Co-reporter:Zhiqiang Liu, Yueying Chu, Xiaomin Tang, Ling Huang, Guangchao Li, Xianfeng Yi, and Anmin Zheng
The Journal of Physical Chemistry C October 19, 2017 Volume 121(Issue 41) pp:22872-22872
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.jpcc.7b07374
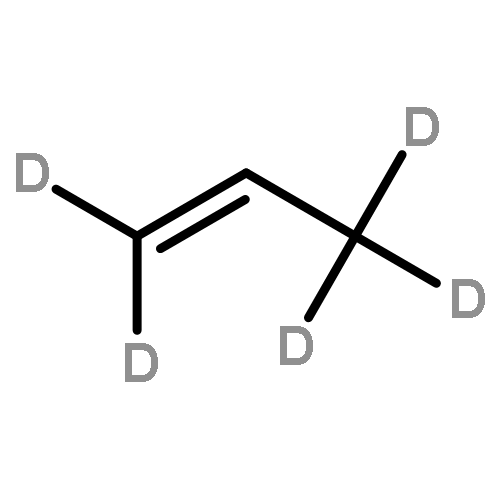
The “dual-cycle” pathway (i.e., olefin-based cycle and aromatic-based cycle) of methanol-to-olefin (MTO) has been generally accepted as hydrocarbon pool mechanism. Understanding the role of diffusion of reactant, intermediate, and product in the MTO process is essential in revealing its reaction mechanism. By using molecular dynamics (MD) simulations for two one-dimensional zeolites (ZSM-12 and ZSM-22) with a channel difference being only 0.3 Å in pore size, the diffusion behaviors of some representative species following “dual-cycle” mechanism (e.g., methanol, polymethylbenzenes, and olefins molecules) have been theoretically investigated in this work. It was found that the diffusion coefficients of methanol and olefins along ZSM-12 were ca. 2–3 times faster than that along ZSM-22 at 673 K. In the aromatic-based cycle, the polymethylbenzenes are crucial intermediates during the MTO reaction. 1,2,3,5-Tetramethylbenzene is almost imprisoned inside ZSM-12; such slower diffusion of tetramethylbenzene offers more opportunities for the geminal methylation reaction to form MTO activated pentamethylbenzenium cation, which would split into olefins through “paring” or “side-chain” pathways. However, in the ZSM-22 zeolite, since 1,2,4-trimethylbenzene is stacked, the following methylation reaction solely results in the formation of tetramethylbenzene, which is not an MTO activated species in ZSM-22 and more bulky polymethylbenzene further blocks the channel more seriously. When it comes to the olefin-based cycle, olefins can diffuse freely inside these two zeolites with methoxide intermediates bound to the zeolite frameworks, which thus facilitates formation of longer-chain olefin through olefin methylation reaction in these two zeolite catalysts. The combination of the higher reaction activity (from DFT calculation) and the longer contact time (from MD simulation) between the olefin and methoxide is apparently illustrated as the olefin-based cycle does more preferentially occur inside ZSM-22 than inside ZSM-12. Apparently, the MTO reaction mechanism is strongly determined by the diffusion behaviors of reaction species inside the zeolite confined pores.
Co-reporter:Anmin Zheng, Shang-Bin Liu, and Feng Deng
Chemical Reviews October 11, 2017 Volume 117(Issue 19) pp:12475-12475
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.chemrev.7b00289
Acid–base catalytic reaction, either in heterogeneous or homogeneous systems, is one of the most important chemical reactions that has provoked a wide variety of industrial catalytic processes for production of chemicals and petrochemicals over the past few decades. In view of the fact that the catalytic performances (e.g., activity, selectivity, and reaction mechanism) of acid-catalyzed reactions over acidic catalysts are mostly dictated by detailed acidic features, viz. type (Brønsted vs Lewis acidity), amount (concentration), strength, and local environments (location) of acid sites, information on and manipulation of their structure–activity correlation are crucial for optimization of catalytic performances as well as innovative design of novel effective catalysts. This review aims to summarize recent developments on acidity characterization of solid and liquid catalysts by means of experimental 31P nuclear magnetic resonance (NMR) spectroscopy using phosphorus probe molecules such as trialkylphosphine (TMP) and trialkylphosphine oxides (R3PO). In particular, correlations between the observed 31P chemical shifts (δ31P) of phosphorus (P)-containing probes and acidic strengths have been established in conjuction with density functional theory (DFT) calculations, rendering practical and reliable acidity scales for Brønsted and Lewis acidities at the atomic level. As illustrated for a variety of different solid and liquid acid systems, such as microporous zeolites, mesoporous molecular sieves, and metal oxides, the 31P NMR probe approaches were shown to provide important acid features of various catalysts, surpassing most conventional methods such as titration, pH measurement, Hammett acidity function, and some other commonly used physicochemical techniques, such as calorimetry, temperature-programmed desorption of ammonia (NH3-TPD), Fourier transformed infrared (FT-IR), and 1H NMR spectroscopies.
Co-reporter:Le Le Gong;Wan Ting Yao;Zhi Qiang Liu;An Min Zheng;Jian Qiang Li;Xue Feng Feng;Lu Fang Ma;Chang Sheng Yan;Ming Biao Luo;Feng Luo
Journal of Materials Chemistry A 2017 vol. 5(Issue 17) pp:7961-7967
Publication Date(Web):2017/05/03
DOI:10.1039/C7TA01388D
MOF materials, as new catalysts, show high catalytic activity and size-, regio-, and stereo-selectivity. However, artificially controlling their catalytic process by convenient external stimulus such as light is still unexploited. Such photocontrol over an organic reaction may enable switchable catalytic activities and/or selectivities, consequently producing desired products from a pool of building blocks according to the order and type of stimuli applied. In this study, we present a novel MOF catalyst, which not only offers ultrahigh photocontrol with the on/off ratio as high as 407, but also displays disparate photomodulation in reaction kinetics towards various aldehyde substrates in light of their sizes, thus creating the first example of MOFs showing photoswitchable catalysis. The origin, as unveiled by photoswitching adsorption experiments and density functional theory calculations, is due to photoswitching storage of guest molecules in the metal–organic framework (MOF).
Co-reporter:Cong Bin Fan;Zhi Qiang Liu;Le Le Gong;An Min Zheng;Le Zhang;Chang Sheng Yan;Hui Qiong Wu;Xue Feng Feng;Feng Luo
Chemical Communications 2017 vol. 53(Issue 4) pp:763-766
Publication Date(Web):2017/01/05
DOI:10.1039/C6CC08982H
The first MOF (metal–organic framework) built on both diarylethene and azobenzene photochromic units is reported here and displays distinct photoresponses for different guest molecules, thus creating an easy-to-use pathway to modulate the adsorption selectivity of MOF materials.
Co-reporter:Xianfeng Yi, Guangchao Li, Ling Huang, Yueying Chu, Zhiqiang Liu, Hongqiang Xia, Anmin Zheng, and Feng Deng
The Journal of Physical Chemistry C 2017 Volume 121(Issue 7) pp:
Publication Date(Web):January 27, 2017
DOI:10.1021/acs.jpcc.6b11518
Pyrrole has been employed as an NMR probe molecule to determine the base strength of solid catalytic materials; however, the quantitative correlation between the 1H chemical shift of adsorbed pyrrole and the intrinsic base strength of solid catalysts is still lacking in the previous work. Here, solid-state NMR experiments and density functional theory (DFT) calculations were employed to explore the adsorption structures and 1H chemical shifts of adsorbed pyrrole molecules over the zeolites with varied base strengths. Based on a generic 8T zeolite ((SiH3)3-Si-X-Si-O-(SiH3)3, X = O or NH), various calculated models with different Si–H bond lengths were constructed to represent the basic sites with varied strengths and used to predict the pyrrole adsorption structures as well as the 1H chemical shifts. The solid-state NMR experimental results demonstrated that a larger 1H chemical shift of adsorbed pyrrole corresponds to a stronger basic site on solid catalysts. A linear correlation between the 1H chemical shift of adsorbed pyrrole and the proton affinity (PA) value of solid bases was theoretically derived, which is independent of the basic central atoms (e.g., O or N). In combination with the available 1H MAS NMR experimental data, it is conclusive that pyrrole could be used as a probe to quantitatively characterize the intrinsic basicity of various solid catalysts, and the 1H chemical shift threshold for superbasicity is 10.0 ppm. In addition, the influence of confinement effect on the adsorption structures and 1H chemical shifts of pyrrole over basic catalysts was investigated as well.
Co-reporter:Chao Zhang;Zengtian Cheng;Zaihui Fu;Yachun Liu;Xianfeng Yi
Cellulose 2017 Volume 24( Issue 1) pp:95-106
Publication Date(Web):2017 January
DOI:10.1007/s10570-016-1118-4
This article first discloses that the fluorine anion-containing ionic liquids-functionalized biochar sulfonic acids (BCSA-IL-F1–3s), which were simply synthesized by an ionic exchange of 1-trimethoxysilylpropyl-3-methylimidazolium chloride (IL-Cl) grafted on the BCSA with CF3SO3H (HF1), HBF4 (HF2), HPF6 (HF3), respectively, can efficiently catalyze cellulose hydrolysis into reducing sugars (RSs) and 5-hydroxymethyl furfural (HMF) in water under microwave irradiation. This process provides a very high catalysis efficiency (turnover numbers, 4.03–4.89) at mild temperature (80 °C) for 3 h, but also possesses an excellent repeatability. More outstandingly, they can achieve much higher HMF yields (12.70–27.94%) compared to the IL-Cl-functionalized BCSA catalyst (HMF yields are lower than 0.1%) under the same reaction conditions. This is likely because the introduction of IL-F1–3s groups can significantly improve the accessibility, acidity and thermal stability of BCSA’s SO3H sites, as supported by evidence from a solid 31P NMR spectrum and thermogravimetric analysis. It is proposed that the good selectivity for HMF perhaps originates from a co-catalysis action of the IL-F1–3s and SO3H groups on BCSA-IL-F1–3s in the further conversion of RSs to HMF.
Co-reporter: Dr. Cong Bin Fan;Le Le Gong;Dr. Ling Huang; Dr. Feng Luo; Dr. Rajamani Krishna;Xian Feng Yi; Dr. An Min Zheng;Le Zhang;Shou Zhi Pu;Xue Feng Feng;Ming Biao Luo; Dr. Guo Cong Guo
Angewandte Chemie 2017 Volume 129(Issue 27) pp:8127-8127
Publication Date(Web):2017/06/26
DOI:10.1002/ange.201704765
Selektive Adsorption und Trennung durch photochrome Metall-organische Gerüste (MOFs) lässt sich bisher nur selten allein durch externe Reize einstellen. In der Zuschrift auf S. 8008 ff. setzen F. Luo, G.-C. Guo et al. eine photochrome Diarylethen-Einheit zur lichtresponsiven Steuerung von Selektivität und Trennverhalten ein. Dadurch erhalten sie eine hoch selektive Adsorption, zum Beispiel im System C2H2/C2H4.
Co-reporter:Yueying Chu;Guangchao Li;Ling Huang;Xianfeng Yi;Hongqiang Xia;Feng Deng
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 12) pp:2512-2523
Publication Date(Web):2017/06/20
DOI:10.1039/C7CY00377C
Zeolites are effective catalysts for amide formation from oxime through the Beckmann rearrangement (BR) reaction; however, debates about which surface (i.e. external or internal) is more effective for the BR reaction over zeolites still remain. In this contribution, the effective rate constants (keff) are used to quantitatively evaluate the dependence of BR reactivity on the Brønsted acid location in H-ZSM-5 zeolite. Based on our theoretical calculations, it was found that in addition to the dimension size of oxime reactants and reaction temperature, the BR reaction is strongly dependent on the location of Brønsted acid sites. For the cyclohexanone oxime rearrangement, the reaction exclusively occurs on the internal surface of ZSM-5 zeolite at room temperature, while the active sites are those located at the pore mouth or on the external surface when the reaction temperature increases to 598 K. In contrast to cyclohexanone oxime, the Brønsted acid sites on the internal surface are kinetically more effective at room temperature or 598 K for the smaller acetoxime BR reaction.
Co-reporter: Dr. Cong Bin Fan;Le Le Gong;Dr. Ling Huang; Dr. Feng Luo; Dr. Rajamani Krishna;Xian Feng Yi; Dr. An Min Zheng;Le Zhang;Shou Zhi Pu;Xue Feng Feng;Ming Biao Luo; Dr. Guo Cong Guo
Angewandte Chemie 2017 Volume 129(Issue 27) pp:8008-8014
Publication Date(Web):2017/06/26
DOI:10.1002/ange.201702484
AbstractA dual temperature- and light-responsive C2H2/C2H4 separation switch in a diarylethene metal–organic framework (MOF) is presented. At 195 K and 100 kPa this MOF shows ultrahigh C2H2/C2H4 selectivity of 47.1, which is almost 21.4 times larger than the corresponding value of 2.2 at 293 K and 100 kPa, or 15.7 times larger than the value of 3.0 for the material under UV at 195 K and 100 kPa. The origin of this unique control in C2H2/C2H4 selectivity, as unveiled by density functional calculations, is due to a guest discriminatory gate-opening effect from the diarylethene unit.
Co-reporter: Dr. Cong Bin Fan;Le Le Gong;Dr. Ling Huang; Dr. Feng Luo; Dr. Rajamani Krishna;Xian Feng Yi; Dr. An Min Zheng;Le Zhang;Shou Zhi Pu;Xue Feng Feng;Ming Biao Luo; Dr. Guo Cong Guo
Angewandte Chemie International Edition 2017 Volume 56(Issue 27) pp:8015-8015
Publication Date(Web):2017/06/26
DOI:10.1002/anie.201704765
Adjusting adsorption selectivity and separation in photochromic metal–organic frameworks (MOFs) just by external stimuli is highly important but still rare. In their Communication on page 7900 ff., F. Luo, G.-C. Guo, and co-workers employ a photochromic diarylethene unit as a light-triggered selectivity and separation regulator, leading to ultrahigh adsorption selectivity, for example, for the mixture C2H2/C2H4.
Co-reporter: Dr. Cong Bin Fan;Le Le Gong;Dr. Ling Huang; Dr. Feng Luo; Dr. Rajamani Krishna;Xian Feng Yi; Dr. An Min Zheng;Le Zhang;Shou Zhi Pu;Xue Feng Feng;Ming Biao Luo; Dr. Guo Cong Guo
Angewandte Chemie International Edition 2017 Volume 56(Issue 27) pp:7900-7906
Publication Date(Web):2017/06/26
DOI:10.1002/anie.201702484
AbstractA dual temperature- and light-responsive C2H2/C2H4 separation switch in a diarylethene metal–organic framework (MOF) is presented. At 195 K and 100 kPa this MOF shows ultrahigh C2H2/C2H4 selectivity of 47.1, which is almost 21.4 times larger than the corresponding value of 2.2 at 293 K and 100 kPa, or 15.7 times larger than the value of 3.0 for the material under UV at 195 K and 100 kPa. The origin of this unique control in C2H2/C2H4 selectivity, as unveiled by density functional calculations, is due to a guest discriminatory gate-opening effect from the diarylethene unit.
Co-reporter:Le Le Gong, Xue Feng Feng, Feng Luo, Xian Feng Yi and An Min Zheng
Green Chemistry 2016 vol. 18(Issue 7) pp:2047-2055
Publication Date(Web):13 Nov 2015
DOI:10.1039/C5GC02182K
Herein, we report a facile and green approach to remove, convert and even release highly toxic allyl alcohol inside a MOF. The MOF used here, namely ECIT-20, barely adsorbs allyl alcohol under ambient conditions, whereas a sharp increase up to 90 mg g−1 (1.03 mol mol−1) under UV (297 nm) irradiation is observed, giving an ultra-big photo-switching behavior of more than 19 times. Thermogravimetric analysis, infrared spectroscopy, and 1H MAS plus 13C CPMAS solid-state NMR spectroscopy showed that a host–guest photochemical [2 + 2] reaction followed by the formation of asymmetric cyclobutanes is responsible for this unique photo-switching behavior towards allyl alcohol. The final conversion capability by means of ECIT-20 from allyl alcohol to asymmetric cyclobutanes is estimated to be 0.5 mol mol−1, which agrees well with both the structural features and the results from density functional theory (DFT) calculations. Moreover, the reuse of this MOF material is also facile and even after four cycles, excellent photo-switching performance towards allyl alcohol could be well maintained. This indicates that the highly promising material of ECIT-20 is suitable for the green removal and safe reuse of highly toxic allyl alcohol.
Co-reporter:Bing Ma, Xianfeng Yi, Li Chen, Anmin Zheng and Chen Zhao
Journal of Materials Chemistry A 2016 vol. 4(Issue 29) pp:11330-11341
Publication Date(Web):17 Jun 2016
DOI:10.1039/C6TA01807F
Hierarchical H-style ultra-stable Y (HUSY) zeolites with abundant interconnected mesopores have been prepared using a sequential post-synthesis strategy that includes steaming dealumination and mixed-alkali desilication. The steaming treatment generates a broad size range of intra-mesopores (around 25 and 45 nm) and a moderate Si/Al ratio of 13.4 in the HUSY, which provides optimal material precursors for the ensuing subsequent alkaline desilication. N2 adsorption–desorption isotherms and X-ray diffractometry results indicate that the sample treated with pyridine/sodium hydroxide (HUSY-4) has a larger external surface area and a higher relative crystallinity. Infrared spectra of adsorbed pyridine show that HUSY-4 contains substantial Brønsted acid sites. The 27Al and 29Si nuclear magnetic resonance spectra show that HUSY-4 possesses few extra-framework alumina species. Infrared spectra in a vacuum show that the peak intensities of HUSY-4 in the bridged hydroxyl group (at 3560 and 3631 cm−1) are much stronger than those of the sample treated with tetrapropylammonium hydroxide (HUSY-3), indicating that the framework integrity of HUSY-4 is better. Differences in treatments with tetrapropylammonium hydroxide/sodium hydroxide and pyridine/sodium hydroxide treatments are attributed to the fact that the pyridine molecule (0.54 nm) can pass through the supercages (0.74 nm) to protect the zeolite framework from deep desilication, whereas the tetrapropylammonium hydroxide molecule (0.85 nm) is adsorbed only on the external surface. Eventually, a HUSY zeolite with a high external surface area, inter-connectedness and hierarchical mesopores (10, 25, and 45 nm) is prepared by initial high-temperature steaming, which is followed by desilication using a mixed alkali solution containing pyridine and sodium hydroxide. High-dispersion (5.5%), high-content (35 wt%), small Ni nanoparticles (4.9 ± 1.2 nm) are loaded onto and into the external surface areas and interpores of the hierarchical HUSY by the deposition–precipitation method. The resultant Ni/HUSY-4 shows an ultra-high efficiency in the hydrodeoxygenation of fatty acids, esters, and palm oil, and achieves high initial rates (60 g g−1 h−1) and a high C18 alkane selectivity (96%), which may be attributed to the enhanced Brønsted acid and adjacent Lewis acid (confirmed by the 1H DQ MAS NMR spectrum) together with the substantial dispersive Ni nanoparticles loaded onto/into the interconnected pores of the hierarchical HUSY support.
Co-reporter:Mengdi Huang, Qiang Wang, Xianfeng Yi, Yueying Chu, Weili Dai, Landong Li, Anmin Zheng and Feng Deng
Chemical Communications 2016 vol. 52(Issue 70) pp:10606-10608
Publication Date(Web):05 Jul 2016
DOI:10.1039/C6CC04943E
Solid-state NMR experiments and DFT calculations have been carried out to determine the complex structures of coadsorbed 13C-labeled tert-butanol and NH3 in acidic H-ZSM-5 zeolite. It is found, besides the physically adsorbed tert-butanol/NH4+ complex on Brønsted acid sites, the tert-butylamine cation is formed as well, confirming the presence of the tert-butyl cation confined in zeolite channels. Furthermore, 13C–27Al double-resonance solid-state NMR spectroscopy is adopted to determine the host/guest interaction between the carbocation and the zeolite framework.
Co-reporter:Yueying Chu, Nianhua Xue, Bolian Xu, Qian Ding, Zhaochi Feng, Anmin Zheng and Feng Deng
Catalysis Science & Technology 2016 vol. 6(Issue 14) pp:5350-5363
Publication Date(Web):05 Apr 2016
DOI:10.1039/C6CY00467A
Theoretical calculations have provided fundamental insights into the possible pathways for the H/D exchange of isobutane with H-ZSM-5 zeolite at room temperature. It is theoretically demonstrated that neither the direct exchange mechanism nor the indirect bimolecular hydride transfer mechanism is an efficient route for isobutane activation due to high activation barriers, which possibly prohibit H/D exchange at low temperatures. It is revealed that a trace of olefin impurities can considerably accelerate the formation of an alkoxyl intermediate which is involved in the bimolecular hydride transfer mechanism. Once the alkoxyl intermediate is generated from the olefin impurities, the catalytic cycle is self-sustaining. On the other hand, the theoretical calculations also illustrate that the extra-framework aluminum (EFAl) species in the dealuminated zeolite has no obvious promotional effect on the H/D exchange. Furthermore, our calculations are also consistent with the experimental results.
Co-reporter:Xianfeng Yi, Lihong Ding, Guangchao Li, Zhiqiang Liu, Hongqiang Xia, Yueying Chu, Anmin Zheng and Feng Deng
Catalysis Science & Technology 2016 vol. 6(Issue 16) pp:6328-6338
Publication Date(Web):24 May 2016
DOI:10.1039/C6CY00757K
A comprehensive understanding of the reaction mechanisms of hydrocarbon conversion over acidic zeolite catalysts would be of great importance to optimize, modify and design more efficient catalytic materials. For this purpose, theoretical calculations based on molecular dynamics (MD) simulations and density functional theory (DFT) calculations have been performed in this work to explore the reaction pathways of propene H/D exchange over deuterated acidic ZSM-5 zeolite (D-ZSM-5). The deuterated propene-D5 is confirmed to be readily formed through the route involving an isopropyl intermediate. With regard to the formation of completely deuterated propene-D6, the propene loading is found to play a crucial role in governing the reaction pathway. The dimerization route (through the dimerization of propene, the intramolecular hydride transfer and then the cracking process) is demonstrated to be predominant with a relatively lower activation energy barrier (12.3 kcal mol−1) at higher propene loading, while the n-propoxy pathway is preferred at lower propene loading. Furthermore, the influences of the acid strength and pore confinement effect of zeolite on the propene H/D exchange reaction activity have been derived as well.
Co-reporter:Fujian Liu, Bojie Li, Chen Liu, Weiping Kong, Xianfeng Yi, Anmin Zheng and Chenze Qi
Catalysis Science & Technology 2016 vol. 6(Issue 9) pp:2995-3007
Publication Date(Web):19 Nov 2015
DOI:10.1039/C5CY01226K
N rich porous carbon based solid acids (NPC-[CxN][X]) have been successfully synthesized by treatment of N rich porous carbon (NPC) with various quaternary ammoniation reagents such as iodomethane, 1,3-propane sultone, and 1,4-butanesultone, and ion exchange with various strong acids such as HSO3CF3, H2SO4, H3PW12O40, HBF4etc. The NPC support was synthesized by carbonization of KOH-activated polypyrrole without using additional templates. Various characterizations showed that NPC-[CxN][X] possesses abundant nanopores, large Brunauer–Emmett–Teller surface areas, good stability, and strong and controllable acid sites with Brønsted characteristics. The immobilized acidic groups were homogeneously dispersed into NPC-[CxN][X]. Notably, NPC-[CxN][X] acted as efficient, reusable and generalized solid acids, which showed excellent activity in various acid-catalyzed reactions such as esterification and transesterification in the synthesis of biodiesel, dehydration of fructose into 5-hydroxymethylfurfural, depolymerization of crystalline cellulose into sugars, and condensation of phenol with acetone in the synthesis of bisphenol A, much higher than that of various solid acids such as Amberlyst 15, H-ZSM-5, H-USY, and sulfonic group functionalized ordered mesoporous silicas. The preparation of NPC-[CxN][X] leads to the development of porous carbon based solid acids with controllable structural characteristics and excellent catalytic activity.
Co-reporter:Wenna Zhang, Yueying Chu, Yingxu Wei, Xianfeng Yi, Shutao Xu, Jindou Huang, Mozhi Zhang, Anmin Zheng, Feng Deng, Zhongmin Liu
Microporous and Mesoporous Materials 2016 Volume 231() pp:216-229
Publication Date(Web):1 September 2016
DOI:10.1016/j.micromeso.2016.05.029
•The reactions involved in the alkenes-based cycle in MTO reaction were discussed in-depth by theoretical calculations.•Confinement effect from the zeolite framework can strongly stabilize the transition states by the vdW interactions.•Acid strength can effectively enhance the catalytic activities by decreasing the reaction barriers.•The reactions related to ethene formation and transform need overcome higher barriers in the alkenes-based cycle.Methanol-to-Olefins (MTO) conversion over acidic zeolite catalysts has become the most important non-petrochemical route for the production of light olefins. The ‘dual-cycle’ mechanism (i.e., alkenes-based cycle and aromatics-based cycle) over H-ZSM-5 zeolite has been generally accepted for olefins generation from methanol conversion. However, the relationship between the catalytic performance and the confinement effect/acid strength of the catalyst is still unclear. Herein, the methylation, isomerization and cracking processes involved in the alkenes-based cycle are discussed in-depth by density functional theory (DFT) calculations. The calculation results predicted that the transition states can be considerably stabilized by the van der Waals (vdW) interactions from the zeolite framework, resulting in the reduction of the activation barriers. And acid strength can also enhance the reaction activities. However, the catalytic reactivity of all elementary steps in the alkenes-based cycle can be improved at a different degree with increasing the acid strength. In addition, the ethene formation, transformation and the precursor of ethene formation need higher energy. And increasing acid strength can sharply decrease the activation barriers of ethene formation of cracking reaction, indicating that ethene formation may need strong acid strength.The methylation, isomerization and cracking processes involved in the alkenes-based cycle of Methanol-to-Olefins conversion (MTO) on H-ZSM-5 are discussed in-depth by density functional theory calculations. The transition states can be considerably strongly stabilized by the van der Waals (vdW) interactions from the zeolite framework, and acid strength can also effectively enhance the reaction activities. In addition, the ethene formation, transform and the precursor of ethene formation need higher energy.
Co-reporter:Kangming Xiong, Fangjun Huo, Caixia Yin, Yueying Chu, Yutao Yang, Jianbin Chao, Anmin Zheng
Sensors and Actuators B: Chemical 2016 Volume 224() pp:307-314
Publication Date(Web):1 March 2016
DOI:10.1016/j.snb.2015.10.047
•We have developed two new colorimetric and fluorescent probes for hypochlorite based on a novel recognition mechanism: amido oxidized nitroso-group by hypochlorites.•The optical properties of the probe 2 and its ClO−-addition product were studied based on DFT and TDDFT were explored at the m062x/6-31+G (d) level.•The probe 2 could be applied in practical applications such as detecting the hypochlorite concentration of sodium hypochlorite disinfectant and bioimagings.We have developed two new colorimetric and fluorescent probes for hypochlorite based on a novel recognition mechanism: amido oxidized nitroso-group by hypochlorites, which realized the preparation of nitroso compounds. Furthermore, 1H NMR, ESI-MS and theoretical calculation proved the recognition mechanism. In addition, the probe 2 was applied in practical applications such as detecting the hypochlorite concentration of sodium hypochlorite disinfectant and bioimagings.In this study, we have developed two new colorimetric and fluorescent probes for hypochlorite based on a novel recognition mechanism: amido oxidized nitroso-group by hypochlorites, which realized the preparation of nitroso compounds. Furthermore, 1H NMR, ESI-MS and TD-DFT calculations proved the novel recognition mechanism. In addition, the probe 2 was applied in practical applications such as detecting the hypochlorite concentration of sodium hypochlorite disinfectant and bioimagings.
Co-reporter:Benteng Song, Yueying Chu, Guangchao Li, Jiqing Wang, An-Ya Lo, Anmin Zheng, and Feng Deng
The Journal of Physical Chemistry C 2016 Volume 120(Issue 48) pp:27349-27363
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.jpcc.6b09059
Our previous work demonstrated that hydrocarbon species can be stabilized in the confined zeolite in the form of an ion pair, π complex, and alkoxy species. Nevertheless, the interaction mechanism between the different reactants/intermediates and the zeolite framework remains undetermined, and thus, the origin of the zeolite confinement effect has not been thoroughly revealed. In this work, a recently developed energy decomposition analysis (EDA) method was applied to theoretically investigate the energy parameters of a series of hydrocarbon species confined in the zeolitic catalysts with different pore diameters. It is demonstrated that for the carbenium ion intermediates, the electrostatic interaction plays a key role in their stabilization; for the alkoxy species, both orbital and electrostatic interactions are the key factors, while for the neutral hydrocarbons, the dispersion interaction favors their stabilization. In addition, the principal components analysis (PCA) reveals that the dispersion interaction does not play a crucial role in improving the reaction activity due to the same extent of stabilization effect for different reaction species (e.g., reactant, transition state, intermediate, or product), and thus, the dispersion contribution would be counteracted in a specific zeolite catalytic reaction. In contrast, the difference in electrostatic interaction caused by the variations of charge characteristics of the various confined species considerably contributes to the decrease of the activation barrier and the increase of the reaction energy, which in turn largely promotes the catalytic performance of zeolite catalysts.
Co-reporter:Jun Xu, Victor V. Terskikh, Yueying Chu, Anmin Zheng, and Yining Huang
Chemistry of Materials 2015 Volume 27(Issue 9) pp:3306
Publication Date(Web):April 13, 2015
DOI:10.1021/acs.chemmater.5b00360
Metal–organic frameworks (MOFs) are important materials with many actual and potential applications. Crystal structure of many MOFs is determined by single-crystal X-ray diffraction. However, due to the inability of XRD to accurately locate hydrogen atoms, the local structures around framework hydrogen are usually poorly characterized even if the overall framework has been accurately determined. 1H solid-state NMR (SSNMR) spectroscopy should, in principle, be used as a complementary method to XRD for characterizing hydrogen local environments. However, the spectral resolution of 1H SSNMR is severely limited by the strong 1H–1H homonuclear dipolar coupling. In this work, we demonstrate that high-resolution 1H MAS spectra of MOF-based material can be obtained by ultrafast sample spinning at high magnetic field in combination with isotopic dilution. In particular, we examined an important MOF, microporous α-Mg3(HCOO)6 and α-Mg3(HCOO)6 in the presence of several guest species. All six chemically very similar, but crystallographically, nonequivalent H sites of these MOFs were resolved in a chemical shift range as small as 0.8 ppm. Although the assignment of 1H peaks due to crystallographically nonequivalent hydrogens is difficult due to that they all have almost identical chemical environments, we are able to show that they can be assigned from 1H–1H proximity maps obtained from 2D 1H–1H double quantum (DQ) experiments in conjunction with theoretical calculations. 1H MAS spectra of framework hydrogen are very sensitive to the guest molecules present inside the pores and they provide insight into host–guest interaction and dynamics of guest molecule. The ability of achieving very high resolution for 1H MAS NMR in MOF-based materials and subsequent spectral assignment demonstrated in this work allows one to obtain new structural information complementary to that obtained from single-crystal XRD.
Co-reporter:Liang Wang, Jian Zhang, Xianfeng Yi, Anmin Zheng, Feng Deng, Chunyu Chen, Yanyan Ji, Fujian Liu, Xiangju Meng, and Feng-Shou Xiao
ACS Catalysis 2015 Volume 5(Issue 5) pp:2727
Publication Date(Web):March 17, 2015
DOI:10.1021/acscatal.5b00083
Zeolite-based catalysts have been widely used in the conversion of biomass recently, but the catalytic yields of the desired products are strongly limited by the relatively small micropores of zeolite. Here, we reported a hierarchically porous ZSM-5 zeolite with micropore and b-axis-aligned mesopore-supported Ru nanoparticles (Ru/HZSM-5-OM) that are highly efficient for the hydrodeoxygenation of both small and bulky phenolic biomolecules to the corresponding alkanes. Compared with the conventional ZSM-5 zeolite-supported Ru catalyst, the high catalytic activities and alkane selectivities over Ru/HZSM-5-OM are attributed to the abundant exposed acidic sites in HZSM-5-OM with open mesopores. This feature is potentially important for future phenolic bio-oil upgrading.Keywords: biomass conversion; hydrodeoxygenation; mesoporous zeolite; phenolic biomolecules; Ru catalyst
Co-reporter:Yueying Chu, Peng Ji, Xianfeng Yi, Shenhui Li, Peng Wu, Anmin Zheng and Feng Deng
Catalysis Science & Technology 2015 vol. 5(Issue 7) pp:3675-3681
Publication Date(Web):18 May 2015
DOI:10.1039/C5CY00619H
The effects of Brønsted acid strength and pore confinement on the Beckmann rearrangement (BR) reaction over solid acid catalysts have been explored. With the help of catalytic evaluation experiments, it is demonstrated that oximes with different size (cyclohexanone oxime and acetoxime) exhibit quite different BR reactivity dependence on the acid strength over microporous and mesoporous zeolites. In order to reveal the origin of such a difference, electronic structure calculations and kinetic analysis were performed. It was theoretically found that the confinement effect from the microporous zeolite framework has a more significant influence on the rate-determining step of the BR reaction when the oxime reactant is well-confined inside the microporous voids, which in turn controls the BR reactivity.
Co-reporter:Yueying Chu, Xianyong Sun, Xianfeng Yi, Lihong Ding, Anmin Zheng and Feng Deng
Catalysis Science & Technology 2015 vol. 5(Issue 7) pp:3507-3517
Publication Date(Web):31 Mar 2015
DOI:10.1039/C5CY00312A
The methanol to olefins (MTO) process, in which low-value carbon-rich feedstocks are converted to value-added petrochemical products, is one of the most prominent alternatives for the production of light olefins. In order to reveal the confinement effect of zeolites on the catalytic reactions, the MTO mechanisms and reactivity over two unidimensional zeolites (H-ZSM-12 and H-ZSM-22) with a channel difference of only 0.3 Å have been systematically explored by DFT calculations in this work. The calculated activation barriers and reaction energies demonstrated that the 0.3 Å channel difference between H-ZSM-12 and H-ZSM-22 zeolites results in a dramatic discrepancy in their transition state selectivity associated with the aromatic-based hydrocarbon pool (HCP) mechanism. For the larger H-ZSM-12 zeolite, the formation of pentamethybenzenium cation was favored, which would be the active HCP species in the MTO reaction. For the H-ZSM-22 zeolite with a 0.3 Å smaller pore structure, the traditional methylation at the C–H sites of polymethylbenzenes occurred exclusively. When the alternative olefin-based cycle is followed for the MTO reaction, both of the zeolites are active catalysts for the formations of butene and propene. A comparison of the activation barriers for the olefin-based cycle revealed that the larger H-ZSM-12 possesses a higher catalytic activity than the H-ZSM-22 zeolite. Our theoretical results demonstrate that both the aromatic-based cycle and the olefin-based cycle can proceed during the MTO reaction over H-ZSM-12, with the latter cycle being predominant.
Co-reporter:Mei Xiang;Xiaojun Ni;Xianfeng Yi; Anmin Zheng;Wenchang Wang; Mingyang He;Jing Xiong;Taotao Liu;Yuli Ma;Pengyuan Zhu;Xiang Zheng;Dr. Tii Tang
ChemCatChem 2015 Volume 7( Issue 3) pp:521-525
Publication Date(Web):
DOI:10.1002/cctc.201402839
Abstract
Developing highly active heterogeneous catalysts for the efficient construction of valuable building blocks is of great importance to synthetic chemistry. For this purpose, a mesoporous zeolite ETS-10 (METS-10) is synthesized by using a mesoscale silane surfactant as a template and applied to achieve highly efficient syntheses of α,β-epoxy ketones by employing simple alkenes and aldehydes as starting materials. The high activity of the METS-10 catalyst is attributed to its unique porous structure and basicity. Electron paramagnetic resonance characterization results and theoretical calculation experimental data reveal that the strong basic sites on METS-10 catalyst can activate the reaction substrate and intermediate. In addition, the mesopores in METS-10 catalyst benefit the mass transfer and further improve the catalytic activity.
Co-reporter:Chin-Te Hung, Ningya Yu, Chia-Ting Chen, Pei-Hao Wu, Xiaoxiang Han, Yu-Siang Kao, Tuan-Chi Liu, Yueying Chu, Feng Deng, Anmin Zheng and Shang-Bin Liu
Journal of Materials Chemistry A 2014 vol. 2(Issue 47) pp:20030-20037
Publication Date(Web):19 Sep 2014
DOI:10.1039/C4TA04403G
A facile method was developed for the synthesis of metal-free, highly N-doped (>7 wt%) mesoscopic carbons (NMCs), which were fabricated by first preparing carbon–silicate (C–Si) composites by co-condensation method using a melamine-formaldehyde resin oligomer as the primary nitrogen and carbon source, and P123 triblock copolymer surfactant and sodium silicate as the soft and hard template, respectively, under microwave irradiation conditions, followed by carbonization and silica-template removal. The NMCs were found to exhibit superior electrocatalytic activity, long-term stability, and excellent tolerance over methanol crossover effect. Such NMCs derived from organic–inorganic hybrids assisted by microwave heating not only possess high surface areas and active quaternary and pyridinic-N species that are favourable for ORR, as verified by DFT calculations, but also render large-scale production and practical applications as cost-effective electrode materials.
Co-reporter:Anmin Zheng, Yueying Chu, Shenhui Li, Dangsheng Su, Feng Deng
Carbon 2014 Volume 77() pp:122-129
Publication Date(Web):October 2014
DOI:10.1016/j.carbon.2014.05.013
The activation and conversion of light alkanes (such as methane, ethane, propane and isobutane) has attracted more and more attention in both industrial and fundamental aspects. Compared to the traditional metal catalysts, carbon catalysts have environmental acceptability with inexhaustible resources. Considerable experimental work concerning the activation of light alkanes by surface-modified carbon nanotubes (CNTs) has been reported, however, both the active centers and the catalytic reaction mechanisms still remain unclear. In our theoretical calculations, a number of possible pathways for the activation of light alkanes over various types of oxygen-containing surface groups of surface-modified CNTs were theoretically explored. It was demonstrated for the first time that the diketone-like carbonyl groups residing in two neighboring phenyl rings at the edges or in the defects of CNTs show reactivity as high as the conventional metal oxide catalysts, and that the metallic CNTs usually have much higher catalytic reactivity with respect to the semi-conducting CNTs.
Co-reporter:Peng He ; Bryan E. G. Lucier ; Victor V. Terskikh ; Qi Shi ; Jinxiang Dong ; Yueying Chu ; Anmin Zheng ; Andre Sutrisno ;Yining Huang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 41) pp:23728-23744
Publication Date(Web):August 6, 2014
DOI:10.1021/jp5063868
Structural characterization of metal–organic frameworks (MOFs) is crucial, since an understanding of the relationship between the macroscopic properties of these industrially relevant materials and their molecular-level structures allows for the development of new applications and improvements in current performance. In many MOFs, the incorporated metal centers dictate the short- and long-range structure and porosity of the material. Here we demonstrate that solid-state NMR (SSNMR) spectroscopy targeting NMR-active metal centers at natural abundance, in concert with ab initio density functional theory (DFT) calculations and X-ray diffraction (XRD), is a powerful tool for elucidating the molecular-level structure of MOFs. 91Zr SSNMR experiments on MIL-140A are paired with DFT calculations and geometry optimizations in order to detect inaccuracies in the reported powder XRD crystal structure. 115In and 139La SSNMR experiments on sets of related MOFs at two different magnetic fields illustrate the sensitivity of the 115In/139La electric field gradient tensors to subtle differences in coordination, bond length distribution, and ligand geometry about the metal center. 47/49Ti SSNMR experiments reflect the presence or absence of guest solvent in MIL-125(Ti), and when combined with DFT calculations, these SSNMR experiments permit the study of local hydroxyl group configurations within the MOF channels. 67Zn SSNMR experiments and DFT calculations are also used to explore the geometry near Zn within a set of four MOFs as well as local disordering caused by distributions of different linkers around the metal. SSNMR spectroscopy of metal centers offers an impressive addition to the arsenal of techniques for MOF characterization and is particularly useful in cases where XRD information may be ambiguous, incomplete, or unavailable.
Co-reporter:Fangjun Huo, Long Wang, Yutao Yang, Yueyin Chu, Caixia Yin, Jianbin Chao, Yongbin Zhang, Xuxiu Yan, Anmin Zheng, Shuo Jin and Peng Zhi
Analyst 2013 vol. 138(Issue 3) pp:813-818
Publication Date(Web):19 Nov 2012
DOI:10.1039/C2AN36492A
The acetate derivatives of coumarin exhibited a prominent turn-on type signaling behavior toward BO3− ions over other common anions. Signaling is based on the selective deprotection of acetate groups by perborate, which resulted in significant fluorogenic signaling in an acetate buffered solution (pH 5.0). Interestingly, the detection process makes the raw material of 7-hydroxycoumarin regenerate, and the probe could be applied for the detection of BO3− of thimerosal. Furthermore, the optical properties of the probe and its BO3−-induced product were theoretically studied based on density functional theory (DFT) and the time-dependent DFT (TDDFT) explored at the B3LYP/6-311+G (d, p) level.
Co-reporter:Haihong Zhao, Linxiang Zeng, Yunlong Li, Cheng Liu, Bo Hou, Dong Wu, Ningdong Feng, Anmin Zheng, Xianglin Xie, Shengpei Su, Ningya Yu
Microporous and Mesoporous Materials 2013 Volume 172() pp:67-76
Publication Date(Web):15 May 2013
DOI:10.1016/j.micromeso.2012.12.040
Fabrication of 12-tungstophosphoric acid-(dihydro)imidazolium cation ionic complexes incorporated into SBA-15 mesoporous silicas was achieved via a one-pot procedure including prehydrolyzation of tetraethoxysilane in the presence of surfactant (EO20PO70EO20), addition of 1-trimethoxysilylpropyl-3-methylimidazolium chloride or N-(3-triethoxysilylpropyl), N(3)-(3-trimethoxysilylpropyl-4,5-dihydroimidazolium chloride, and successive addition of 12-tungstophosphoric acid. The obtained composite materials were well characterized. XRD, TEM, and nitrogen adsorption–desorption results indicated that the composite materials possessed good mesoporous character. FT-IR, ICP, and solid-state NMR analyses showed 12-tungstophosphoric acid-(dihydro)imidazolium cation ionic complexes had been incorporated into SBA-15 mesoporous silicas with intact structure of organic moieties and Keggin units. Using H2O2-mediated selective alcohol oxidations in aqueous media as test reactions, it was found that the occasion of adding HPW, together with the location of the (dihydro)imidazolium cations in the mesostructures, played a crucial role in 12-tungstophosphoric acid loading and ultimate catalytic performance. Under optimum conditions, our catalysts showed better catalytic efficiency and reusability than their analog prepared by a multistep process due to better dispersion of the Keggin units, higher structural stability, and lower leaching level of active Keggin units.Graphical abstractHighlights► Incorporation of HPW-onium cation complexes into mesoporous silicas was achieved. ► H2O2-mediated alcohol oxidation in aqueous media was used as test reaction. ► The catalysts prepared by one-pot procedure showed superior activity.
Co-reporter:Xiao-Liu Wu; Dr. Feng Luo;Dr. Gong-Ming Sun; Dr. An-Min Zheng; Dr. Jian Zhang; Dr. Ming-Biao Luo; Dr. Wen-Yuan Xu;Yan Zhu;Xiao-Min Zhang;Shu-Yun Huang
ChemPhysChem 2013 Volume 14( Issue 15) pp:3594-3599
Publication Date(Web):
DOI:10.1002/cphc.201300564
Abstract
Three new metal-organic frameworks (MOFs) were prepared by solvo(hydro)thermolysis and further characterized as framework isomers. The structural transformation from non-porous to porous MOFs and the purity of these products can be modulated by controlling the reaction temperature. The periodic-increased porosity observed was further confirmed by CO2 adsorption isotherms. Owing to the presence of acylamide groups in the pore walls and the flexible nature of the skeleton of these MOFs, highly selective CO2 adsorption over N2 was observed, as well as structure-dependent periodic varieties in luminescence properties.
Co-reporter:Xianfeng Yi, Youngchul Byun, Yueying Chu, Anmin Zheng, Suk Bong Hong, and Feng Deng
The Journal of Physical Chemistry C 2013 Volume 117(Issue 45) pp:23626-23637
Publication Date(Web):October 18, 2013
DOI:10.1021/jp4089386
The strain energies of the main reaction intermediates (i.e., monoethylated diphenylethane (mEDPE) and diethylated diphenylethane (dEDPE) derivatives), which can be formed during ethylbenzene (EB) disproportionation over six 10-ring zeolites with different framework topologies, as well as over the large-pore zeolite Y, were determined by the density functional theory calculations in order to more precisely investigate the effects of the pore structure of medium-pore zeolites on their formations. It was found that while the strain energies of mEDPE and dEDPE intermediates in zeolite Y, MCM-22 and TNU-9, were always lower than 19.6 kJ mol–1, some of them were characterized by considerably higher energies (>32.8 kJ mol–1) when positioned in the intersection channels of ZSM-5 and ZSM-57. As expected, in addition, all the mEDPE and dEDPE derivatives embedded in TNU-10 and ZSM-22 with narrower 10-ring channels were strongly distorted, giving them much higher strain energies (>37.7 kJ mol–1), which were in excellent agreements with our recently reported experimental results ( J. Phys. Chem. C 2010, 115, 16124). This led us to conclude that the size and shape of void spaces in the medium-pore zeolites play a crucial role in governing the type of mEDPE and dEDPE formations during the EB disproportionation. Our work also shows that the strain energies of various reaction intermediates confined within zeolites with different pore topologies could be regarded as a useful quantitative means in better understanding the shape-selective nature of this important class of microporous crystalline catalysts.
Co-reporter:Yutao Yang, Caixia Yin, Fangjun Huo, Yueyin Chu, Hongbo Tong, Jianbin Chao, Fangqin Cheng, Anmin Zheng
Sensors and Actuators B: Chemical 2013 Volume 186() pp:212-218
Publication Date(Web):September 2013
DOI:10.1016/j.snb.2013.06.013
The salicylaldehyde derivative, 9-formyl-8-hydroxyjulolidine was developed as a fluorescent probe to detect weak acid ions. With the salicylaldehyde moiety as the chromophore, the probe exhibits pH sensitive detection of hydrogen sulfide and silicate in aqueous solution. It displays an excellent selectivity for hydrogen sulfide and silicate over other weak acid salts. Time-dependent density functional theory calculations confirmed that the fluorescence turn-on mechanism involved blocking intramolecular charge transfer. The probe detects S2− in live cells, providing a powerful method to study H2S chemistry in biological systems.9-Formyl-8-hydroxyjulolidine, with the salicylaldehyde moiety as the chromophore, was developed as a fluorescent probe to detect hydrogen sulfide and silicate in aqueous solution. Time-dependent density functional theory calculations confirmed that the fluorescence turn-on mechanism involved blocking intramolecular charge transfer. The probe detects S2− in live cells, providing a powerful method to study H2S chemistry in biological systems.
Co-reporter:Yueying Chu ; Bing Han ; Anmin Zheng ; Xianfeng Yi ;Feng Deng
The Journal of Physical Chemistry C 2013 Volume 117(Issue 5) pp:2194-2202
Publication Date(Web):January 17, 2013
DOI:10.1021/jp311264u
The activity of olefins protonated by Brønsted acid sties in different pore structures (12-MR and 8-MR channel) of mordenite (MOR) zeolite is investigated by the B3LYP+D/6-31G(D,P)//ONIOM(B3LYP/6-31G(D,P):MNDO) method to reveal the pore selectivity for the protonation reaction. It is demonstrated that for ethene, the size of the molecule is smaller compared with the zeolite pores (both 12-MR and 8-MR channels), the pore confinement effect is weak, and the intrinsic acid strength of the solid acid plays a key role in the reaction, resulting in that the ethene protonation occurs preferentially at the strong acid sites within the 12-MR channel. For propene, the reaction can occur inside both channels (12-MR and 8-MR) due to the reactant being well fitted into the 8-MR channel of MOR zeolite. In this case, the confinement effect that stabilizes the intermediates and transition states compensates the deficiency of acid sites in the 8-MR channel. However, for the bulky isobutene, the protonation reaction occurs selectively in the 12-MR channel as the size of the reactant is larger than the size of the 8-MR channel, which results in a pronounced destabilizing effect due to the steric constraint.
Co-reporter:Yueqin Li, Ye Li, Yueying Chu, Xian Tao, Huihua Xu, Yingzhong Shen, Anmin Zheng
Journal of Luminescence 2012 Volume 132(Issue 7) pp:1663-1667
Publication Date(Web):July 2012
DOI:10.1016/j.jlumin.2012.02.023
A new ternary samarium complex Sm(β-NBM)3·(PD) has been synthesized by the reaction of SmCl3·6H2O with β-naphthoylbenzoylmethane (β-HNBM) and 1,10-phenanthroline-5,6-dione (PD) in stoichiometry. The new samarium complex obtained was characterized by elemental analysis, 1H NMR, and FT-IR spectroscopy. The absorption and emission of this complex were systematically investigated. Photoluminescence studies indicated that the energy absorbed by the organic ligands was efficiently transferred to the central Sm3+ ions and the complex showed intensely and characteristically orange emission due to the 4G5/2→6Hj transitions of the central Sm3+ ions. The energy levels (HOMO and LUMO) of the ligands and the complex were further confirmed by computer simulation and cyclic voltammetry, respectively. All the results suggested that the synthesized Sm(β-NBM)3·(PD) would have the potential application for organic light-emitting diodes.Highlights► Rare-earth ternary complexes Sm(β-NBM)3·(PD) are synthesized and characterized. ► Sm(β-NBM)3·(PD) shows orange emission due to the 4G5/2→6Hj transitions of the central Sm3+ ions. ► HOMO and LUMO energy levels of Sm(β-NBM)3·(PD) are of 6.20 and 3.25 eV. ► “Antenna effect” exists in the luminescence of Sm(β-NBM)3·(PD).
Co-reporter:Yueying Chu, Bing Han, Hanjun Fang, Anmin Zheng, Feng Deng
Microporous and Mesoporous Materials 2012 Volume 151() pp:241-249
Publication Date(Web):15 March 2012
DOI:10.1016/j.micromeso.2011.10.030
The influence of Brønsted acid strength on the reactivities of alkane activations has been systematically studied by density functional theory (DFT) calculations. Some typical reactions, such as methane hydrogen exchange (C–H bond activation), propane dehydrogenation (C–H bond dissociation and H–H bond formation), and propane cracking (C–C cleavage), were investigated on 8T solid acid models with varying acid strengths from weak-, strong-, to super-acid. According to our calculational results, it was revealed that the activation barriers (Eact) of all the reactions decrease linearly, while the rate coefficients (log k) increase linearly with increasing the acid strength, being indicative of the enhancement of reactivities. However, the reactivity of different reactions exhibits different sensitivity to acid strength: the propane cracking is most sensitive to acid strength, while the methane hydrogen exchange reaction is least sensitive to acid strength. On the basis of the natural charge on the organic fragment in the transition state, it was revealed that the sensitivity of the reactivity to acid strength could be related to the ionic character of the transition state. The propane cracking reaction possesses a more ionic transition state (0.913–0.964 |e|), while the methane hydrogen exchange has a less ionic transition state (0.646–0.773 |e|).Graphical abstractThe rate coefficients (log k) of alkane activation are all increasing with the increase of acid strength (decreasing DPE value). And the slope of the line, d(log k)/d(DPE), can represent the sensitivity of various alkane activations to acid strength. It can be seen that the propane cracking is most sensitive to acid strength.Highlights► Afford a quantitative description of the relationship between reactivities of alkane activation and acidity. ► Stronger acidity could improve the reactivity of all the reactions. ► Propane cracking is most sensitivity to acid strength while methane hydrogen exchange is the least one.
Co-reporter:Yueqin Li, Yueying Chu, Runchen Fang, Shijin Ding, Yulong Wang, Yingzhong Shen, Anmin Zheng
Polymer 2012 Volume 53(Issue 1) pp:229-240
Publication Date(Web):5 January 2012
DOI:10.1016/j.polymer.2011.11.044
A series of soluble aromatic polyimides were prepared from 2,2′-diphenyl-4,4′-biphenyl diamine (DPBD), 2,2′-bis(biphenyl)-4,4′-biphenyl diamine (BBPBD), 2,2′-bis[4-(naphthalen-1-yl)phenyl]-4,4′-biphenyl diamine (BNPBD) and 2,2′-bis(3,5-dimethoxyphenyl)-4,4′-biphenyl diamine (BMPBD) by polycondensation with 2,2′-bis[4′-(3′′,4′′,5′′-trifluorophenyl)phenyl]-4,4′,5,5′-biphenyl tetracarboxylic dianhydride via a two-step procedure. The resulting polymers were fully characterized and they exhibited excellent organosolubility, high thermal and dimensional stability. Resistive switching devices with the configuration of Al/polymer/ITO were constructed from these polyimides by using conventional solution coating process. Devices with the active layer based on DPBD, BBPBD and BNPBD exhibited nonvolatile and rewritable flash type memory characteristics with the turn-on voltage at −2.0 to −3.0 V and the turn-off voltage at 2.0–3.0 V. Whereas, device based on BMPBD demonstrated a bipolar write-once read-many times (WORM) memory capability with the writing voltage around ±3.0 V. The ON/OFF current ratio of these devices was of about 106 and the retention times can be as long as 104 s.
Co-reporter:Yueying Chu, Bing Han, Anmin Zheng, and Feng Deng
The Journal of Physical Chemistry C 2012 Volume 116(Issue 23) pp:12687-12695
Publication Date(Web):May 24, 2012
DOI:10.1021/jp302960w
The influence of both Brønsted acid strength and pore confinement effect on the ethylene dimerization reaction has been systematically studied by density functional theory (DFT) calculations. In the theoretical calculations, both stepwise and concerted reaction mechanisms are considered. It is demonstrated that the reactivity of the ethylene dimerization reaction can be significantly enhanced by increasing acid strength no matter which mechanism is included, while on the basis of activated barriers, the concerted mechanism is preferred on weak acids and two mechanisms are competitive when the acid strength increases to a medium–strong acid. Due to the pore confinement effect that can effectively stabilize the ionic transition states of the dimerization reaction, the activity of the dimerization reaction is considerably improved inside the zeolite pore. Compared with the reaction on the isolated acid sites, the transition states of the stepwise reaction are more effectively stabilized than those of the concerted reaction inside the zeolite confined pore, resulting in the former reaction being preferred when the dimerization reaction occurs inside the zeolite confinement spaces. Additionally, on the basis of the systematic investigations on the alkene dimerization reactions over zeolites with varying pore sizes (such as ZSM-22, ZSM-5, and SSZ-13), it is demonstrated that ZSM-22 and ZSM-5 zeolites are effective catalysts for the ethylene dimerization.
Co-reporter:Hanjun Fang ; Anmin Zheng ; Jun Xu ; Shenhui Li ; Yueying Chu ; Lei Chen ;Feng Deng
The Journal of Physical Chemistry C 2011 Volume 115(Issue 15) pp:7429-7439
Publication Date(Web):March 24, 2011
DOI:10.1021/jp1097316
Twenty carbenium ions in various zeolite models (8T HZSM-5, 72T HZSM-5, 84T HY, and 80T Hβ) are investigated by theoretical calculation methods in order to reveal the effects of the zeolite framework on the stability of carbenium ion intermediates. Most of the carbenium ions are found to be stable in these zeolite models and exist in the form of an ion pair with the zeolite conjugate bases. The carbenium ions can also be transformed into π complexes and, in some cases, into alkoxy species if the steric constraint around the C−O bond is not pronounced. It is found that zeolite framework inclusion facilitates the formation of ion pairs and concurrently can increase the relative stability of ion pairs compared to π complexes and alkoxy species no matter which effect (electrostatic stabilization or steric constraint destabilization) is predominant. It is also found that bulkier carbenium ions could be accommodated well in the zeolite frameworks with larger channels or cages, and vice versa. This is relevant to the shape selectivity of zeolites. It is shown that the energy difference between the ion pair and the π complex for the carbenium ions in the three zeolite frameworks is related to the proton affinity (PA) of the corresponding neutral hydrocarbons. In general, the larger the PA value, the more negative the energy difference, and thus the higher the stability of the carbenium ion, which is consistent with the experimental observations for many carbenium ions.
Co-reporter:Yueying Chu ; Zhiwu Yu ; Anmin Zheng ; Hanjun Fang ; Hailu Zhang ; Shing-Jong Huang ; Shang-Bin Liu ;Feng Deng
The Journal of Physical Chemistry C 2011 Volume 115(Issue 15) pp:7660-7667
Publication Date(Web):March 28, 2011
DOI:10.1021/jp200811b
The validity of using 31P NMR of adsorbed trimethylphosphine (TMP) as a probe molecule for discerning the types (Brønsted vs Lewis) and strengths of acid sites in solid acid catalysts have been studied by density functional theory (DFT) calculations. Brønsted acid sites with varied acidic strengths covering from weak, strong, to superacid, mimicked by 8T zeolite cluster models having different Si−H bond lengths and hence proton affinities, were examined together with Lewis acid systems having different metallic centers, e.g., BClnF3−n (n = 0−3), AlClnF3−n (n = 0−3), and TiClnF4−n (n = 0−4) and their mixed halides. The theoretical 31P chemical shifts predicted for the hydrogen-bonded TMP···H complex and the TMPH+ adducts were −61 ± 1 and −3 ± 1 ppm, respectively, in good agreement with the experimental data. For the TMP−Lewis acid complex, a linear correlation between the calculated 31P chemical shifts and corresponding binding energies was observed for the B-, Al-, and Ti-containing Lewis acids, respectively, indicating the feasibility of using the 31P chemical shift of adsorbed TMP as a scale for Lewis acidic strength.
Co-reporter:Hanjun Fang, Anmin Zheng, Yueying Chu and Feng Deng
The Journal of Physical Chemistry C 2010 Volume 114(Issue 29) pp:12711-12718
Publication Date(Web):July 2, 2010
DOI:10.1021/jp1044749
Adsorption of basic probe molecules is one of the widely used methods to characterize the acid strength of solid acids. In this contribution, the adsorptions of acetone on various Brønsted and Lewis acid sites (from weak acid to superacid) are theoretically studied, in order to elucidate the quantitative relationships between 13C chemical shifts of acetone and intrinsic acid strength of solid acids. The Brønsted acid sites are represented by a series of 8T zeolite models with varying terminal Si−H bond lengths, and the different extents of acidic proton transfer from these acid sites to acetone are revealed explicitly. We found that three adsorption conformations (hydrogen-bonded, proton-shared, and ion-pair) exist for acetone, and concurrently, a correlation of three-broken lines is obtained for the 13C chemical shift of acetone versus the deprotonation energy (DPE). The correlation can be used as a scale for quantitatively measuring the Brønsted acid strength of solid acids. A threshold of 245 ppm is determined for superacidity, in good agreement with the experimental value (244 ppm). The Lewis acid sites are modeled by tricoordinate framework aluminum species and various extra-framework aluminum cations or neutral species such as Al3+, AlO+, AlOH2+, Al(OH)2+, Al(OH)3, and AlOOH. We found that acetone is coordinately adsorbed on the aluminum atoms of Lewis acid sites and that the 13C chemical shift of acetone is almost linear to the lowest unoccupied molecular orbital (LUMO) energy of the acid sites.
Co-reporter:Hanjun Fang, Anmin Zheng, Shenhui Li, Jun Xu, Lei Chen and Feng Deng
The Journal of Physical Chemistry C 2010 Volume 114(Issue 22) pp:10254-10264
Publication Date(Web):May 14, 2010
DOI:10.1021/jp103247f
The protonation reactions of propene, isobutene, styrene, and α-methylstyrene on 8T solid acid models with varying acid strengths are theoretically studied in order to reveal the effects of acid strength on the solid acid-catalyzed reactions. It is shown that with the increase of acid strength (evaluated by deprotonation energy) the ion pair intermediates are preferentially formed, and their stabilities relative to the π complex and alkoxy species intermediates are considerably increased. Some alkoxy species for the bulkier olefinic hydrocarbons (i.e., isobutene, styrene, and α-methylstyrene) cannot exist as stable intermediates on strong solid acid models. It is further found that with the increase of acid strength the extent of energy reduction generally follows the order of ion pair > transition states (TS, TS′) > (π complex, alkoxy species), which is in line with the ionic character of these species. The ionic species are much more sensitive to acid strength than the covalent species regardless of intermediates or transition states. This can explain the increase of the stability of ion pair intermediates on strong solid acid models. In addition, the results obtained here can be used to interpret the effects of acid strength on the catalytic activities for other solid acid-catalyzed reactions, such as alkanol dehydration, alkane isomerization, and Beckmann rearrangement.
Co-reporter:Ningdong Feng ; Anmin Zheng ; Shing-Jong Huang ; Hailu Zhang ; Ningya Yu ; Chih-Yi Yang ; Shang-Bin Liu ;Feng Deng
The Journal of Physical Chemistry C 2010 Volume 114(Issue 36) pp:15464-15472
Publication Date(Web):August 20, 2010
DOI:10.1021/jp105683y
The strength and distribution of Brønsted acidic protons in anhydrous phosphomolybdic acid (H3PMo12O40, HPMo) have been studied by solid-state magic-angle-spinning (MAS) NMR, using trimethylphosphine oxide (TMPO) as the probe molecule in conjunction with density functional theory (DFT) calculations. Brønsted acid sties with strengths exceeding the threshold of superacidity (Zheng, A. et al. J. Phys. Chem. B 2008, 112, 4496) were observed for HPMo. In addition, the locations and adsorption structures of Brønsted protons on various oxygen sites in HPMo were also identified. The preferred location of the acidic proton was found to follow the trend: corner-sharing (Ob) > edge-sharing (Oc) ≫ terminal (Od) sites. Moreover, a tendency of hybridization among Brønsted protons residing at Ob and Oc sites of HPMo was inferred by experimental as well as theoretical 31P chemical shifts of the adsorbed TMPO.
Co-reporter:Anmin Zheng, Shang-Bin Liu, Feng Deng
Microporous and Mesoporous Materials 2009 Volume 121(1–3) pp:158-165
Publication Date(Web):1 May 2009
DOI:10.1016/j.micromeso.2009.01.026
A comprehensive study has been made on product selectivity and detailed reaction pathway invoked during propene hydrogenation reaction over H-ZSM-5 zeolite by DFT calculations based on an extended 64T cluster model. Accordingly, structural parameters and activation barrier heights as well as reaction energies can be deduced from the optimized adsorption complexes. By comparing with the results predicted based on simplified 8T cluster models, it is indicative that the carbonium ion transition states are effectively stabilized by the long-range electrostatic effects provoked by the zeolite framework. The stability of the propoxide complexes and the effects of acid strength on reaction mechanism were also investigated. It is found that the propene hydrogenation reaction is strictly kinetically controlled and the formation of i-propoxide is intrinsically much more favorable than n-propoxide over zeolites, particularly those with higher acidic strengths.
Co-reporter:Hongchuan Xin, Xiangping Li, Yuan Fang, Xianfeng Yi, Wenhui Hu, Yueying Chu, Feng Zhang, Anmin Zheng, Hongpeng Zhang, Xuebing Li
Journal of Catalysis (April 2014) Volume 312() pp:204-215
Publication Date(Web):1 April 2014
DOI:10.1016/j.jcat.2014.02.003
•Hierarchically porous structures and varying acidity of ZSM-5 were obtained by facile desilication and/or dealumination.•Amount of weak acid sites of acidic ZSM-5 was correlated with ethylene selectivity over various ZSM-5 catalysts.•Weak acid sites facilitated ethylene production, probably because decreased catalytic Brønsted acidity led to decreased diethyl ether formation.•Stable ethanol conversion and ethylene selectivity over post-treated ZSM-5 within time-on-stream of around 12 h.•Reaction pathways for diethyl ether and ethylene formations from ethanol were investigated by theoretical calculation.Microporous ZSM-5 zeolite was post-treated by desilication with sodium hydroxide, dealumination with oxalic acid, or both of them in a sequential way to finely tune the zeolite catalysts with hierarchically porous structure and varying acidity. In the catalytic dehydration of ethanol, diethyl ether and ethylene were two main products competitively formed at 200 °C and atmospheric pressure. The post-treated ZSM-5 catalysts could display stable ethanol conversion and ethylene selectivity within time-on-stream of around 12 h. The correlation between the steady-state ethylene selectivity and the amount of weak acid sites from ammonia temperature-programmed desorption (NH3-TPD) indicated that the weak acid sites facilitated the ethylene production during ethanol transformation under present reaction conditions. The reaction pathways for diethyl ether and ethylene formations from ethanol were investigated by theoretical calculation. Both the activation energies and natural charges of the transition states strongly supported that the selectivity for the diethyl ether tended to deteriorate with decreasing catalytic Brønsted acidity.Graphical abstractDownload high-res image (103KB)Download full-size image
Co-reporter:Le Le Gong, Wan Ting Yao, Zhi Qiang Liu, An Min Zheng, Jian Qiang Li, Xue Feng Feng, Lu Fang Ma, Chang Sheng Yan, Ming Biao Luo and Feng Luo
Journal of Materials Chemistry A 2017 - vol. 5(Issue 17) pp:NaN7967-7967
Publication Date(Web):2017/03/28
DOI:10.1039/C7TA01388D
MOF materials, as new catalysts, show high catalytic activity and size-, regio-, and stereo-selectivity. However, artificially controlling their catalytic process by convenient external stimulus such as light is still unexploited. Such photocontrol over an organic reaction may enable switchable catalytic activities and/or selectivities, consequently producing desired products from a pool of building blocks according to the order and type of stimuli applied. In this study, we present a novel MOF catalyst, which not only offers ultrahigh photocontrol with the on/off ratio as high as 407, but also displays disparate photomodulation in reaction kinetics towards various aldehyde substrates in light of their sizes, thus creating the first example of MOFs showing photoswitchable catalysis. The origin, as unveiled by photoswitching adsorption experiments and density functional theory calculations, is due to photoswitching storage of guest molecules in the metal–organic framework (MOF).
Co-reporter:Yueying Chu, Guangchao Li, Ling Huang, Xianfeng Yi, Hongqiang Xia, Anmin Zheng and Feng Deng
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 12) pp:NaN2523-2523
Publication Date(Web):2017/04/25
DOI:10.1039/C7CY00377C
Zeolites are effective catalysts for amide formation from oxime through the Beckmann rearrangement (BR) reaction; however, debates about which surface (i.e. external or internal) is more effective for the BR reaction over zeolites still remain. In this contribution, the effective rate constants (keff) are used to quantitatively evaluate the dependence of BR reactivity on the Brønsted acid location in H-ZSM-5 zeolite. Based on our theoretical calculations, it was found that in addition to the dimension size of oxime reactants and reaction temperature, the BR reaction is strongly dependent on the location of Brønsted acid sites. For the cyclohexanone oxime rearrangement, the reaction exclusively occurs on the internal surface of ZSM-5 zeolite at room temperature, while the active sites are those located at the pore mouth or on the external surface when the reaction temperature increases to 598 K. In contrast to cyclohexanone oxime, the Brønsted acid sites on the internal surface are kinetically more effective at room temperature or 598 K for the smaller acetoxime BR reaction.
Co-reporter:Fujian Liu, Bojie Li, Chen Liu, Weiping Kong, Xianfeng Yi, Anmin Zheng and Chenze Qi
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 9) pp:NaN3007-3007
Publication Date(Web):2015/11/19
DOI:10.1039/C5CY01226K
N rich porous carbon based solid acids (NPC-[CxN][X]) have been successfully synthesized by treatment of N rich porous carbon (NPC) with various quaternary ammoniation reagents such as iodomethane, 1,3-propane sultone, and 1,4-butanesultone, and ion exchange with various strong acids such as HSO3CF3, H2SO4, H3PW12O40, HBF4etc. The NPC support was synthesized by carbonization of KOH-activated polypyrrole without using additional templates. Various characterizations showed that NPC-[CxN][X] possesses abundant nanopores, large Brunauer–Emmett–Teller surface areas, good stability, and strong and controllable acid sites with Brønsted characteristics. The immobilized acidic groups were homogeneously dispersed into NPC-[CxN][X]. Notably, NPC-[CxN][X] acted as efficient, reusable and generalized solid acids, which showed excellent activity in various acid-catalyzed reactions such as esterification and transesterification in the synthesis of biodiesel, dehydration of fructose into 5-hydroxymethylfurfural, depolymerization of crystalline cellulose into sugars, and condensation of phenol with acetone in the synthesis of bisphenol A, much higher than that of various solid acids such as Amberlyst 15, H-ZSM-5, H-USY, and sulfonic group functionalized ordered mesoporous silicas. The preparation of NPC-[CxN][X] leads to the development of porous carbon based solid acids with controllable structural characteristics and excellent catalytic activity.
Co-reporter:Chin-Te Hung, Ningya Yu, Chia-Ting Chen, Pei-Hao Wu, Xiaoxiang Han, Yu-Siang Kao, Tuan-Chi Liu, Yueying Chu, Feng Deng, Anmin Zheng and Shang-Bin Liu
Journal of Materials Chemistry A 2014 - vol. 2(Issue 47) pp:NaN20037-20037
Publication Date(Web):2014/09/19
DOI:10.1039/C4TA04403G
A facile method was developed for the synthesis of metal-free, highly N-doped (>7 wt%) mesoscopic carbons (NMCs), which were fabricated by first preparing carbon–silicate (C–Si) composites by co-condensation method using a melamine-formaldehyde resin oligomer as the primary nitrogen and carbon source, and P123 triblock copolymer surfactant and sodium silicate as the soft and hard template, respectively, under microwave irradiation conditions, followed by carbonization and silica-template removal. The NMCs were found to exhibit superior electrocatalytic activity, long-term stability, and excellent tolerance over methanol crossover effect. Such NMCs derived from organic–inorganic hybrids assisted by microwave heating not only possess high surface areas and active quaternary and pyridinic-N species that are favourable for ORR, as verified by DFT calculations, but also render large-scale production and practical applications as cost-effective electrode materials.
Co-reporter:Cong Bin Fan, Zhi Qiang Liu, Le Le Gong, An Min Zheng, Le Zhang, Chang Sheng Yan, Hui Qiong Wu, Xue Feng Feng and Feng Luo
Chemical Communications 2017 - vol. 53(Issue 4) pp:NaN766-766
Publication Date(Web):2016/12/15
DOI:10.1039/C6CC08982H
The first MOF (metal–organic framework) built on both diarylethene and azobenzene photochromic units is reported here and displays distinct photoresponses for different guest molecules, thus creating an easy-to-use pathway to modulate the adsorption selectivity of MOF materials.
Co-reporter:Bing Ma, Xianfeng Yi, Li Chen, Anmin Zheng and Chen Zhao
Journal of Materials Chemistry A 2016 - vol. 4(Issue 29) pp:NaN11341-11341
Publication Date(Web):2016/06/17
DOI:10.1039/C6TA01807F
Hierarchical H-style ultra-stable Y (HUSY) zeolites with abundant interconnected mesopores have been prepared using a sequential post-synthesis strategy that includes steaming dealumination and mixed-alkali desilication. The steaming treatment generates a broad size range of intra-mesopores (around 25 and 45 nm) and a moderate Si/Al ratio of 13.4 in the HUSY, which provides optimal material precursors for the ensuing subsequent alkaline desilication. N2 adsorption–desorption isotherms and X-ray diffractometry results indicate that the sample treated with pyridine/sodium hydroxide (HUSY-4) has a larger external surface area and a higher relative crystallinity. Infrared spectra of adsorbed pyridine show that HUSY-4 contains substantial Brønsted acid sites. The 27Al and 29Si nuclear magnetic resonance spectra show that HUSY-4 possesses few extra-framework alumina species. Infrared spectra in a vacuum show that the peak intensities of HUSY-4 in the bridged hydroxyl group (at 3560 and 3631 cm−1) are much stronger than those of the sample treated with tetrapropylammonium hydroxide (HUSY-3), indicating that the framework integrity of HUSY-4 is better. Differences in treatments with tetrapropylammonium hydroxide/sodium hydroxide and pyridine/sodium hydroxide treatments are attributed to the fact that the pyridine molecule (0.54 nm) can pass through the supercages (0.74 nm) to protect the zeolite framework from deep desilication, whereas the tetrapropylammonium hydroxide molecule (0.85 nm) is adsorbed only on the external surface. Eventually, a HUSY zeolite with a high external surface area, inter-connectedness and hierarchical mesopores (10, 25, and 45 nm) is prepared by initial high-temperature steaming, which is followed by desilication using a mixed alkali solution containing pyridine and sodium hydroxide. High-dispersion (5.5%), high-content (35 wt%), small Ni nanoparticles (4.9 ± 1.2 nm) are loaded onto and into the external surface areas and interpores of the hierarchical HUSY by the deposition–precipitation method. The resultant Ni/HUSY-4 shows an ultra-high efficiency in the hydrodeoxygenation of fatty acids, esters, and palm oil, and achieves high initial rates (60 g g−1 h−1) and a high C18 alkane selectivity (96%), which may be attributed to the enhanced Brønsted acid and adjacent Lewis acid (confirmed by the 1H DQ MAS NMR spectrum) together with the substantial dispersive Ni nanoparticles loaded onto/into the interconnected pores of the hierarchical HUSY support.
Co-reporter:Yueying Chu, Nianhua Xue, Bolian Xu, Qian Ding, Zhaochi Feng, Anmin Zheng and Feng Deng
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 14) pp:NaN5363-5363
Publication Date(Web):2016/04/05
DOI:10.1039/C6CY00467A
Theoretical calculations have provided fundamental insights into the possible pathways for the H/D exchange of isobutane with H-ZSM-5 zeolite at room temperature. It is theoretically demonstrated that neither the direct exchange mechanism nor the indirect bimolecular hydride transfer mechanism is an efficient route for isobutane activation due to high activation barriers, which possibly prohibit H/D exchange at low temperatures. It is revealed that a trace of olefin impurities can considerably accelerate the formation of an alkoxyl intermediate which is involved in the bimolecular hydride transfer mechanism. Once the alkoxyl intermediate is generated from the olefin impurities, the catalytic cycle is self-sustaining. On the other hand, the theoretical calculations also illustrate that the extra-framework aluminum (EFAl) species in the dealuminated zeolite has no obvious promotional effect on the H/D exchange. Furthermore, our calculations are also consistent with the experimental results.
Co-reporter:Mengdi Huang, Qiang Wang, Xianfeng Yi, Yueying Chu, Weili Dai, Landong Li, Anmin Zheng and Feng Deng
Chemical Communications 2016 - vol. 52(Issue 70) pp:NaN10608-10608
Publication Date(Web):2016/07/05
DOI:10.1039/C6CC04943E
Solid-state NMR experiments and DFT calculations have been carried out to determine the complex structures of coadsorbed 13C-labeled tert-butanol and NH3 in acidic H-ZSM-5 zeolite. It is found, besides the physically adsorbed tert-butanol/NH4+ complex on Brønsted acid sites, the tert-butylamine cation is formed as well, confirming the presence of the tert-butyl cation confined in zeolite channels. Furthermore, 13C–27Al double-resonance solid-state NMR spectroscopy is adopted to determine the host/guest interaction between the carbocation and the zeolite framework.
Co-reporter:Yueying Chu, Xianyong Sun, Xianfeng Yi, Lihong Ding, Anmin Zheng and Feng Deng
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 7) pp:NaN3517-3517
Publication Date(Web):2015/03/31
DOI:10.1039/C5CY00312A
The methanol to olefins (MTO) process, in which low-value carbon-rich feedstocks are converted to value-added petrochemical products, is one of the most prominent alternatives for the production of light olefins. In order to reveal the confinement effect of zeolites on the catalytic reactions, the MTO mechanisms and reactivity over two unidimensional zeolites (H-ZSM-12 and H-ZSM-22) with a channel difference of only 0.3 Å have been systematically explored by DFT calculations in this work. The calculated activation barriers and reaction energies demonstrated that the 0.3 Å channel difference between H-ZSM-12 and H-ZSM-22 zeolites results in a dramatic discrepancy in their transition state selectivity associated with the aromatic-based hydrocarbon pool (HCP) mechanism. For the larger H-ZSM-12 zeolite, the formation of pentamethybenzenium cation was favored, which would be the active HCP species in the MTO reaction. For the H-ZSM-22 zeolite with a 0.3 Å smaller pore structure, the traditional methylation at the C–H sites of polymethylbenzenes occurred exclusively. When the alternative olefin-based cycle is followed for the MTO reaction, both of the zeolites are active catalysts for the formations of butene and propene. A comparison of the activation barriers for the olefin-based cycle revealed that the larger H-ZSM-12 possesses a higher catalytic activity than the H-ZSM-22 zeolite. Our theoretical results demonstrate that both the aromatic-based cycle and the olefin-based cycle can proceed during the MTO reaction over H-ZSM-12, with the latter cycle being predominant.
Co-reporter:Yueying Chu, Peng Ji, Xianfeng Yi, Shenhui Li, Peng Wu, Anmin Zheng and Feng Deng
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 7) pp:NaN3681-3681
Publication Date(Web):2015/05/18
DOI:10.1039/C5CY00619H
The effects of Brønsted acid strength and pore confinement on the Beckmann rearrangement (BR) reaction over solid acid catalysts have been explored. With the help of catalytic evaluation experiments, it is demonstrated that oximes with different size (cyclohexanone oxime and acetoxime) exhibit quite different BR reactivity dependence on the acid strength over microporous and mesoporous zeolites. In order to reveal the origin of such a difference, electronic structure calculations and kinetic analysis were performed. It was theoretically found that the confinement effect from the microporous zeolite framework has a more significant influence on the rate-determining step of the BR reaction when the oxime reactant is well-confined inside the microporous voids, which in turn controls the BR reactivity.
Co-reporter:Xianfeng Yi, Lihong Ding, Guangchao Li, Zhiqiang Liu, Hongqiang Xia, Yueying Chu, Anmin Zheng and Feng Deng
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 16) pp:NaN6338-6338
Publication Date(Web):2016/05/24
DOI:10.1039/C6CY00757K
A comprehensive understanding of the reaction mechanisms of hydrocarbon conversion over acidic zeolite catalysts would be of great importance to optimize, modify and design more efficient catalytic materials. For this purpose, theoretical calculations based on molecular dynamics (MD) simulations and density functional theory (DFT) calculations have been performed in this work to explore the reaction pathways of propene H/D exchange over deuterated acidic ZSM-5 zeolite (D-ZSM-5). The deuterated propene-D5 is confirmed to be readily formed through the route involving an isopropyl intermediate. With regard to the formation of completely deuterated propene-D6, the propene loading is found to play a crucial role in governing the reaction pathway. The dimerization route (through the dimerization of propene, the intramolecular hydride transfer and then the cracking process) is demonstrated to be predominant with a relatively lower activation energy barrier (12.3 kcal mol−1) at higher propene loading, while the n-propoxy pathway is preferred at lower propene loading. Furthermore, the influences of the acid strength and pore confinement effect of zeolite on the propene H/D exchange reaction activity have been derived as well.





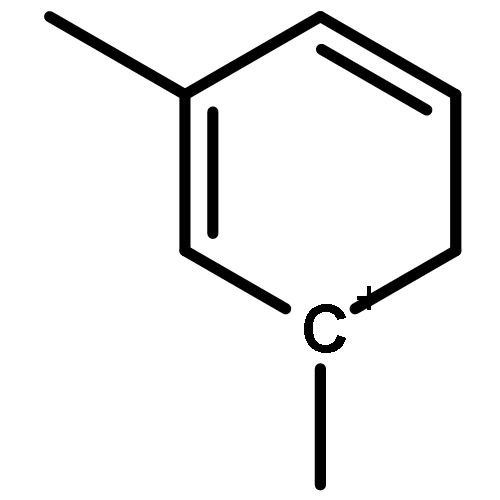
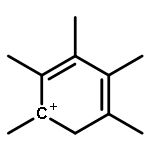
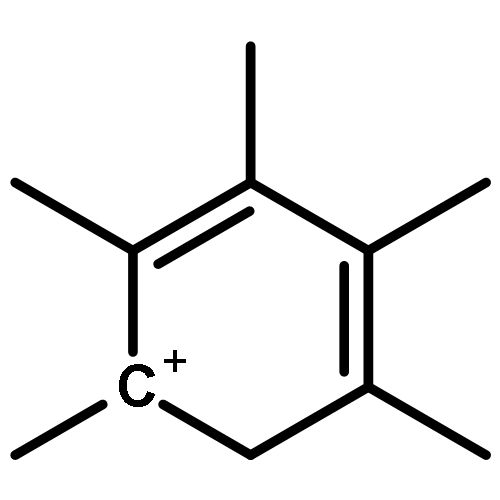
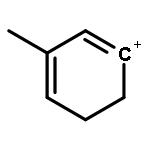
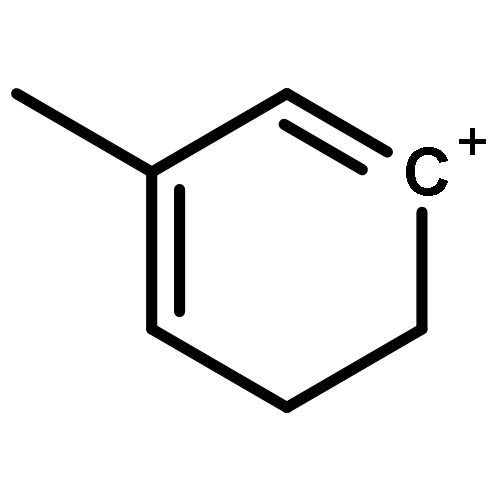


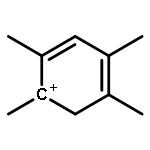
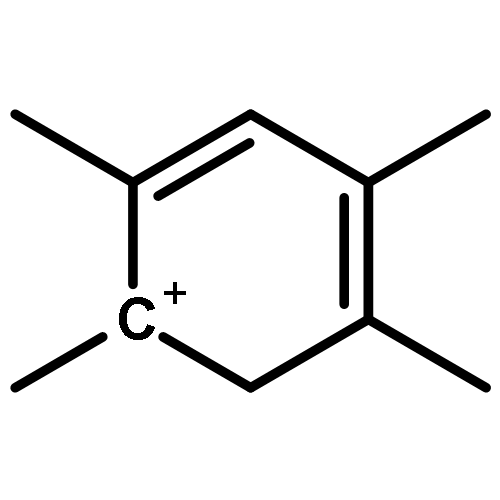


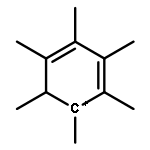
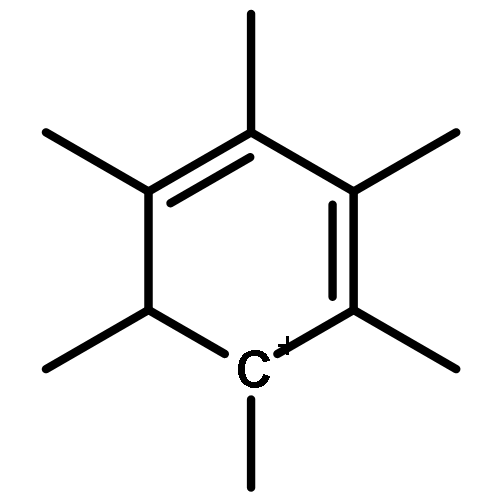





![8-FLUORO-1H-BENZO[D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/20800/20717-73-1.png)
![8-FLUORO-1H-BENZO[D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/20800/20717-73-1_b.png)