosynthetic pathways responsible for the synthesis of two natural products, curacin and jamaicamide, are examined; several domains are characterized to help determine how, although the two pathways have a high sequence identity, they are responsible for the production of two such dramatically different chemical motifs.
Co-reporter:Shengying Li;Yojiro Anzai Dr.;Kenji Kinoshita ;Fumio Kato
ChemBioChem 2009 Volume 10( Issue 8) pp:1297-1301
Publication Date(Web):
DOI:10.1002/cbic.200900088
Co-reporter:Patrick J. Hu
PNAS 2009 Volume 106 (Issue 19 ) pp:7708-7713
Publication Date(Web):2009-05-12
DOI:10.1073/pnas.0903021106
Co-reporter:Shengying Li;Mani Raj Chaulagain;Allison R. Knauff;Larissa M. Podust;John Montgomery
PNAS 2009 Volume 106 (Issue 44 ) pp:18463-18468
Publication Date(Web):2009-11-03
DOI:10.1073/pnas.0907203106
Regio- and stereoselective oxidation of an unactivated C–H bond remains a central challenge in organic chemistry. Considerable
effort has been devoted to identifying transition metal complexes, biological catalysts, or simplified mimics, but limited
success has been achieved. Cytochrome P450 mono-oxygenases are involved in diverse types of regio- and stereoselective oxidations,
and represent a promising biocatalyst to address this challenge. The application of this class of enzymes is particularly
significant if their substrate spectra can be broadened, selectivity controlled, and reactions catalyzed in the absence of
expensive heterologous redox partners. In this study, we engineered a macrolide biosynthetic P450 mono-oxygenase PikC (PikCD50N-RhFRED) with remarkable substrate flexibility, significantly increased activity compared to wild-type enzyme, and self-sufficiency.
By harnessing its unique desosamine-anchoring functionality via a heretofore under-explored “substrate engineering” strategy,
we demonstrated the ability of PikC to hydroxylate a series of carbocyclic rings linked to the desosamine glycoside via an
acetal linkage (referred to as “carbolides”) in a regioselective manner. Complementary analysis of a number of high-resolution
enzyme-substrate cocrystal structures provided significant insights into the function of the aminosugar-derived anchoring
group for control of reaction site selectivity. Moreover, unexpected biological activity of a select number of these carbolide
systems revealed their potential as a previously unrecorded class of antibiotics.
Co-reporter:Nicole B. Lopanik, Jennifer A. Shields, Tonia J. Buchholz, Christopher M. Rath, Joanne Hothersall, Margo G. Haygood, Kristina Håkansson, Christopher M. Thomas, David H. Sherman
Chemistry & Biology 2008 Volume 15(Issue 11) pp:1175-1186
Publication Date(Web):24 November 2008
DOI:10.1016/j.chembiol.2008.09.013
The putative modular polyketide synthase (PKS) that prescribes biosynthesis of the bryostatin natural products from the uncultured bacterial symbiont of the marine bryozoan Bugula neritina possesses a discrete open reading frame (ORF) (bryP) that encodes a protein containing tandem acyltransferase (AT) domains upstream of the PKS ORFs. BryP is hypothesized to catalyze in trans acylation of the PKS modules for polyketide chain elongation. To verify conservation of function, bryP was introduced into AT-deletion mutant strains of a heterologous host containing a PKS cluster with similar architecture, and polyketide production was partially rescued. Biochemical characterization demonstrated that BryP catalyzes selective malonyl-CoA acylation of native and heterologous acyl carrier proteins and complete PKS modules in vitro. The results support the hypothesis that BryP loads malonyl-CoA onto Bry PKS modules, and provide the first biochemical evidence of the functionality of the bry cluster.
Co-reporter:Yojiro Anzai, Shengying Li, Mani Raj Chaulagain, Kenji Kinoshita, Fumio Kato, John Montgomery, David H. Sherman
Chemistry & Biology 2008 Volume 15(Issue 9) pp:950-959
Publication Date(Web):22 September 2008
DOI:10.1016/j.chembiol.2008.07.014
Macrolides are a class of valuable antibiotics that include a macrolactone ring, at least one appended sugar unit, and, in most cases, additional hydroxyl or epoxide groups installed by cytochrome P450 enzymes. These functional groups contribute to structural diversification and serve to improve the bioactivity profiles of natural products. Here, we have characterized in vitro two P450 enzymes from the mycinamicin biosynthetic pathway of Micromonospora griseorubida. First, MycCI was characterized as the C21 methyl hydroxylase of mycinamicin VIII, the earliest macrolide form in the postpolyketide synthase tailoring pathway. Moreover, we established that optimal activity of MycCI depends on the native ferredoxin MycCII. Second, MycG P450 catalyzes consecutive hydroxylation and epoxidation reactions with mycinamicin IV as initial substrate. These reactions require prior dimethylation of 6-deoxyallose to mycinose for effective conversion by the dual function MycG enzyme.
Co-reporter:Brian F. Pfleger;Natalia Maltseva;Christopher M. Rath;Jung Yeop Lee;Youngchang Kim;Tyler D. Nusca;Jamie B. Scaglione;Brian K. Janes;Erica C. Anderson;Nicholas H. Bergman;Philip C. Hanna;Andrzej Joachimiak
PNAS 2008 Volume 105 (Issue 44 ) pp:17133-17138
Publication Date(Web):2008-11-04
DOI:10.1073/pnas.0808118105
Petrobactin, a virulence-associated siderophore produced by Bacillus anthracis, chelates ferric iron through the rare 3,4-isomer of dihydroxybenzoic acid (3,4-DHBA). Most catechol siderophores, including
bacillibactin and enterobactin, use 2,3-DHBA as a biosynthetic subunit. Significantly, siderocalin, a factor involved in human
innate immunity, sequesters ferric siderophores bearing the more typical 2,3-DHBA moiety, thereby impeding uptake of iron
by the pathogenic bacterial cell. In contrast, the unusual 3,4-DHBA component of petrobactin renders the siderocalin system
incapable of obstructing bacterial iron uptake. Although recent genetic and biochemical studies have revealed selected early
steps in petrobactin biosynthesis, the origin of 3,4-DHBA as well as the function of the protein encoded by the final gene
in the B. anthracis siderophore biosynthetic (asb) operon, asbF (BA1986), has remained unclear. In this study we demonstrate that 3,4-DHBA is produced through conversion of the common bacterial
metabolite 3-dehydroshikimate (3-DHS) by AsbF—a 3-DHS dehydratase. Elucidation of the cocrystal structure of AsbF with 3,4-DHBA,
in conjunction with a series of biochemical studies, supports a mechanism in which an enolate intermediate is formed through
the action of this 3-DHS dehydratase metalloenzyme. Structural and functional parallels are evident between AsbF and other
enzymes within the xylose isomerase TIM-barrel family. Overall, these data indicate that microbial species shown to possess
homologs of AsbF may, like B. anthracis, also rely on production of the unique 3,4-DHBA metabolite to achieve full viability in the environment or virulence within
the host.
Co-reporter:Jeffrey D. Kittendorf, Brian J. Beck, Tonia J. Buchholz, Wolfgang Seufert, David H. Sherman
Chemistry & Biology 2007 Volume 14(Issue 8) pp:944-954
Publication Date(Web):24 August 2007
DOI:10.1016/j.chembiol.2007.07.013
The pikromycin polyketide synthase (PKS) is unique in its ability to generate both 12 and 14 membered ring macrolactones. As such, dissection of the molecular basis for controlling metabolic diversity in this system remains an important objective for understanding modular PKS function and expanding chemical diversity. Here, we describe a series of experiments designed to probe the importance of the protein-protein interaction that occurs between the final two monomodules, PikAIII (module 5) and PikAIV (module 6), for the production of the 12 membered ring macrolactone 10-deoxymethynolide. The results obtained from these in vitro studies demonstrate that PikAIII and PikAIV generate the 12 membered ring macrocycle most efficiently when engaged in their native protein-protein interaction. Accordingly, the data are consistent with PikAIV adopting an alternative conformation that enables the terminal thioesterase domain to directly off-load the PikAIII-bound hexaketide intermediate for macrocyclization.
Co-reporter:Todd W. Geders;Liangcai Gu;Bo Wang;William H. Gerwick;Kristina Håkansson;Janet L. Smith
Science 2007 Volume 318(Issue 5852) pp:970-974
Publication Date(Web):09 Nov 2007
DOI:10.1126/science.1148790
Abstract
An unexpected biochemical strategy for chain initiation is described for the loading module of the polyketide synthase of curacin A, an anticancer lead derived from the marine cyanobacterium Lyngbya majuscula. A central GCN5-related N-acetyltransferase (GNAT) domain bears bifunctional decarboxylase/S-acetyltransferase activity, both unprecedented for the GNAT superfamily. A CurA loading tridomain, consisting of an adaptor domain, the GNAT domain, and an acyl carrier protein, was assessed biochemically, revealing that a domain showing homology to GNAT (GNATL) catalyzes (i) decarboxylation of malonyl-coenzyme A (malonyl-CoA) to acetyl-CoA and (ii) direct S-acetyl transfer from acetyl-CoA to load an adjacent acyl carrier protein domain (ACPL). Moreover, the N-terminal adapter domain was shown to facilitate acetyl-group transfer. Crystal structures of GNATL were solved at 1.95 angstroms (ligand-free form) and 2.75 angstroms (acyl-CoA complex), showing distinct substrate tunnels for acyl-CoA and holo-ACPL binding. Modeling and site-directed mutagenesis experiments demonstrated that histidine-389 and threonine-355, at the convergence of the CoA and ACP tunnels, participate in malonyl-CoA decarboxylation but not in acetyl-group transfer. Decarboxylation precedes acetyl-group transfer, leading to acetyl-ACPL as the key curacin A starter unit.
Co-reporter:Wolfgang Seufert Dr.;Zachary Q. Beck Dr.;David H. Sherman Dr.
Angewandte Chemie 2007 Volume 119(Issue 48) pp:
Publication Date(Web):2 NOV 2007
DOI:10.1002/ange.200703665
Es müssen nicht immer Thioester sein: Cryptophycin-Thioesterase (Crp TE) spaltet lineare Cryptophycine, die an aktivierte PEGA-Harze gebunden sind, und überführt sie in Makrolactone. Diese neuartige enzymatische Umwandlung an der Festphase wurde verwendet, um die Toleranz von Crp TE in Bezug auf Änderungen in der Substratstruktur zu untersuchen.
Co-reporter:Wolfgang Seufert Dr.;Zachary Q. Beck Dr.;David H. Sherman Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 48) pp:
Publication Date(Web):2 NOV 2007
DOI:10.1002/anie.200703665
Thioesters need not apply: Cryptophycin thioesterase (Crp TE) cleaves and macrolactonizes linear cryptophycin substrates bound to activated safety-catch PEGA resin. This novel enzymatic solid-phase approach was used to further investigate the tolerance of Crp TE for structural variations of substrates.
Co-reporter:Zachary Q. Beck Dr.;Douglas A. Burr Dr. Dr.
ChemBioChem 2007 Volume 8(Issue 12) pp:
Publication Date(Web):28 JUN 2007
DOI:10.1002/cbic.200700162
More pieces for the puzzle. The β-methylaspartate-α-decarboxylase (CrpG) from the cryptophycin biosynthetic pathway was cloned, over-expressed, and purified. We found that CrpG decarboxylates (2S,3R)-3-methylaspartic acid to form 3-amino-2(R)-methylpropionic acid, which is subsequently incorporated into Unit C of cryptophycins (see scheme).
Co-reporter:J. L. Fortman
ChemBioChem 2005 Volume 6(Issue 6) pp:
Publication Date(Web):6 MAY 2005
DOI:10.1002/cbic.200400428
Marine organisms are a rich source of secondary metabolites. They have yielded thousands of compounds with a broad range of biomedical applications. Thus far, samples required for preclinical and clinical studies have been obtained by collection from the wild, by mariculture, and by total chemical synthesis. However, for a number of complex marine metabolites, none of these options is feasible for either economic or environmental reasons. In order to proceed with the development of many of these promising therapeutic compounds, a reliable and renewable source must be found. Over the last twenty years, the study of microbial secondary metabolites has greatly advanced our understanding of how nature utilizes simple starting materials to yield complex small molecules. Much of this work has focused on polyketides and nonribosomal peptides, two classes of molecules that are prevalent in marine micro- and macroorganisms. The lessons learned from the study of terrestrial metabolite biosynthesis are now being applied to the marine world. As techniques for cloning and heterologous expression of biosynthetic pathways continue to improve, they may provide our greatest hope for bridging the gap between the promise and application of many marine natural products.
Co-reporter:Christine E. Salomon, Nathan A. Magarvey and David H. Sherman
Natural Product Reports 2004 vol. 21(Issue 1) pp:105-121
Publication Date(Web):15 Dec 2003
DOI:10.1039/B301384G
Covering: 1999–2003
Co-reporter:Michael M Schofield, David H Sherman
Current Opinion in Biotechnology (December 2013) Volume 24(Issue 6) pp:1151-1158
Publication Date(Web):1 December 2013
DOI:10.1016/j.copbio.2013.05.001
•Meta-omics permits the study of the biosynthetic gene clusters of uncultivable microbes.•Unculturable microbe genomes can be sequenced via libraries, directly, or by single cell methods.•Natural product-specific bioinformatic tools assist metagenomic sequencing data analysis.•OASIS, PrISM, and derivatives help pave the way for metaproteomic methodologies.•New techniques link metabolomics to natural products research.Microorganisms produce a remarkable selection of bioactive small molecules. The study and exploitation of these secondary metabolites have traditionally been restricted to the cultivable minority of bacteria. Rapid advances in meta-omics challenge this paradigm. Breakthroughs in metagenomic library methodologies, direct sequencing, single cell genomics, and natural product-specific bioinformatic tools now facilitate the retrieval of previously inaccessible biosynthetic gene clusters. Similarly, metaproteomic developments enable the direct study of biosynthetic enzymes from complex microbial communities. Additional methods within and beyond meta-omics are also in development. This review discusses recent reports in these arenas and how they can be utilized to characterize natural product biosynthetic gene clusters and pathways.Download high-res image (203KB)Download full-size image
Co-reporter:Kyle L. Bolduc, Scott D. Larsen and David H. Sherman
Chemical Communications 2012 - vol. 48(Issue 51) pp:NaN6416-6416
Publication Date(Web):2012/05/02
DOI:10.1039/C2CC32417B
A flexible and divergent synthesis of cryptophycin unit A analogues is described. This method relies on iridium-catalysed stereo- and enantioselective crotylation and chemoselective one-pot oxidative olefination to access common intermediate 8. Heck, cross metathesis, and Suzuki-Miyaura reactions are illustrated for the generation of methyl ester unit A analogues 10a–d.
.jpg)
![Propanamide,2-amino-N-[[(5S,6S,7S,9R,14aS,15R)-6,7,9,10,13,14,14a,15-octahydro-1,4,7-trihydroxy-2,5,11-trimethoxy-3,12,16-trimethyl-10,13-dioxo-6,15-imino-5H-isoquino[3,2-b][3]benzazocin-9-yl]methyl]-,(2S)-](http://img.cochemist.com/ccimg/113100/113036-78-5.png)
![Propanamide,2-amino-N-[[(5S,6S,7S,9R,14aS,15R)-6,7,9,10,13,14,14a,15-octahydro-1,4,7-trihydroxy-2,5,11-trimethoxy-3,12,16-trimethyl-10,13-dioxo-6,15-imino-5H-isoquino[3,2-b][3]benzazocin-9-yl]methyl]-,(2S)-](http://img.cochemist.com/ccimg/113100/113036-78-5_b.png)
![Naphth[1,2,3-cd]indole,9-ethenyl-2,6,6a,7,8,9,10,10a-octahydro-10-isocyano-6,6,9-trimethyl-,(6aS,9R,10R,10aS)-](http://img.cochemist.com/ccimg/107000/106928-30-7.png)
![Naphth[1,2,3-cd]indole,9-ethenyl-2,6,6a,7,8,9,10,10a-octahydro-10-isocyano-6,6,9-trimethyl-,(6aS,9R,10R,10aS)-](http://img.cochemist.com/ccimg/107000/106928-30-7_b.png)


![Benzene, 1-[[(methylthio)methoxy]methyl]-2-nitro-](http://img.cochemist.com/ccimg/500800/500776-06-7.png)
![Benzene, 1-[[(methylthio)methoxy]methyl]-2-nitro-](http://img.cochemist.com/ccimg/500800/500776-06-7_b.png)






![PENTAFLUOROPHENYL N-[(9H-FLUOREN-9-YLMETHOXY)CARBONYL]-D-PHENYLAL<WBR />ANINATE](http://img.cochemist.com/ccimg/73700/73684-69-2.png)
![PENTAFLUOROPHENYL N-[(9H-FLUOREN-9-YLMETHOXY)CARBONYL]-D-PHENYLAL<WBR />ANINATE](http://img.cochemist.com/ccimg/73700/73684-69-2_b.png)

![Tylonolide,5-O-[3,6-dideoxy-3-(dimethylamino)-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/61300/61257-02-1.png)
![Tylonolide,5-O-[3,6-dideoxy-3-(dimethylamino)-b-D-glucopyranosyl]-](http://img.cochemist.com/ccimg/61300/61257-02-1_b.png)

![2H-Pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4(3H,5aH)-dione,10b-(1,1-dimethyl-2-propen-1-yl)-6,10b,11,11a-tetrahydro-3-(1H-imidazol-5-ylmethylene)-,(3E,5aS,10bR,11aS)-](http://img.cochemist.com/ccimg/58800/58735-64-1.png)
![2H-Pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4(3H,5aH)-dione,10b-(1,1-dimethyl-2-propen-1-yl)-6,10b,11,11a-tetrahydro-3-(1H-imidazol-5-ylmethylene)-,(3E,5aS,10bR,11aS)-](http://img.cochemist.com/ccimg/58800/58735-64-1_b.png)










![Erythromycin,3-de[(2,6-dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-10,11-didehydro-10-demethyl-6,11,12-trideoxy-3-oxo-,(10E)-](http://img.cochemist.com/ccimg/6100/6036-25-5.png)
![Erythromycin,3-de[(2,6-dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-10,11-didehydro-10-demethyl-6,11,12-trideoxy-3-oxo-,(10E)-](http://img.cochemist.com/ccimg/6100/6036-25-5_b.png)




![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0.png)
![(1AR,4R,4AR,7S,7AS,7BR)-1,1,4,7-TETRAMETHYLDECAHYDRO-1H-CYCLOPROP<WBR />A[E]AZULENE-4,7-DIOL](http://img.cochemist.com/ccimg/800/763-10-0_b.png)



![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)



























































































































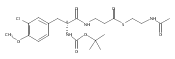
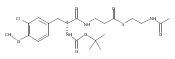


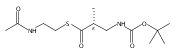
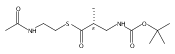

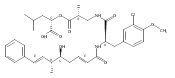
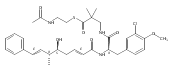
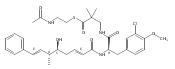
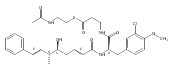
























































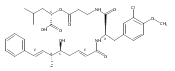


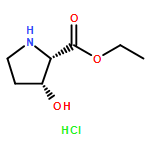

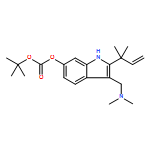
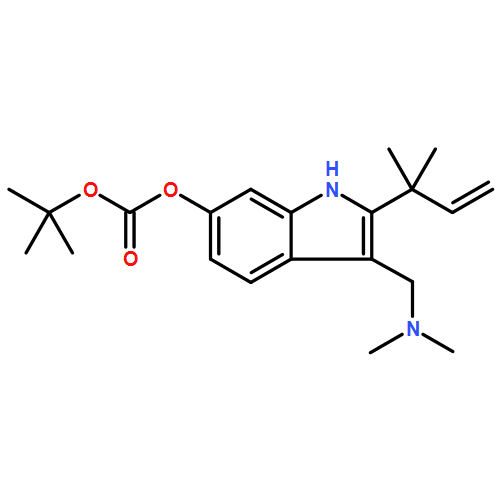
































![Cyclo[2,2-dimethyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/186300/186256-67-7.png)
![Cyclo[2,2-dimethyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/186300/186256-67-7_b.png)


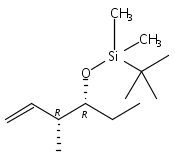
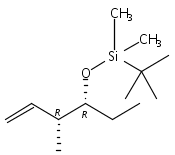
![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methyl-,(3S,4R)-](/data/chemimg/115900/168777-81-9.png)
![5-Hexen-3-ol, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-methyl-,(3S,4R)-](/data/chemimg/115900/168777-81-9_b.png)










![Cyclo[b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/162700/162600-54-6.png)
![Cyclo[b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/162700/162600-54-6_b.png)
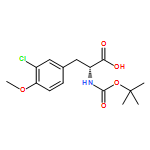

![4-Thiazolecarboxylic acid, 2-[(2S)-2-[(4R)-2-[(4R)-4,5-dihydro-2-(2-hydroxyphenyl)-4-thiazolyl]-4-thiazolidinyl]-2-hydroxy-1,1-dimethylethyl]-4,5-dihydro-4-methyl-, (4S)-](/data/chemimg/111600/152166-40-0.png)
![4-Thiazolecarboxylic acid, 2-[(2S)-2-[(4R)-2-[(4R)-4,5-dihydro-2-(2-hydroxyphenyl)-4-thiazolyl]-4-thiazolidinyl]-2-hydroxy-1,1-dimethylethyl]-4,5-dihydro-4-methyl-, (4S)-](/data/chemimg/111600/152166-40-0_b.png)
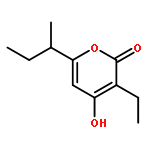







![1-Propanol, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/136400/136320-64-4.png)
![1-Propanol, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/136400/136320-64-4_b.png)
![Propanoic acid, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, methyl ester,(2S)-](http://img.cochemist.com/ccimg/133000/132969-71-2.png)
![Propanoic acid, 3-[(4-methoxyphenyl)methoxy]-2-methyl-, methyl ester,(2S)-](http://img.cochemist.com/ccimg/133000/132969-71-2_b.png)
![Cyclo[(2R)-2-methyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6R,7E)-5-hydroxy-6-methyl-8-phenyl-2,7-octadienoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/124700/124689-64-1.png)
![Cyclo[(2R)-2-methyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6R,7E)-5-hydroxy-6-methyl-8-phenyl-2,7-octadienoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/124700/124689-64-1_b.png)

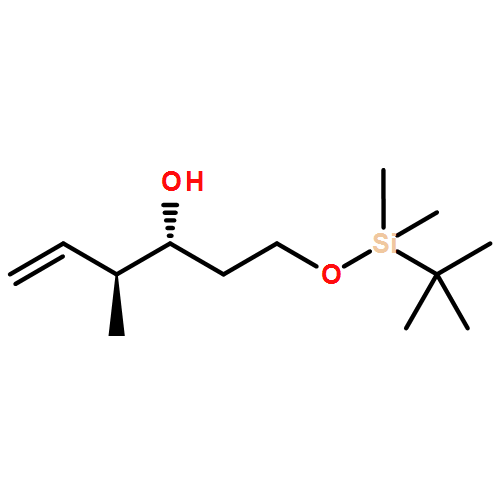




![Pentanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2S,3R)-](http://img.cochemist.com/ccimg/114200/114181-55-4.png)
![Pentanal, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-methyl-, (2S,3R)-](http://img.cochemist.com/ccimg/114200/114181-55-4_b.png)




![(2S)-3-{[tert-Butyl(dimethyl)silyl]oxy}-2-methylpropan-1-ol](http://img.cochemist.com/ccimg/105900/105859-45-8.png)
![(2S)-3-{[tert-Butyl(dimethyl)silyl]oxy}-2-methylpropan-1-ol](http://img.cochemist.com/ccimg/105900/105859-45-8_b.png)






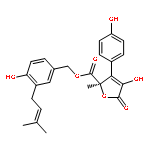
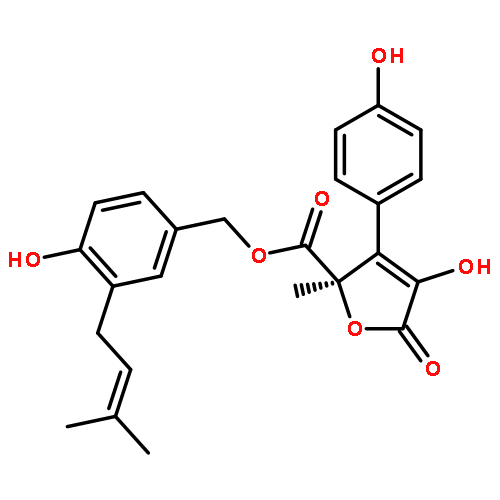
![2,5-Dihydro-2-[3-(3,3-dimethyloxiranylmethyl)-4-hydroxybenzyl]-4-hydroxy-3-(4-hydroxyphenyl)-5-oxofuran-2-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/87500/87414-45-7.png)
![2,5-Dihydro-2-[3-(3,3-dimethyloxiranylmethyl)-4-hydroxybenzyl]-4-hydroxy-3-(4-hydroxyphenyl)-5-oxofuran-2-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/87500/87414-45-7_b.png)
![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6.png)
![BRYOSTATIN 1;(1S,3S,5Z,7R,8E,11S,12S,13E,15S,17R,21R,23R,25S)-25-(ACETYLOXY)-1,11,21-TRIHYDROXY-17-[(1R)-1-HYDROXYETHYL]-5,13-BIS(2-METHOXY-2-OXOETHYLIDENE)-10,10,26,26-TETRAMETHYL-19-OXO-18,27,28,29-TETRAOXATETRACYCLO[21.3.1.13,7.111,15]NONACOS-8-EN-12-Y](http://img.cochemist.com/ccimg/83400/83314-01-6_b.png)

![(3E,11E,13E)-15-{[(6-deoxy-2,3-di-O-methylhexopyranosyl)oxy]methyl}-16-ethyl-15-hydroxy-5,7,9-trimethyl-2,10-dioxooxacyclohexadeca-3,11,13-trien-6-yl 3,4,6-trideoxy-3-(dimethylamino)hexopyranoside](http://img.cochemist.com/ccimg/73700/73684-72-7.png)
![(3E,11E,13E)-15-{[(6-deoxy-2,3-di-O-methylhexopyranosyl)oxy]methyl}-16-ethyl-15-hydroxy-5,7,9-trimethyl-2,10-dioxooxacyclohexadeca-3,11,13-trien-6-yl 3,4,6-trideoxy-3-(dimethylamino)hexopyranoside](http://img.cochemist.com/ccimg/73700/73684-72-7_b.png)
![Oxacyclohexadeca-3,11,13-triene-2,10-dione, 15-[[(6-deoxy-2,3-di-O-methyl-β-D-allopyranosyl)oxy]methyl]-16-ethyl-5,7,9-trimethyl-6-[[3,4,6-trideoxy-3-](/data/chemimg/1955000/73684-71-6.png)
![Oxacyclohexadeca-3,11,13-triene-2,10-dione, 15-[[(6-deoxy-2,3-di-O-methyl-β-D-allopyranosyl)oxy]methyl]-16-ethyl-5,7,9-trimethyl-6-[[3,4,6-trideoxy-3-](/data/chemimg/1955000/73684-71-6_b.png)
![Oxacyclohexadeca-3,11,13-triene-2,10-dione, 15-[[(6-deoxy-2-O-methyl-β-D-allopyranosyl)oxy]methyl]-16-ethyl-5,7,9-trimethyl-6-[[3,4,6-trideoxy-3-](/data/chemimg/1957100/73684-70-5.png)
![Oxacyclohexadeca-3,11,13-triene-2,10-dione, 15-[[(6-deoxy-2-O-methyl-β-D-allopyranosyl)oxy]methyl]-16-ethyl-5,7,9-trimethyl-6-[[3,4,6-trideoxy-3-](/data/chemimg/1957100/73684-70-5_b.png)




![(2Z)-5-hydroxy-4-[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]-2-{[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]methylidene}furan-3(2H)-one](http://img.cochemist.com/ccimg/57800/57744-69-1.png)
![(2Z)-5-hydroxy-4-[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]-2-{[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]methylidene}furan-3(2H)-one](http://img.cochemist.com/ccimg/57800/57744-69-1_b.png)
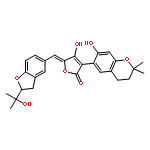
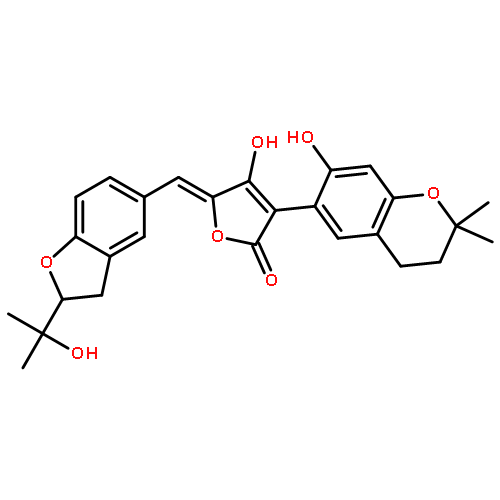

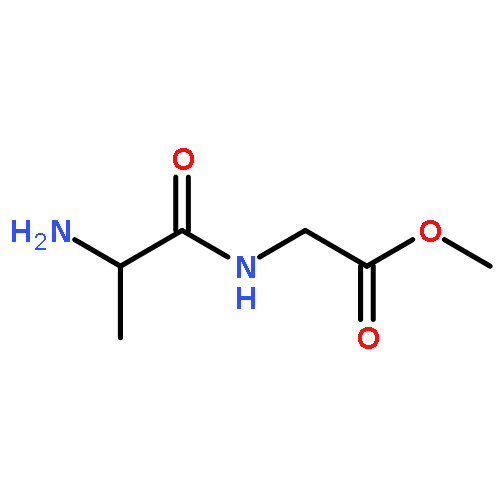



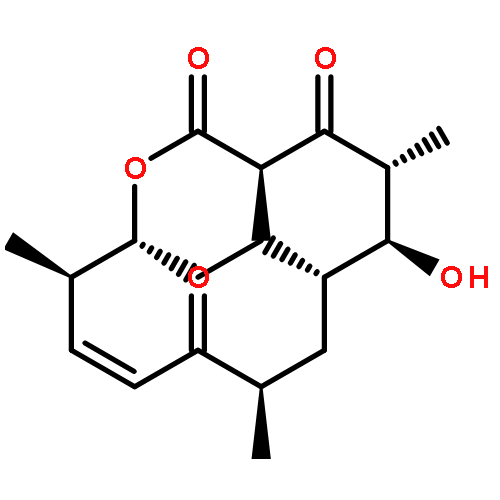








![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)


![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,hexahydro-3-(phenylmethyl)-, (3S,8aS)-](http://img.cochemist.com/ccimg/3800/3705-26-8_b.png)






![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9.png)
![Butanoic acid, 2(or3)-methyl-,(2R,3S,6S,7R,8R)-3-[[3-(formylamino)-2-hydroxybenzoyl]amino]-8-hexyl-2,6-dimethyl-4,9-dioxo-1,5-dioxonan-7-ylester (9CI)](http://img.cochemist.com/ccimg/700/642-15-9_b.png)
![Benzoic acid,2-hydroxy-3-[(2-hydroxy-4-methoxy-6-propylbenzoyl)oxy]-4-methoxy-6-propyl-](http://img.cochemist.com/ccimg/700/607-11-4.png)
![Benzoic acid,2-hydroxy-3-[(2-hydroxy-4-methoxy-6-propylbenzoyl)oxy]-4-methoxy-6-propyl-](http://img.cochemist.com/ccimg/700/607-11-4_b.png)


![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7.png)
![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7_b.png)









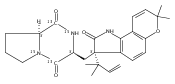

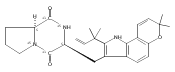

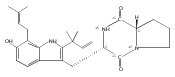
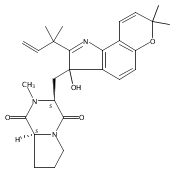


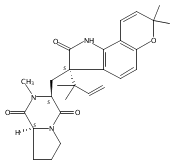

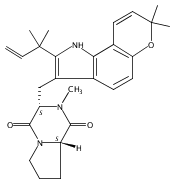


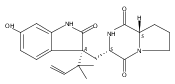
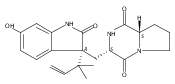
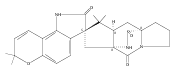
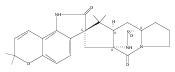


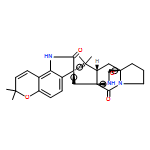


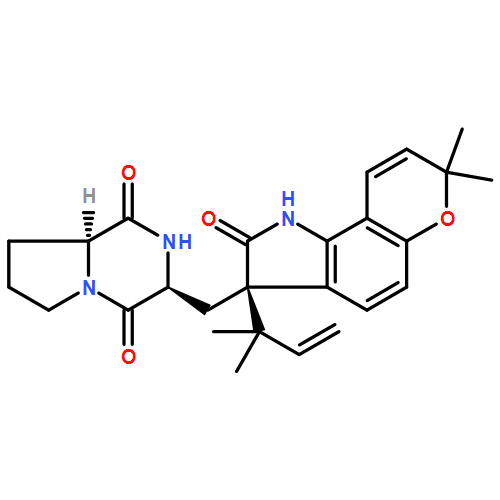

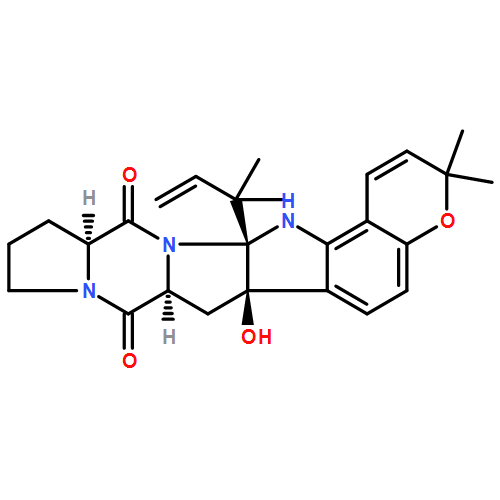
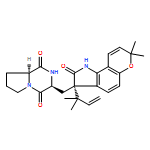


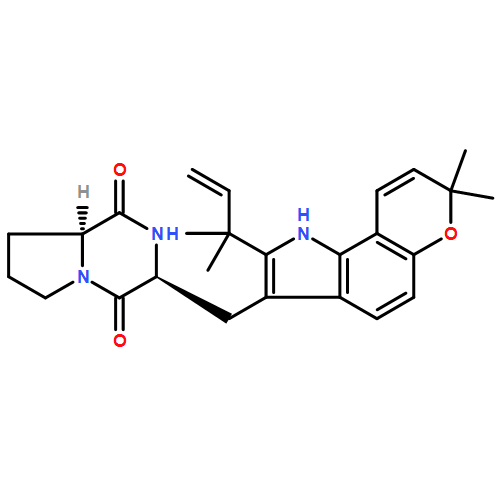




![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6.png)
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6_b.png)






![11H-Dibenzo[b,e][1,4]dioxepin-7-carboxylicacid, 8-hydroxy-3-methoxy-11-oxo-1-(1-oxopentyl)-6-pentyl-](http://img.cochemist.com/ccimg/600/522-53-2.png)
![11H-Dibenzo[b,e][1,4]dioxepin-7-carboxylicacid, 8-hydroxy-3-methoxy-11-oxo-1-(1-oxopentyl)-6-pentyl-](http://img.cochemist.com/ccimg/600/522-53-2_b.png)

![Oxacyclododec-9-ene-2,8-dione,12-ethyl-11-hydroxy-3,5,7,11-tetramethyl-4-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-,(3R,4S,5S,7R,9E,11S,12R)-](http://img.cochemist.com/ccimg/500/497-72-3.png)
![Oxacyclododec-9-ene-2,8-dione,12-ethyl-11-hydroxy-3,5,7,11-tetramethyl-4-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-,(3R,4S,5S,7R,9E,11S,12R)-](http://img.cochemist.com/ccimg/500/497-72-3_b.png)
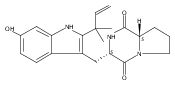
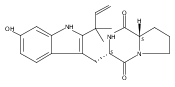
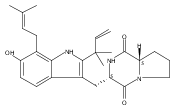
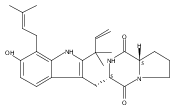


![Cyclo[(2R)-2-methyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/124700/124689-65-2.png)
![Cyclo[(2R)-2-methyl-b-alanyl-(2S)-2-hydroxy-4-methylpentanoyl-(2E,5S,6S)-5-hydroxy-6-[(2R,3R)-3-phenyl-2-oxiranyl]-2-heptenoyl-3-chloro-O-methyl-D-tyrosyl]](http://img.cochemist.com/ccimg/124700/124689-65-2_b.png)
![5-hydroxy-4-{(2E,4E)-6-[2-(hydroxymethyl)-1,7-dimethyl-5-oxo-3,9,10-trioxatricyclo[4.3.1.0~2,4~]dec-8-yl]-4-methylhepta-2,4-dienoyl}-1,2-dihydro-3H-pyrrol-3-one (non-preferred name)](http://img.cochemist.com/ccimg/60600/60587-14-6.png)
![5-hydroxy-4-{(2E,4E)-6-[2-(hydroxymethyl)-1,7-dimethyl-5-oxo-3,9,10-trioxatricyclo[4.3.1.0~2,4~]dec-8-yl]-4-methylhepta-2,4-dienoyl}-1,2-dihydro-3H-pyrrol-3-one (non-preferred name)](http://img.cochemist.com/ccimg/60600/60587-14-6_b.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8.png)
![(3S,8aS)-3-(1H-indol-3-ylmethyl)hexahydropyrrolo[1,2-a]pyrazine-1,4-dione](http://img.cochemist.com/ccimg/38200/38136-70-8_b.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,3-[[2-(1,1- dimethyl-2-propenyl)-1H-indol-3-yl]methyl]- hexahydro-,(3S,8aS)-](http://img.cochemist.com/ccimg/34700/34610-68-9.png)
![Pyrrolo[1,2-a]pyrazine-1,4-dione,3-[[2-(1,1- dimethyl-2-propenyl)-1H-indol-3-yl]methyl]- hexahydro-,(3S,8aS)-](http://img.cochemist.com/ccimg/34700/34610-68-9_b.png)








![Thiazole,4,5-dihydro-4-[(1Z,5E,7E,11R)-11-methoxy-8-methyl-1,5,7,13-tetradecatetraen-1-yl]-2-[(1R,2S)-2-methylcyclopropyl]-,(4R)-](http://img.cochemist.com/ccimg/155300/155233-30-0.png)
![Thiazole,4,5-dihydro-4-[(1Z,5E,7E,11R)-11-methoxy-8-methyl-1,5,7,13-tetradecatetraen-1-yl]-2-[(1R,2S)-2-methylcyclopropyl]-,(4R)-](http://img.cochemist.com/ccimg/155300/155233-30-0_b.png)