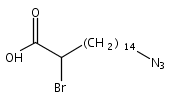
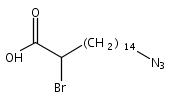
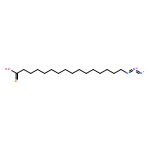
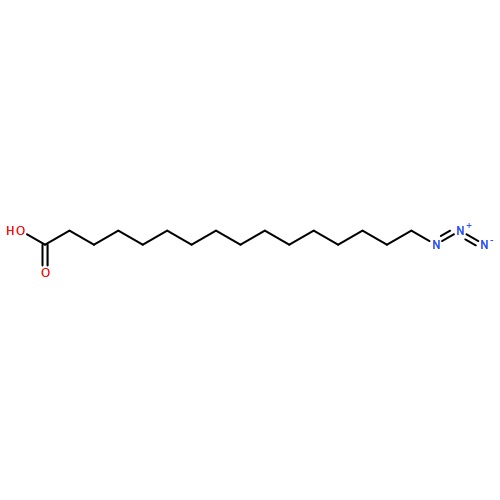
Co-reporter:Lei Shen, McKenna Schroeder, Tadeusz L. Ogorzalek, Pei Yang, Fu-Gen Wu, E. Neil G. Marsh, and Zhan Chen
Langmuir May 27, 2014 Volume 30(Issue 20) pp:5930-5938
Publication Date(Web):May 6, 2014
DOI:10.1021/la5016862
We demonstrate the control of enzyme orientation for enzymes chemically immobilized on surfaces. Nitro-reductase (NfsB) has the ability to reduce a broad range of nitro-containing compounds and has potential applications in a broad range of areas including the detection and decomposition of explosives. The enzyme was tethered through unique surface cysteine residues to a self-assembled monolayer (SAM) terminated with maleimide groups. One cysteine was introduced close to the active site (V424C), and the other, at a remote site (H360C). The surface-tethered NfsB variants were interrogated by a combination of surface-sensitive sum frequency generation (SFG) vibrational spectroscopy and attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) to determine how the mode of attachment altered the enzyme’s orientation. The activities of the two immobilized NfsB variants were measured and can be well correlated to the deduced orientations. The relationships among enzyme engineering, surface immobilization, enzyme orientation, and enzyme activity were revealed.
Co-reporter:Somayesadat Badieyan, Qiuming Wang, Xingquan Zou, Yaoxin Li, Maggie Herron, Nicholas L. Abbott, Zhan Chen, and E. Neil G. Marsh
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:2872-2872
Publication Date(Web):February 13, 2017
DOI:10.1021/jacs.6b12174
In the absence of aqueous buffer, most enzymes retain little or no activity; however, “water-free” enzymes would have many diverse applications. Here, we describe the chemically precise immobilization of an enzyme on an engineered surface designed to support catalytic activity in air at ambient humidity. Covalent immobilization of haloalkane dehalogenase on a surface support displaying poly(sorbitol methacrylate) chains resulted in ∼40-fold increase in activity over lyophilized enzyme powders for the gas-phase dehalogenation of 1-bromopropane. The activity of the immobilized enzyme in air approaches 25% of the activity obtained in buffer for the immobilized enzyme. Poly(sorbitol methacrylate) appears to enhance activity by replacing protein–water interactions, thereby preserving the protein structure.
Co-reporter:McKenna M. Schroeder, Qiuming Wang, Somayesadat Badieyan, Zhan Chen, and E. Neil G. Marsh
Langmuir July 18, 2017 Volume 33(Issue 28) pp:7152-7152
Publication Date(Web):June 27, 2017
DOI:10.1021/acs.langmuir.7b00646
We have investigated two surface properties that are generally thought to have an important influence of enzyme activity and stability: surface hydrophobicity and surface crowding. Here two variants of an engineered bacterial nitro-reductase were covalently tethered to orient the protein’s pseudo-2-fold symmetry axis either parallel or perpendicular to the surface. The surface hydrophobicity was systematically varied by changing the ratio of methyl- to hydroxyl-groups displayed on the SAM surface, and the effects on enzyme activity, thermal stability, and structure investigated. Increasing surface hydrophobicity progressively decreased enzyme activity, but had no effect on thermal stability. Surface-sensitive sum frequency generation and attenuated total reflectance Fourier transform IR spectroscopies indicated that the enzyme is not denatured by the more hydrophobic surface, but is more likely trapped in less active conformations by transient hydrophobic interactions. In contrast, increasing enzyme surface concentration increased the specific activity of the parallel oriented enzyme, but had no effect on the activity of the perpendicularly oriented enzyme, suggesting that crowding effects are highly context dependent.
Co-reporter:Kyle L. Ferguson, Joseph D. Eschweiler, Brandon T. Ruotolo, and E. Neil G. Marsh
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:10972-10972
Publication Date(Web):July 28, 2017
DOI:10.1021/jacs.7b05060
Ferulic acid decarboxylase catalyzes the decarboxylation of phenylacrylic acid using a newly identified cofactor, prenylated flavin mononucleotide (prFMN). The proposed mechanism involves the formation of a putative pentacyclic intermediate formed by a 1,3 dipolar cyclo-addition of prFMN with the α–β double bond of the substrate, which serves to activate the substrate toward decarboxylation. However, enzyme-catalyzed 1,3 dipolar cyclo-additions are unprecedented and other mechanisms are plausible. Here we describe the use of a mechanism-based inhibitor, 2-fluoro-2-nitrovinylbenzene, to trap the putative cyclo-addition intermediate, thereby demonstrating that prFMN can function as a dipole in a 1,3 dipolar cyclo-addition reaction as the initial step in a novel type of enzymatic reaction.
Co-reporter:Marie Hoarau;Somayesadat Badieyan
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 45) pp:9539-9551
Publication Date(Web):2017/11/22
DOI:10.1039/C7OB01880K
Enzymes immobilized on solid supports have important and industrial and medical applications. However, their uses are limited by the significant reductions in activity and stability that often accompany the immobilization process. Here we review recent advances in our understanding of the molecular level interactions between proteins and supporting surfaces that contribute to changes in stability and activity. This understanding has been facilitated by the application of various surface-sensitive spectroscopic techniques that allow the structure and orientation of enzymes at the solid/liquid interface to be probed, often with monolayer sensitivity. An appreciation of the molecular interactions between enzyme and surface support has allowed the surface chemistry and method of enzyme attachement to be fine-tuned such that activity and stability can be greatly enhanced. These advances suggest that a much wider variety of enzymes may eventually be amenable to immobilization as green catalysts.
Co-reporter:Ajitha S. Cristie-David;Aaron Sciore;Somayesadat Badieyan;Joseph D. Escheweiler;Philipp Koldewey;James C. A. Bardwell;Brandon T. Ruotolo
Molecular Systems Design & Engineering (2016-Present) 2017 vol. 2(Issue 2) pp:140-148
Publication Date(Web):2017/05/09
DOI:10.1039/C7ME00012J
Coiled-coil domains are attractive modular components for assembling individual protein subunits into higher order structures because they can be designed de novo with well-defined oligomerization states, topologies, and dissociation energies. However, the utility of coiled-coil designs as plug-and-play components for synthetic biology applications depends critically on them robustly maintaining their oligomerization states when fused to larger proteins of interest. Here, we investigate the ability of a series of well-characterized de novo-designed parallel coiled coils, with oligomerization states ranging from dimer to pentamer, to mediate the oligomerization of a model monomeric protein, green fluorescent protein (GFP). Six coiled-coil GFP fusion proteins were initially constructed and their oligomerization states investigated using size exclusion chromatography, analytical ultracentrifugation, and native mass spectrometry. Somewhat surprisingly, only two of these initial designs adopted their intended oligomerization states. However, with minor refinements, the intended oligomerization states of two of the four other constructs could be achieved. Parameters found to influence the oligomerization state of the GFP fusions included the number of heptad repeats and the length of the linker sequence separating GFP from the coiled coil. These results demonstrate that even for stable, well-designed coiled coils, the oligomerization state is subject to unanticipated changes when connected to larger protein components. Therefore, although coiled coils can be successfully used as components in protein designs their ability to achieve the desired oligomerization state requires experimental verification.
Co-reporter:Benjamin R. Ellington, Bishwajit Paul, Debasis Das, Andrew K. Vitek, Paul M. Zimmerman, and E. Neil G. Marsh
ACS Catalysis 2016 Volume 6(Issue 5) pp:3293
Publication Date(Web):April 22, 2016
DOI:10.1021/acscatal.6b00592
The development of new pathways for next-generation biofuel production has spurred recent investigations into the mechanisms of enzymes that biosynthesize hydrocarbons. One widely distributed group of enzymes, aldehyde decarbonylases, catalyze unusual deformylation reactions in which long-chain fatty aldehydes are converted to alkanes. These enzymes are all iron-dependent and in insects are represented by a cytochrome P450 enzyme that releases the aldehyde carbon as CO2. Here we describe a novel nonenzymatic reaction of an α-cyclopropyl-substituted aldehyde that mimics the enzyme reaction. This aldehyde is oxidatively deformylated in the presence of aqueous iron(II) salts and oxygen to yield an alkyl-substituted cyclopropane and CO2, in a reaction that competes with the more conventional oxidation of the aldehyde to the carboxylic acid. Like the enzymatic reaction, the reaction occurs with retention of the aldehyde proton in the alkane product and probably proceeds through an iron–peroxo species. Computational reaction discovery tools were used to search for potential reaction pathways and investigate their energetic feasibility. These identified a plausible reaction pathway leading to the experimentally observed products and reproduced the transfer of the aldehyde proton to the cyclopropane product. These studies provide further insight into how enzymes may control reactive iron–oxo species to catalyze the diverse range of iron-dependent oxidative transformations observed in biology.Keywords: aldehyde decarbonylase; computational reaction discovery; cytochrome p450; enzyme models; isotope effects; nonheme iron oxygenases
Co-reporter:Kyle L. Ferguson, Nattapol Arunrattanamook, and E. Neil G. Marsh
Biochemistry 2016 Volume 55(Issue 20) pp:2857-2863
Publication Date(Web):April 27, 2016
DOI:10.1021/acs.biochem.6b00170
Ferulic acid decarboxylase from Saccharomyces cerevisiae catalyzes the decarboxylation of phenylacrylic acid to form styrene using a newly described prenylated flavin mononucleotide cofactor. A mechanism has been proposed, involving an unprecedented 1,3-dipolar cyclo-addition of the prenylated flavin with the α═β bond of the substrate that serves to activate the substrate toward decarboxylation. We measured a combination of secondary deuterium kinetic isotope effects (KIEs) at the α- and β-positions of phenylacrylic acid together with solvent deuterium KIEs. The solvent KIE is 3.3 on Vmax/KM but is close to unity on Vmax, indicating that proton transfer to the product occurs before the rate-determining step. The secondary KIEs are normal at both the α- and β-positions but vary in magnitude depending on whether the reaction is performed in H2O or D2O. In D2O, the enzyme catalyzed the exchange of deuterium into styrene; this reaction was dependent on the presence of bicarbonate. This observation implies that CO2 release must occur after protonation of the product. Further information was obtained from a linear free-energy analysis of the reaction through the use of a range of para- and meta-substituted phenylacrylic acids. Log(kcat/KM) for the reaction correlated well with the Hammett σ– parameter with ρ = −0.39 ± 0.03; r2 = 0.93. The negative ρ value and secondary isotope effects are consistent with the rate-determining step being the formation of styrene from the prenylated flavin–product adduct through a cyclo-elimination reaction.
Co-reporter:Lei Shen, Kenneth Chun Kuen Cheng, McKenna Schroeder, Pei Yang, E. Neil G. Marsh, Joerg Lahann, Zhan Chen
Surface Science 2016 Volume 648() pp:53-59
Publication Date(Web):June 2016
DOI:10.1016/j.susc.2015.10.046
•Enzymes were successfully immobilized on polymer surfaces.•Orientations of immobilized enzymes on polymers were determined.•Enzyme orientations on polymer and on SAM may or may not be similar.We successfully immobilized enzymes onto polymer surfaces via covalent bonds between cysteine groups of the enzyme and dibromomaleimide functionalities present at the polymer surface. In this work, we used nitroreductase (NfsB) as a model enzyme molecule. The polymers were prepared by chemical vapor deposition (CVD) polymerization, resulting in surfaces with dibromomaleimide groups. NfsB variants were engineered so that each NfsB molecule only has one cysteine group on the enzyme surface. Two different NfsB constructs were studied, with cysteines at the positions of H360 and V424, respectively. A combination of sum frequency generation (SFG) vibrational and attenuated total reflectance Fourier transformed infrared (ATR-FTIR) spectroscopies were used to deduce the orientation of the immobilized enzymes on the surface. It was found that the orientation of the immobilized enzymes is controlled by the position of the cysteine residue in the protein. The NfsB H360C construct exhibited a similar orientational behavior on the polymer surface as compared to that on the self-assembled monolayer surface, but the NsfB V424C construct showed markedly different orientations on the two surfaces.
Co-reporter:Junjie Chen, Mallory A. van Dongen, Rachel L. Merzel, Casey A. Dougherty, Bradford G. Orr, Ananda Kumar Kanduluru, Philip S. Low, E. Neil G. Marsh, and Mark M. Banaszak Holl
Biomacromolecules 2016 Volume 17(Issue 3) pp:
Publication Date(Web):January 27, 2016
DOI:10.1021/acs.biomac.5b01586
Polymer–ligand conjugates are designed to bind proteins for applications as drugs, imaging agents, and transport scaffolds. In this work, we demonstrate a folic acid (FA)-triggered exosite binding of a generation five poly(amidoamine) (G5 PAMAM) dendrimer scaffold to bovine folate binding protein (bFBP). The protein exosite is a secondary binding site on the protein surface, separate from the FA binding pocket, to which the dendrimer binds. Exosite binding is required to achieve the greatly enhanced binding constants and protein structural change observed in this study. The G5Ac-COG-FA1.0 conjugate bound tightly to bFBP, was not displaced by a 28-fold excess of FA, and quenched roughly 80% of the initial fluorescence. Two-step binding kinetics were measured using the intrinsic fluorescence of the FBP tryptophan residues to give a KD in the low nanomolar range for formation of the initial G5Ac-COG-FA1.0/FBP* complex, and a slow conversion to the tight complex formed between the dendrimer and the FBP exosite. The extent of quenching was sensitive to the choice of FA-dendrimer linker chemistry. Direct amide conjugation of FA to G5-PAMAM resulted in roughly 50% fluorescence quenching of the FBP. The G5Ac-COG-FA, which has a longer linker containing a 1,2,3-triazole ring, exhibited an ∼80% fluorescence quenching. The binding of the G5Ac-COG-FA1.0 conjugate was compared to poly(ethylene glycol) (PEG) conjugates of FA (PEGn-FA). PEG2k-FA had a binding strength similar to that of FA, whereas other PEG conjugates with higher molecular weight showed weaker binding. However, no PEG conjugates gave an increased degree of total fluorescence quenching.
Co-reporter:Aaron Sciore;Min Su;Philipp Koldewey;Joseph D. Eschweiler;Kelsey A. Diffley;Brian M. Linhares;Brandon T. Ruotolo;James C. A. Bardwell;Georgios Skiniotis;
Proceedings of the National Academy of Sciences 2016 113(31) pp:8681-8686
Publication Date(Web):July 18, 2016
DOI:10.1073/pnas.1606013113
Co-reporter:Fengming Lin, Kyle L. Ferguson, David R. Boyer, Xiaoxia Nina Lin, and E. Neil G. Marsh
ACS Chemical Biology 2015 Volume 10(Issue 4) pp:1137
Publication Date(Web):February 3, 2015
DOI:10.1021/cb5008103
The decarboxylation of antimicrobial aromatic acids such as phenylacrylic acid (cinnamic acid) and ferulic acid by yeast requires two enzymes described as phenylacrylic acid decarboxylase (PAD1) and ferulic acid decarboxylase (FDC). These enzymes are of interest for various biotechnological applications, such as the production of chemical feedstocks from lignin under mild conditions. However, the specific role of each protein in catalyzing the decarboxylation reaction remains unknown. To examine this, we have overexpressed and purified both PAD1 and FDC from E. coli. We demonstrate that PAD1 is a flavin mononucleotide (FMN)-containing protein. However, it does not function as a decarboxylase. Rather, PAD1 catalyzes the formation of a novel, diffusible cofactor required by FDC for decarboxylase activity. Coexpression of FDC and PAD1 results in the production of FDC with high levels cofactor bound. Holo-FDC catalyzes the decarboxylation of phenylacrylic acid, coumaric acid and ferulic acid with apparent kcat ranging from 1.4–4.6 s–1. The UV-visible and mass spectra of the cofactor indicate that it appears to be a novel, modified form of reduced FMN; however, its instability precluded determination of its structure. The E. coli enzymes UbiX and UbiD are related by sequence to PAD1 and FDC respectively and are involved in the decarboxylation of 4-hydroxy-3-octaprenylbenzoic acid, an intermediate in ubiquinone biosynthesis. We found that endogenous UbiX can also activate FDC. This implies that the same cofactor is required for decarboxylation of 4-hydroxy-3-polyprenylbenzoic acid by UbiD and suggests a wider role for this cofactor in metabolism.
Co-reporter:Feng-Ming Lin, E. Neil G. Marsh, Xiaoxia Nina Lin
Chinese Chemical Letters 2015 Volume 26(Issue 4) pp:431-434
Publication Date(Web):April 2015
DOI:10.1016/j.cclet.2015.03.018
Biofuels derived from hydrocarbon biosynthetic pathways have attracted increasing attention. Routes to hydrocarbon biofuels are emerging and mainly fall into two categories based on the metabolic pathways utilized: Fatty acid pathway-based alkanes/alkenes and isoprenoid biosynthetic pathway based terpenes. The primary focus of this review is on recent progress in the application of hydrocarbon biosynthetic pathways for hydrocarbon biofuel production, together with studies on enzymes, including efforts to engineering them for improved performance.The primary focus of this review is on recent progress in the application of hydrocarbon biosynthetic pathways for hydrocarbon biofuel production, together with studies on enzymes, including efforts to engineering them for improved performance.
Co-reporter:Tadeusz L. Ogorzalek, Shuai Wei, Yuwei Liu, Quiming Wang, Charles L. Brooks III, Zhan Chen, and E. Neil G. Marsh
Langmuir 2015 Volume 31(Issue 22) pp:6145-6153
Publication Date(Web):May 14, 2015
DOI:10.1021/acs.langmuir.5b01735
Surface-immobilized enzymes are important for a wide range of technological applications, including industrial catalysis, drug delivery, medical diagnosis, and biosensors; however, our understanding of how enzymes and proteins interact with abiological surfaces on the molecular level remains extremely limited. We have compared the structure, activity, and thermal stability of two variants of a β-galactosidase attached to a chemically well-defined maleimide-terminated self-assembled monolayer surface through a unique cysteinyl residue. In one case the enzyme is attached through an α helix and in the other case through an adjacent loop. Both enzymes exhibit similar specific activities and adopt similar orientations with respect to the surface normal, as determined by sum-frequency generation and attenuated total reflectance FT-IR spectroscopies. Surprisingly, however, the loop-tethered enzyme exhibits a thermal stability 10 °C lower than the helix-tethered enzyme and 13 °C lower than the enzyme in free solution. Using coarse-grain models, molecular dynamics simulations of the thermal unfolding of the surface-tethered enzymes were able to reproduce these differences in stability. Thus, revealing that tethering through the more flexible loop position provides more opportunity for surface residues on the protein to interact with the surface and undergo surface-induced unfolding. These observations point to the importance of the location of the attachment point in determining the performance of surface-supported biocatalysts and suggest strategies for optimizing their activity and thermal stability through molecular simulations.
Co-reporter:E. Neil G. Marsh
Accounts of Chemical Research 2014 Volume 47(Issue 10) pp:2878
Publication Date(Web):June 2, 2014
DOI:10.1021/ar500125m
Fluorine is all but absent from biology; however, it has proved to be a remarkably useful element with which to modulate the activity of biological molecules and to study their mechanism of action. Our laboratory’s interest in incorporating fluorine into proteins was stimulated by the unusual physicochemical properties exhibited by perfluorinated small molecules. These include extreme chemical inertness and thermal stability, properties that have made them valuable as nonstick coatings and fire retardants. Fluorocarbons also exhibit an unusual propensity to phase segregation. This phenomenon, which has been termed the “fluorous effect”, has been effectively exploited in organic synthesis to purify compounds from reaction mixtures by extracting fluorocarbon-tagged molecules into fluorocarbon solvents. As biochemists, we were curious to explore whether the unusual physicochemical properties of perfluorocarbons could be engineered into proteins.To do this, we developed a synthesis of a highly fluorinated amino acid, hexafluoroleucine, and designed a model 4-helix bundle protein, α4H, in which the hydrophobic core was packed exclusively with leucine. We then investigated the effects of repacking the hydrophobic core of α4H with various combinations of leucine and hexafluoroleucine. These initial studies demonstrated that fluorination is a general and effective strategy for enhancing the stability of proteins against chemical and thermal denaturation and proteolytic degradation.We had originally envisaged that the “fluorous interactions”, postulated from the self-segregating properties of fluorous solvents, might be used to mediate specific protein–protein interactions orthogonal to those of natural proteins. However, various lines of evidence indicate that no special, favorable fluorine–fluorine interactions occur in the core of the fluorinated α4 protein. This makes it unlikely that fluorinated amino acids can be used to direct protein–protein interactions.More recent detailed thermodynamic and structural studies in our laboratory have uncovered the basis for the remarkably general ability of fluorinated side chains to stabilize protein structure. Crystal structures of α4H and its fluorinated analogues show that the fluorinated residues fit into the hydrophobic core with remarkably little perturbation to the structure. This is explained by the fact that fluorinated side chains, although larger, very closely preserve the shape of the hydrophobic amino acids they replace. Thus, an increase in buried hydrophobic surface area in the folded state is responsible for the additional thermodynamic stability of the fluorinated protein. Measurements of ΔG°, ΔH°, ΔS°, and ΔCp° for unfolding demonstrate that the “fluorous” stabilization of these protein arises from the hydrophobic effect in the same way that hydrophobic partitioning stabilizes natural proteins.
Co-reporter:Debasis Das, Benjamin Ellington, Bishwajit Paul, and E. Neil G. Marsh
ACS Chemical Biology 2014 Volume 9(Issue 2) pp:570
Publication Date(Web):December 6, 2013
DOI:10.1021/cb400772q
The biosynthesis of long-chain aliphatic hydrocarbons, which are derived from fatty acids, is widespread in Nature. The last step in this pathway involves the decarbonylation of fatty aldehydes to the corresponding alkanes or alkenes. In cyanobacteria, this is catalyzed by an aldehyde deformylating oxygenase. We have investigated the mechanism of this enzyme using substrates bearing an oxirane ring adjacent to the aldehyde carbon. The enzyme catalyzed the deformylation of these substrates to produce the corresponding oxiranes. Performing the reaction in D2O allowed the facial selectivity of proton addition to be examined by 1H NMR spectroscopy. The proton is delivered with equal probability to either face of the oxirane ring, indicating the formation of an oxiranyl radical intermediate that is free to rotate during the reaction. Unexpectedly, the enzyme also catalyzes a side reaction in which oxiranyl-aldehydes undergo tandem deformylation to furnish alkanes two carbons shorter. We present evidence that this involves the rearrangement of the intermediate oxiranyl radical formed in the first step, resulting in aldehyde that is further deformylated in a second step. These observations provide support for a radical mechanism for deformylation and, furthermore, allow the lifetime of the radical intermediate to be estimated based on prior measurements of rate constants for the rearrangement of oxiranyl radicals.
Co-reporter:E. Neil G. Marsh and Yuta Suzuki
ACS Chemical Biology 2014 Volume 9(Issue 6) pp:1242
Publication Date(Web):April 24, 2014
DOI:10.1021/cb500111u
Fluorine is a valuable probe for investigating the interactions of biological molecules because of its favorable NMR characteristics, its small size, and its near total absence from biology. Advances in biosynthetic methods allow fluorine to be introduced into peptides and proteins with high precision, and the increasing sensitivity of NMR spectrometers has facilitated the use of 19F NMR to obtain molecular-level insights into a wide range of often-complex biological interactions. Here, we summarize the advantages of solution-state 19F NMR for studying the interactions of peptides and proteins with other biological molecules, review methods for the production of fluorine-labeled materials, and describe some representative recent examples in which 19F NMR has been used to study conformational changes in peptides and proteins and their interactions with other biological molecules.
Co-reporter:Jiarui Wang, Rory P. Woldring, Gabriel D. Román-Meléndez, Alan M. McClain, Brian R. Alzua, and E. Neil G. Marsh
ACS Chemical Biology 2014 Volume 9(Issue 9) pp:1929
Publication Date(Web):July 10, 2014
DOI:10.1021/cb5004674
The radical S-adenosylmethionine (SAM) superfamily of enzymes catalyzes an amazingly diverse variety of reactions ranging from simple hydrogen abstraction to complicated multistep rearrangements and insertions. The reactions they catalyze are important for a broad range of biological functions, including cofactor and natural product biosynthesis, DNA repair, and tRNA modification. Generally conserved features of the radical SAM superfamily include a CX3CX2C motif that binds an [Fe4S4] cluster essential for the reductive cleavage of SAM. Here, we review recent advances in our understanding of the structure and mechanisms of these enzymes that, in some cases, have overturned widely accepted mechanisms.
Co-reporter:Benjamin C. Buer, Bishwajit Paul, Debasis Das, Jeanne A. Stuckey, and E. Neil G. Marsh
ACS Chemical Biology 2014 Volume 9(Issue 11) pp:2584
Publication Date(Web):September 15, 2014
DOI:10.1021/cb500343j
The nonheme diiron enzyme cyanobacterial aldehyde deformylating oxygenase, cADO, catalyzes the highly unusual deformylation of aliphatic aldehydes to alkanes and formate. We have determined crystal structures for the enzyme with a long-chain water-soluble aldehyde and medium-chain carboxylic acid bound to the active site. These structures delineate a hydrophobic channel that connects the solvent with the deeply buried active site and reveal a mode of substrate binding that is different from previously determined structures with long-chain fatty acids bound. The structures also identify a water channel leading to the active site that could facilitate the entry of protons required in the reaction. NMR studies examining 1-[13C]-octanal binding to cADO indicate that the enzyme binds the aldehyde form rather than the hydrated form. Lastly, the fortuitous cocrystallization of the metal-free form of the protein with aldehyde bound has revealed protein conformation changes that are involved in binding iron.
Co-reporter:Gabriel D. Román-Meléndez, Patrick von Glehn, Jeremy N. Harvey, Adrian J. Mulholland, and E. Neil G. Marsh
Biochemistry 2014 Volume 53(Issue 1) pp:
Publication Date(Web):December 16, 2013
DOI:10.1021/bi4012644
Adenosylcobalamin (AdoCbl) serves as a source of reactive free radicals that are generated by homolytic scission of the coenzyme’s cobalt–carbon bond. AdoCbl-dependent enzymes accelerate AdoCbl homolysis by ∼1012-fold, but the mechanism by which this is accomplished remains unclear. We have combined experimental and computational approaches to gain molecular-level insight into this process for glutamate mutase. Two residues, glutamate 330 and lysine 326, form hydrogen bonds with the adenosyl group of the coenzyme. A series of mutations that impair the enzyme’s ability to catalyze coenzyme homolysis and tritium exchange with the substrate by 2–4 orders of magnitude were introduced at these positions. These mutations, together with the wild-type enzyme, were also characterized in silico by molecular dynamics simulations of the enzyme–AdoCbl–substrate complex with AdoCbl modeled in the associated (Co–C bond formed) or dissociated [adenosyl radical with cob(II)alamin] state. The simulations reveal that the number of hydrogen bonds between the adenosyl group and the protein side chains increases in the homolytically dissociated state, with respect to the associated state, for both the wild-type and mutant enzymes. The mutations also cause a progressive increase in the mean distance between the 5′-carbon of the adenosyl radical and the abstractable hydrogen of the substrate. Interestingly, the distance between the 5′-carbon and substrate hydrogen, determined computationally, was found to inversely correlate with the log k for tritium exchange (r = 0.93) determined experimentally. Taken together, these results point to a dual role for these residues: they both stabilize the homolytic state through electrostatic interactions between the protein and the dissociated coenzyme and correctly position the adenosyl radical to facilitate the abstraction of hydrogen from the substrate.
Co-reporter:Matthew W. Waugh and E. Neil G. Marsh
Biochemistry 2014 Volume 53(Issue 34) pp:
Publication Date(Web):August 21, 2014
DOI:10.1021/bi5005766
The reaction catalyzed by cyanobacterial aldehyde deformylating oxygenase is of interest both because of its potential application to the production of biofuels and because of the highly unusual nature of the deformylation reaction it catalyzes. Whereas the proton in the product alkane derives ultimately from the solvent, the identity of the proton donor in the active site remains unclear. To investigate the proton transfer step, solvent isotope effect (SIE) studies were undertaken. The rate of alkane formation was found to be maximal at pH 6.8 and to be the same in D2O or H2O within experimental error, implying that proton transfer is not a kinetically significant step. However, when the ratio of protium to deuterium in the product alkane was measured as a function of the mole fraction of D2O, a D2OSIEobs of 2.19 ± 0.02 was observed. The SIE was invariant with the mole fraction of D2O, indicating the involvement of a single protic site in the reaction. We interpret this SIE as most likely arising from a reactant state equilibrium isotope effect on a proton donor with an inverse fractionation factor, for which Φ = 0.45. These observations are consistent with an iron-bound water molecule being the proton donor to the alkane in the reaction.
Co-reporter:Lei Shen, McKenna Schroeder, Tadeusz L. Ogorzalek, Pei Yang, Fu-Gen Wu, E. Neil G. Marsh, and Zhan Chen
Langmuir 2014 Volume 30(Issue 20) pp:5930-5938
Publication Date(Web):May 6, 2014
DOI:10.1021/la5016862
We demonstrate the control of enzyme orientation for enzymes chemically immobilized on surfaces. Nitro-reductase (NfsB) has the ability to reduce a broad range of nitro-containing compounds and has potential applications in a broad range of areas including the detection and decomposition of explosives. The enzyme was tethered through unique surface cysteine residues to a self-assembled monolayer (SAM) terminated with maleimide groups. One cysteine was introduced close to the active site (V424C), and the other, at a remote site (H360C). The surface-tethered NfsB variants were interrogated by a combination of surface-sensitive sum frequency generation (SFG) vibrational spectroscopy and attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) to determine how the mode of attachment altered the enzyme’s orientation. The activities of the two immobilized NfsB variants were measured and can be well correlated to the deduced orientations. The relationships among enzyme engineering, surface immobilization, enzyme orientation, and enzyme activity were revealed.
Co-reporter:Bishwajit Paul ; Debasis Das ; Benjamin Ellington
Journal of the American Chemical Society 2013 Volume 135(Issue 14) pp:5234-5237
Publication Date(Web):March 20, 2013
DOI:10.1021/ja3115949
Cyanobacterial aldehyde decarbonylase (cAD) is a non-heme diiron oxygenase that catalyzes the conversion of fatty aldehydes to alkanes and formate. The mechanism of this chemically unusual reaction is poorly understood. We have investigated the mechanism of C1–C2 bond cleavage by cAD using a fatty aldehyde that incorporates a cyclopropyl group, which can act as a radical clock. When reacted with cAD, the cyclopropyl aldehyde produces 1-octadecene as the rearranged product, providing evidence for a radical mechanism for C–C bond scission. In an alternate pathway, the cyclopropyl aldehyde acts as a mechanism-based irreversible inhibitor of cAD through covalent binding of the alkyl chain to the enzyme.
Co-reporter:Yuwei Liu ; Tadeusz L. Ogorzalek ; Pei Yang ; McKenna M. Schroeder ; E. Neil G. Marsh ;Zhan Chen
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12660-12669
Publication Date(Web):July 24, 2013
DOI:10.1021/ja403672s
The immobilization of enzymes on solid supports is widely used in many applications, including biosensors, antifouling coatings, food packaging materials, and biofuel cells. Enzymes tend to lose their activity when in contact with a support surface, a phenomenon that has been attributed to unfavorable orientation and (partial) unfolding. In this work, specific immobilization of 6-phospho-β-galactosidase (β-Gal) on a self-assembled monolayer (SAM) containing maleimide end groups and oligo(ethylene glycol) spacer segments was achieved through a unique cysteinyl residue. A systematic means to characterize the interfacial orientation of immobilized enzymes has been developed using a combination of sum frequency generation vibrational spectroscopy and attenuated total reflectance FTIR-spectroscopy. The possible orientations of the immobilized β-Gal were determined and found to be well-correlated with the tested activity of β-Gal. This study will impact the development of an increasingly wide range of devices that use surface-immobilized enzymes as integral components with improved functions, better sensitivity, enhanced stability, and longer shelf life.
Co-reporter:E. Neil G. Marsh and Matthew W. Waugh
ACS Catalysis 2013 Volume 3(Issue 11) pp:2515
Publication Date(Web):September 18, 2013
DOI:10.1021/cs400637t
Co-reporter:Yuta Suzuki, Jeffrey R. Brender, Molly T. Soper, Janarthanan Krishnamoorthy, Yunlong Zhou, Brandon T. Ruotolo, Nicholas A. Kotov, Ayyalusamy Ramamoorthy, and E. Neil G. Marsh
Biochemistry 2013 Volume 52(Issue 11) pp:
Publication Date(Web):February 27, 2013
DOI:10.1021/bi400027y
In the commonly used nucleation-dependent model of protein aggregation, aggregation proceeds only after a lag phase in which the concentration of energetically unfavorable nuclei reaches a critical value. The formation of oligomeric species prior to aggregation can be difficult to detect by current spectroscopic techniques. By using real-time 19F NMR along with other techniques, we are able to show that multiple oligomeric species can be detected during the lag phase of Aβ1–40 fiber formation, consistent with a complex mechanism of aggregation. At least six types of oligomers can be detected by 19F NMR. These include the reversible formation of large β-sheet oligomer immediately after solubilization at high peptide concentration, a small oligomer that forms transiently during the early stages of the lag phase, and four spectroscopically distinct forms of oligomers with molecular weights between ∼30 and 100 kDa that appear during the later stages of aggregation. The ability to resolve individual oligomers and track their formation in real-time should prove fruitful in understanding the aggregation of amyloidogenic proteins and in isolating potentially toxic nonamyloid oligomers.
Co-reporter:Benjamin C. Buer ; Benjamin J. Levin
Journal of the American Chemical Society 2012 Volume 134(Issue 31) pp:13027-13034
Publication Date(Web):July 16, 2012
DOI:10.1021/ja303521h
The introduction of highly fluorinated analogues of hydrophobic amino acid residues into proteins has proved an effective and general strategy for increasing protein stability toward both chemical denaturants and heat. However, the thermodynamic basis for this stabilizing effect, whether enthalpic or entropic in nature, has not been extensively investigated. Here we describe studies in which the values of ΔH°, ΔS°, and ΔCp° have been determined for the unfolding of a series of 12 small, de novo-designed proteins in which the hydrophobic core is packed with various combinations of fluorinated and non-fluorinated amino acid residues. The increase in the free energy of unfolding with increasing fluorine content is associated with increasingly unfavorable entropies of unfolding and correlates well with calculated changes in apolar solvent-accessible surface area. ΔCp° for unfolding is positive for all the proteins and, similarly, correlates with changes in apolar solvent-accessible surface area. ΔH° for unfolding shows no correlation with either fluorine content or changes in apolar solvent-accessible surface area. We conclude that conventional hydrophobic effects adequately explain the enhanced stabilities of most highly fluorinated proteins. The extremely high thermal stability of these proteins results, in part, from their very low per-residue ΔCp°, as has been observed for natural thermostable proteins.
Co-reporter:Yuta Suzuki, Jeffrey R. Brender, Kevin Hartman, Ayyalusamy Ramamoorthy, and E. Neil G. Marsh
Biochemistry 2012 Volume 51(Issue 41) pp:
Publication Date(Web):September 21, 2012
DOI:10.1021/bi3012548
Amyloid formation, a complex process involving many intermediate states, is proposed to be the driving force for amyloid-related toxicity in common degenerative diseases. Unfortunately, the details of this process have been obscured by the limitations in the methods that can follow this reaction in real time. We show that alternative pathways of aggregation can be distinguished by using 19F nuclear magnetic resonance (NMR) to monitor monomer consumption along with complementary measurements of fibrillogenesis. The utility of this technique is demonstrated by tracking amyloid formation in the diabetes-related islet amyloid polypeptide (IAPP). Using this technique, we show IAPP fibrillizes without an appreciable buildup of nonfibrillar intermediates, in contrast to the well-studied Aβ and α-synuclein proteins. To further develop the usage of 19F NMR, we have tracked the influence of the polyphenolic amyloid inhibitor epigallocatechin gallate (EGCG) on the aggregation pathway. Polyphenols have been shown to strongly inhibit amyloid formation in many systems. However, spectroscopic measurements of amyloid inhibition by these compounds can be severely compromised by background signals and competitive binding with extrinsic probes. Using 19F NMR, we show that thioflavin T strongly competes with EGCG for binding sites on IAPP fibers. By comparing the rates of monomer consumption and fiber formation, we are able to show that EGCG stabilizes nonfibrillar large aggregates during fibrillogenesis.
Co-reporter:Benjamin C. Buer;Jennifer L. Meagher;Jeanne A. Stuckey
PNAS 2012 Volume 109 (Issue 13 ) pp:4810-4815
Publication Date(Web):2012-03-27
DOI:10.1073/pnas.1120112109
Noncanonical amino acids have proved extremely useful for modifying the properties of proteins. Among them, extensively fluorinated
(fluorous) amino acids seem particularly effective in increasing protein stability; however, in the absence of structural
data, the basis of this stabilizing effect remains poorly understood. To address this problem, we solved X-ray structures
for three small proteins with hydrophobic cores that are packed with either fluorocarbon or hydrocarbon side chains and compared
their stabilities. Although larger, the fluorinated residues are accommodated within the protein with minimal structural perturbation,
because they closely match the shape of the hydrocarbon side chains that they replace. Thus, stability increases seem to be
better explained by increases in buried hydrophobic surface area that accompany fluorination than by specific fluorous interactions
between fluorinated side chains. This finding is illustrated by the design of a highly fluorinated protein that, by compensating
for the larger volume and surface area of the fluorinated side chains, exhibits similar stability to its nonfluorinated counterpart.
These structure-based observations should inform efforts to rationally modulate protein function using noncanonical amino
acids.
Co-reporter:E. Neil G. Marsh, Gabriel D. Román Meléndez
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2012 Volume 1824(Issue 11) pp:1154-1164
Publication Date(Web):November 2012
DOI:10.1016/j.bbapap.2012.03.012
Adenosylcobalamin (coenzyme B12) serves as the cofactor for a group of enzymes that catalyze unusual rearrangement or elimination reactions. The role of the cofactor as the initiator of reactive free radicals needed for these reactions is well established. Less clear is how these enzymes activate the coenzyme towards homolysis and control the radicals once generated. The availability of high resolution X-ray structures combined with detailed kinetic and spectroscopic analyses have allowed several adenosylcobalamin enzymes to be computationally modeled in some detail. Computer simulations have generally obtained good agreement with experimental data and provided valuable insight into the mechanisms of these unusual reactions. Importantly, atomistic modeling of the enzymes has allowed the role of specific interactions between protein, substrate and coenzyme to be explored, leading to mechanistic predictions that can now be tested experimentally. This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.Highlights► AdoCbl-dependent enzymes catalyze unusual rearrangement and elimination reactions. ► Experiments point to profound stabilization of radical intermediates by the protein. ► Recent computational modeling informs mechanistic understanding of these enzymes.
Co-reporter:Yuta Suzuki, Benjamin C. Buer, Hashim M. Al-Hashimi, and E. Neil G. Marsh
Biochemistry 2011 Volume 50(Issue 27) pp:
Publication Date(Web):June 6, 2011
DOI:10.1021/bi200639c
The antimicrobial peptide MSI-78 serves as a model system for studying interactions of bioactive peptides with membranes. Using a series of MSI-78 peptides that incorporate l-4,4,4-trifluoroethylglycine, a small and sensitive 19F nuclear magnetic resonance probe, we investigated how the local structure and dynamics of the peptide change when it binds to the lipid bilayer. The fluorinated MSI-78 analogues exhibited position-specific changes in 19F chemical shift ranging from 1.28 to −1.35 ppm upon binding to lipid bicelles. The largest upfield shifts are associated with the most hydrophobic positions in the peptide. Changes in solvent isotope effects (H2O/D2O) on 19F chemical shifts were observed for the peptides that are consistent with the MSI-78 solvent-inaccessible hydrophobic core upon binding bicelles. Transverse relaxation measurements of the 19F nucleus, using the Carr–Purcell–Meiboom–Gill pulse sequence, were used to examine changes in the local mobility of MSI-78 that occur upon binding to the lipid bilayer. Positions in the hydrophobic core of peptide–membrane complex show the greatest decrease in mobility upon binding of the lipid bilayer, whereas residues that interact with lipid headgroups are more mobile. The most mobile positions are at the N- and C-termini of the peptide. These results provide support for the proposed mechanism of membrane disruption by MSI-78 and reveal new details about the dynamic changes that accompany membrane binding.
Co-reporter:Bekir E. Eser, Debasis Das, Jaehong Han, Patrik R. Jones, and E. Neil G. Marsh
Biochemistry 2011 Volume 50(Issue 49) pp:
Publication Date(Web):November 10, 2011
DOI:10.1021/bi2012417
Cyanobacterial aldehyde decarbonylase (cAD) is, structurally, a member of the di-iron carboxylate family of oxygenases. We previously reported that cAD from Prochlorococcus marinus catalyzes the unusual hydrolysis of aldehydes to produce alkanes and formate in a reaction that requires an external reducing system but does not require oxygen [Das et al. (2011) Angew. Chem. 50, 7148–7152]. Here we demonstrate that cADs from divergent cyanobacterial classes, including the enzyme from N. puntiformes that was reported to be oxygen dependent, catalyze aldehyde decarbonylation at a much faster rate under anaerobic conditions and that the oxygen in formate derives from water. The very low activity (<1 turnover/h) of cAD appears to result from inhibition by the ferredoxin reducing system used in the assay and the low solubility of the substrate. Replacing ferredoxin with the electron mediator phenazine methosulfate allowed the enzyme to function with various chemical reductants, with NADH giving the highest activity. NADH is not consumed during turnover, in accord with the proposed catalytic role for the reducing system in the reaction. With octadecanal, a burst phase of product formation, kprod = 3.4 ± 0.5 min–1, is observed, indicating that chemistry is not rate-determining under the conditions of the assay. With the more soluble substrate, heptanal, kcat = 0.17 ± 0.01 min–1 and no burst phase is observed, suggesting that a chemical step is limiting in the reaction of this substrate.
Co-reporter:Dustin P. Patterson, Ankur M. Desai, Mark M. Banaszak Holl and E. Neil G. Marsh
RSC Advances 2011 vol. 1(Issue 6) pp:1004-1012
Publication Date(Web):20 Sep 2011
DOI:10.1039/C1RA00282A
We evaluate a strategy for assembling proteins into large cage-like structures, based on the symmetry associated with the native protein's quaternary structure. Using a trimeric protein, KDPG aldolase, as a building block, two fusion proteins were designed that could assemble together upon mixing. The fusion proteins, designated A-(+) and A-(−), comprise the aldolase domain, a short, flexible spacer sequence, and a sequence designed to form a heterodimeric antiparallel coiled-coil between A-(+) and A-(−). The flexible spacer is included to minimize constraints on the ability of the fusion proteins to assemble into larger structures. On incubating together, A-(+) and A-(−) assembled into a mixture of complexes that were analyzed by size exclusion chromatography coupled to multi-angle laser light scattering, analytical ultracentrifugation, transmission electron microscopy and atomic force microscopy. Our analysis indicates that, despite the inherent flexibility of the assembly strategy, the proteins assemble into a limited number of globular structures. Dimeric and tetrameric complexes of A-(+) and A-(−) predominate, with some evidence for the formation of larger assemblies; e.g. octameric A-(+) : A-(−) complexes.
Co-reporter:Debasis Das;Dr. Bekir E. Eser; Jaehong Han;Aaron Sciore; E. Neil G. Marsh
Angewandte Chemie 2011 Volume 123( Issue 31) pp:7286-7290
Publication Date(Web):
DOI:10.1002/ange.201101552
Co-reporter:Debasis Das;Dr. Bekir E. Eser; Jaehong Han;Aaron Sciore; E. Neil G. Marsh
Angewandte Chemie International Edition 2011 Volume 50( Issue 31) pp:7148-7152
Publication Date(Web):
DOI:10.1002/anie.201101552
Co-reporter:Miri Yoon, Hangtian Song, Kristina Håkansson and E. Neil G. Marsh
Biochemistry 2010 Volume 49(Issue 14) pp:
Publication Date(Web):March 12, 2010
DOI:10.1021/bi1001695
Hydrogen atom transfer reactions between the substrate and coenzyme are key mechanistic features of all adenosylcobalamin-dependent enzymes. For one of these enzymes, glutamate mutase, we have investigated whether hydrogen tunneling makes a significant contribution to the mechanism by examining the temperature dependence of the deuterium kinetic isotope effect associated with the transfer of a hydrogen atom from methylaspartate to the coenzyme. To do this, we designed a novel intramolecular competition experiment that allowed us to measure the intrinsic kinetic isotope effect, even though hydrogen transfer may not be rate-determining. From the Arrhenius plot of the kinetic isotope effect, the ratio of the pre-exponential factors (AH/AD) was 0.17 ± 0.04 and the isotope effect on the activation energy [ΔEa(D−H)] was 1.94 ± 0.13 kcal/mol. The results imply that a significant degree of hydrogen tunneling occurs in glutamate mutase, even though the intrinsic kinetic isotope effects are well within the semiclassical limit and are much smaller than those measured for other AdoCbl enzymes and model reactions for which hydrogen tunneling has been implicated.
Co-reporter:Benjamin C. Buer, Jeetender Chugh, Hashim M. Al-Hashimi and E. Neil G. Marsh
Biochemistry 2010 Volume 49(Issue 27) pp:
Publication Date(Web):June 8, 2010
DOI:10.1021/bi100605e
A variety of biologically active peptides exert their function through direct interactions with the lipid membrane of the cell. These surface interactions are generally transient and highly dynamic, making them hard to study. Here we have examined the feasibility of using solution phase 19F nuclear magnetic resonance (NMR) to study peptide−membrane interactions. Using the antimicrobial peptide MSI-78 as a model system, we demonstrate that peptide binding to either small unilamellar vesicles (SUVs) or bicelles can readily be detected by simple one-dimensional 19F NMR experiments with peptides labeled with l-4,4,4-trifluoroethylglycine. The 19F chemical shift associated with the peptide−membrane complex is sensitive both to the position of the trifluoromethyl reporter group (whether in the hydrophobic face or positively charged face of the amphipathic peptide) and to the curvature of the lipid bilayer (whether the peptide is bound to SUVs or bicelles). 19F spin echo experiments using the Carr−Purcell−Meiboom−Gill pulse sequence were used to measure the transverse relaxation (T2) of the nucleus and thereby examine the local mobility of the MSI-78 analogues bound to bicelles. The fluorine probe positioned in the hydrophobic face of the peptide relaxes at a rate that correlates with the tumbling of the bicelle, suggesting that it is relatively immobile, whereas the probe at the positively charged face relaxes more slowly, indicating this position is much more dynamic. These results are in accord with structural models of MSI-78 bound to lipids and point to the feasibility of using fluorine-labeled peptides to monitor peptide−membrane interactions in living cells.
Co-reporter:E. Neil G. Marsh ;Dustin P. Patterson;Lei Li
ChemBioChem 2010 Volume 11( Issue 5) pp:604-621
Publication Date(Web):
DOI:10.1002/cbic.200900777
Abstract
Adenosine is undoubtedly an ancient biological molecule that is a component of many enzyme cofactors: ATP, FADH, NAD(P)H, and coenzyme A, to name but a few, and, of course, of RNA. Here we present an overview of the role of adenosine in its most reactive form: as an organic radical formed either by homolytic cleavage of adenosylcobalamin (coenzyme B12, AdoCbl) or by single-electron reduction of S-adenosylmethionine (AdoMet) complexed to an iron–sulfur cluster. Although many of the enzymes we discuss are newly discovered, adenosine's role as a radical cofactor most likely arose very early in evolution, before the advent of photosynthesis and the production of molecular oxygen, which rapidly inactivates many radical enzymes. AdoCbl-dependent enzymes appear to be confined to a rather narrow repertoire of rearrangement reactions involving 1,2-hydrogen atom migrations; nevertheless, mechanistic insights gained from studying these enzymes have proved extremely valuable in understanding how enzymes generate and control highly reactive free radical intermediates. In contrast, there has been a recent explosion in the number of radical-AdoMet enzymes discovered that catalyze a remarkably wide range of chemically challenging reactions; here there is much still to learn about their mechanisms. Although all the radical-AdoMet enzymes so far characterized come from anaerobically growing microbes and are very oxygen sensitive, there is tantalizing evidence that some of these enzymes might be active in aerobic organisms including humans.
Co-reporter:Lei Li, Dustin P. Patterson, Christel C. Fox, Brian Lin, Peter W. Coschigano and E. Neil G. Marsh
Biochemistry 2009 Volume 48(Issue 6) pp:
Publication Date(Web):January 21, 2009
DOI:10.1021/bi801766g
Benzylsuccinate synthase is a member of the glycyl radical family of enzymes. It catalyzes the addition of toluene to fumarate to form benzylsuccinate as the first step in the anaerobic pathway of toluene fermentation. The enzyme comprises three subunits, α, β, and γ, that in Thauera aromatica strain T1 are encoded by the tutD, tutG, and tutF genes, respectively. The large α-subunit contains the essential glycine and cysteine residues that are conserved in all glycyl radical enzymes. However, the function of the small β- and γ-subunits has remained unclear. We have overexpressed all three subunits of benzylsuccinate synthase in Escherichia coli, both individually and in combination. Coexpression of the γ-subunit (but not the β-subunit) is essential for efficient expression of the α-subunit. The benzylsuccinate synthase complex lacking the glycyl radical could be purified as an α2β2γ2 hexamer by nickel affinity chromatography through a “His6” affinity tag engineered onto the C-terminus of the α-subunit. Unexpectedly, BSS was found to contain two iron−sulfur clusters, one associated with the β-subunit and the other with the γ-subunit that appear to be necessary for the structural integrity of the complex. The spectroscopic properties of these clusters suggest that they are most likely [4Fe-4S] clusters. Removal of iron with chelating agents results in dissociation of the complex; similarly, a mutant γ-subunit lacking the [4Fe-4S] cluster is unable to stabilize the α-subunit when the proteins are coexpressed.
Co-reporter:Benjamin C. Buer, Roberto de la Salud-Bea, Hashim M. Al Hashimi and E. Neil G. Marsh
Biochemistry 2009 Volume 48(Issue 45) pp:
Publication Date(Web):October 13, 2009
DOI:10.1021/bi901481k
The incorporation of extensively fluorinated, or fluorous, analogues of hydrophobic amino acids into proteins potentially provides the opportunity to modulate the physicochemical properties of proteins in a predictable manner. On the basis of the properties of small fluorocarbon molecules, extensively fluorinated proteins should be both more thermodynamically stable and self-segregate through “fluorous” interactions between fluorinated amino acids. We have examined the effects of introducing the fluorous leucine analogue l-5,5,5,5′,5′,5′,-hexafluoroleucine (hFLeu) at various positions within the hydrophobic core of a de novo-designed four-α-helix bundle protein, α4. The stabilizing effect of hFLeu is strongly dependent on the positions at which it is incorporated, with per-residue ΔΔGo(fold) ranging from −0.09 to −0.8 kcal mol−1 residue−1. In particular, incorporating hFLeu at all the “a” positions or all the “d” positions of the hydrophobic core, thereby creating an alternating packing arrangement of leucine and hFLeu, leads to very stably folded proteins that exhibit higher per-residue ΔΔGo(fold) values than proteins that are packed entirely with hFleu. We conclude that efficient packing of the fluorous amino acid within the hydrophobic core provides a more important contribution to enhancing protein stability than do fluorocarbon−fluorocarbon interactions between fluorinated protein side chains.
Co-reporter:E. Neil G. Marsh, Benjamin C. Buer and Ayyalusamy Ramamoorthy
Molecular BioSystems 2009 vol. 5(Issue 10) pp:1143-1147
Publication Date(Web):28 Jul 2009
DOI:10.1039/B909864J
Antimicrobial peptides (also known as genetically encoded peptide antibiotics) are a diverse class of short cationic amphipathic polypeptides that exhibit a broad-spectrum of antimicrobial activities by selectively disrupting the bacterial cell membrane. In this review article, we present the use of fluorinated amino acids in the design of antimicrobial peptides and other membrane-active peptides.
Co-reporter:Hyang-Yeol Lee, Jeffery W. Kampf, Kyo Sung Park and E. Neil G. Marsh
Crystal Growth & Design 2008 Volume 8(Issue 1) pp:296
Publication Date(Web):November 20, 2007
DOI:10.1021/cg700724h
Peptides are attractive ligands for the design of metal organic frameworks, with the potential to confer chirality and biological activity on these materials. However, very few such materials have been reported. Here we describe the X-ray structures of four extended molecular framework compounds formed by the complexation of di- and tripeptides with cadmium ions. The tripeptide complex of cadmium with glycine, Cd(Gly3)2·H2O, forms a two-dimensional complex in which the carboxylate group of the peptide bridges between Cd ions. In contrast, the tripeptide complex of cadmium with alanine, Cd(l-Ala3)2, forms a one-dimensional extended molecular chain comprised of an infinite series of rings linked together through the Cd ions. The dipeptide complexes Cd(l-Ala2)2 and Cd(l-Ala,l-Thr)2·4H2O form covalently linked two-dimensional square lattices. Hydrogen bonding between peptide amide groups and hydrophobic interactions between side chains are seen to play important roles in defining these extended structures. Most interestingly, substitution of the more hydrophilic threonine side chain in place of alanine introduces a layer of water molecules into the crystal lattice. These results suggest that it may be possible to engineer the properties of these extended networks through judicious choice of amino acid side chains.
Co-reporter:Lindsey M. Gottler, Roberto de la Salud-Bea and E. Neil G. Marsh
Biochemistry 2008 Volume 47(Issue 15) pp:
Publication Date(Web):March 25, 2008
DOI:10.1021/bi702476f
To test the prediction that extensively fluorinated (fluorous) proteins should be more stable and exhibit novel self-segregating behavior, the properties of the de novo designed model 4-α-helix bundle protein, α4F6, in which the hydrophobic core is packed entirely with the extensively fluorinated amino acid l-5,5,5,5′,5′,5′-hexafluoroleucine, have been compared with its nonfluorinated counterpart, α4H, in which the core is packed with leucine. α4F6 exhibits much greater resistance to proteolysis by either chymotrypsin or trypsin than α4H and resists unfolding by organic solvents far better than α4H. Whereas increasing concentrations of ethanol or 2-propanol cause the helices of the α4H tetramer first to dissociate into monomeric helices and then to completely unfold, these solvents have little effect on the structure of α4F6. In contrast, increasing the concentrations of the fluorinated alcohol trifluoroethanol promotes dissociation of both α4H and α4F6 to monomeric helices, whereas the secondary structure of both peptides remains intact. 19F NMR experiments indicate that the two peptides can form mixed α-helical α4F6:α4H bundles and thus do not exhibit the self-segregating behavior predicted by the fluorous effect. We conclude that the properties of α4F6 are best explained by the more hydrophobic nature of the hexafluoroleucine side chain, rather than the low solubility of fluorocarbons in hydrocarbon solvents that forms the basis of the fluorous effect.
Co-reporter:Lindsey M. Gottler, Roberto de la Salud Bea, Charles E. Shelburne, Ayyalusamy Ramamoorthy and E. Neil G. Marsh
Biochemistry 2008 Volume 47(Issue 35) pp:
Publication Date(Web):August 12, 2008
DOI:10.1021/bi801045n
Protegrins are potent members of the β-hairpin-forming class of antimicrobial peptides. Key to their antimicrobial activity is their assembly into oligomeric structures upon binding to the bacterial membrane. To examine the relationship between the physicochemical properties of the peptide and its biological activity, we have synthesized variants of protegrin-1 in which key residues in the hydrophobic core, valine-14 and -16, are changed to leucine and to the extensively fluorinated analogue hexafluoroleucine. These substitutions have the effect of making the peptide progressively more hydrophobic while minimally perturbing the secondary structure. The leucine-containing peptide was significantly more active than wild-type protegrin against several common pathogenic bacterial strains, whereas the hexafluoroleucine-substituted peptide, in contrast, showed significantly diminished activity against several bacterial strains. Isothermal titration calorimetry measurements revealed significant changes in the interaction of the peptides binding to small unilamelar vesicles that mimic the lipid composition of the bacterial membrane. The binding isotherms for wild-type and leucine-substituted protegrins indicate that electrostatic interactions dominate the membrane−peptide interaction, whereas the isotherm for the hexafluoroleucine-substituted protegrin suggests a diminished electrostatic component to binding. Notably both of these substitutions appear to alter the stoichiometry of the lipid−peptide interaction, suggesting that these substitutions may stabilize oligomerized forms of protegrin that are postulated to be intermediates in the assembly of the β-barrel membrane pore structure.
Co-reporter:Lindsey M. Gottler;Hyang-Yeol Lee;Charles E. Shelburne;Ayyalusamy Ramamoorthy Dr.
ChemBioChem 2008 Volume 9( Issue 3) pp:370-373
Publication Date(Web):
DOI:10.1002/cbic.200700643
Co-reporter:Miri Yoon;Anastasia Kalli;Hyang-Yeol Lee;Kristina Håkansson ;E. Neil G. Marsh
Angewandte Chemie 2007 Volume 119(Issue 44) pp:
Publication Date(Web):1 OCT 2007
DOI:10.1002/ange.200702448
Die intramolekulare Konkurrenz zwischen Wasserstoff- und Deuteriumatomen in der Methylgruppe von Methylaspartat wurde genutzt, um den primären Deuteriumisotopeneffekt für die Bildung eines 5′-Desoxyadenosins in einem B12-Enzym zu messen. Der Wert ist sehr viel kleiner als aus Studien anderer B12-Enzyme und Modellsysteme erwartet wurde, woraus geschlossen wird, dass Glutamatmutase den Übergangszustand des Wasserstofftransfers moduliert.
Co-reporter:Miri Yoon;Anastasia Kalli;Hyang-Yeol Lee;Kristina Håkansson ;E. Neil G. Marsh
Angewandte Chemie International Edition 2007 Volume 46(Issue 44) pp:
Publication Date(Web):1 OCT 2007
DOI:10.1002/anie.200702448
Intramolecular competition between hydrogen and deuterium atoms at the methyl group of methylaspartate was used to measure the intrinsic primary deuterium isotope effect for 5′-deoxyadenosine formation in a B12 enzyme. The value is much smaller than expected based on measurements on other B12 enzymes and model systems. This strongly suggests that glutamate mutase modulates the transition state for hydrogen transfer.
Co-reporter:E. Neil G. Marsh;William F. DeGrado
PNAS 2002 Volume 99 (Issue 8 ) pp:5150-5154
Publication Date(Web):2002-04-16
DOI:10.1073/pnas.052023199
Diiron and dimanganese proteins catalyze a wide range of hydrolytic and oxygen-dependent reactions. To probe the mechanisms
by which individual members of this class of proteins are able to catalyze such a wide range of reactions, we have prepared
a model four-helix bundle with a diiron site located near the center of the bundle. The four-helix bundle is constructed by
the noncovalent self-assembly of three different chains (Aa, Ab, and B) that self-assemble into the desired heterotetramer when mixed in a 1:1:2 molar ratio. On addition of ferrous ions
and oxygen, the protein forms a complex with a UV-visible spectrum closely resembling that of peroxo-bridged diferric species
in natural proteins and model compounds. By combining a small collection of n variants of these peptides, it should now be possible to prepare an n3 member library, which will allow systematic exploration of the features giving rise to the catalytic properties of this class
of proteins.
Co-reporter:E.Neil G Marsh, Catherine L Drennan
Current Opinion in Chemical Biology 2001 Volume 5(Issue 5) pp:499-505
Publication Date(Web):1 October 2001
DOI:10.1016/S1367-5931(00)00238-6
Adenosylcobalamin-dependent isomerases catalyze a variety of chemically difficult 1,2-rearrangements that proceed through a mechanism involving free radical intermediates. These radicals are initially generated by homolysis of the cobalt–carbon bond of the coenzyme. Recently, the crystal structures of several of these enzymes have been solved, revealing two modes of coenzyme binding and highlighting the role of the protein in controlling the rearrangement of reactive substrate radical intermediates. Complementary data from kinetic, spectroscopic and theoretical studies have produced insights into the mechanism by which substrate radicals are generated at the active site, and the pathways by which they rearrange.
Co-reporter:Anjali Patwardhan, E. Neil G. Marsh
Archives of Biochemistry and Biophysics (15 May 2007) Volume 461(Issue 2) pp:194-199
Publication Date(Web):15 May 2007
DOI:10.1016/j.abb.2007.01.010
.jpg)