Co-reporter:Zhiqian Wang, Brandon J. Reinus, and Guangbin Dong
Journal of the American Chemical Society August 29, 2012 Volume 134(Issue 34) pp:13954-13957
Publication Date(Web):August 29, 2012
DOI:10.1021/ja306123m
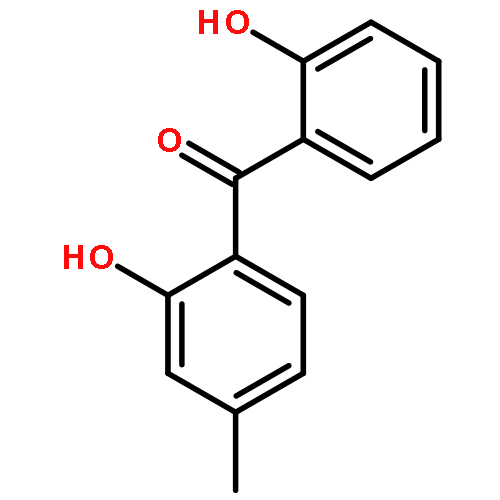
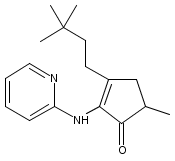
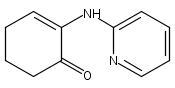
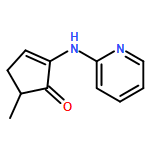
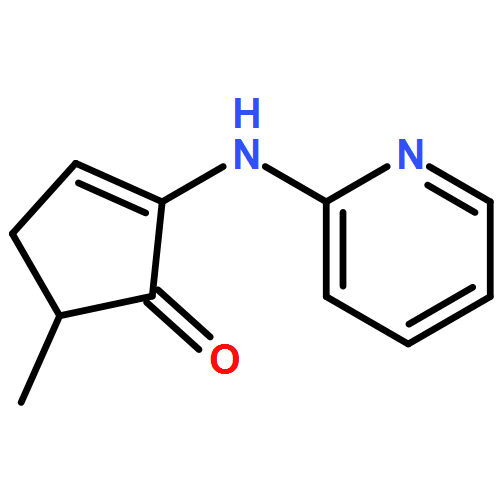
We describe the first example of Rh-catalyzed intermolecular C-alkylation of cyclic 1,2-diketones using simple terminal olefins as alkylating agents. Aminopyridine is employed as a recyclable directing group. First, it reacts with ketones to give enamines and delivers Rh to activate the vinyl C–H bonds in the same pot; second, it can be cleaved off and recovered via hydrolysis. A broad range of olefins can be utilized as substrates, including aliphatic, aromatic olefins and vinyl esters. The efficiency of this method is also demonstrated in the synthesis of a natural flavoring compound, 3-ethyl-5-methyl-1,2-cyclopentadione (one-pot 53% yield vs a previous four-step route 16% yield from the same starting material). This work is expected to serve as a seminal study toward catalytic ketone α-alkylation with unactivated olefins.
Co-reporter:Rong Zeng, Peng-hao Chen, and Guangbin Dong
ACS Catalysis 2016 Volume 6(Issue 2) pp:969
Publication Date(Web):December 30, 2015
DOI:10.1021/acscatal.5b02532
The first decarbonylative cycloaddition of less-strained cyclic ketones (isatins) with isocyanates is reported. Initiated by C–C activation, this distinct [5 – 2 + 2] transformation provides a rapid entry to access various benzimidazolidinone derivatives, and a wide range of isocyanates can be efficiently coupled with broad functional group tolerance. A modified one-pot process combining a Curtius rearrangement and C–C activation was also achieved by using acyl azides as the starting materials. A detailed mechanistic study revealed a surprising double-decarbonylative reaction pathway. The novel reactivity discovered in this basic research is expected to shed light on the development of new heterocycle formation methods through C–C/isocyanate coupling.Keywords: cycloaddition; C−C activation; isatin; isocyanate; rhodium
Co-reporter:Hee Nam Lim and Guangbin Dong
Organic Letters 2016 Volume 18(Issue 5) pp:1104-1107
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.orglett.6b00207
A general and practical catalytic method has been developed for the rapid synthesis of HCTD (heptacyclo[6.6.0.02,6.03,13.04,11.05,9.010,14]tetradecanes) and various new 7,12-disubstituted HCTDs from norbornadienes. Compared to the known approaches, this new protocol avoids stoichiometric metals, utilizes commercially available reagents as catalysts, and affords higher yields and significantly improved selectivity. In addition, quadracyclane was discovered for the first time to undergo a similar endo,cis,endo cycloaddition to give HCTD in a good yield. Derivatization of HCTDs led to efficient preparation of a range of novel homo- and heterobifunctional scaffolds that hold potentials for biological and material applications.
Co-reporter:Guangbin Dong
Tetrahedron 2016 Volume 72(Issue 22) pp:2717
Publication Date(Web):2 June 2016
DOI:10.1016/j.tet.2016.03.089
Co-reporter:Guangbin Dong
Tetrahedron 2016 Volume 72(Issue 22) pp:2718
Publication Date(Web):2 June 2016
DOI:10.1016/j.tet.2016.03.090
Co-reporter:Yan Xu;Dr. Michael C. Young;Chengpeng Wang;David M. Magness ; Guangbin Dong
Angewandte Chemie 2016 Volume 128( Issue 31) pp:9230-9233
Publication Date(Web):
DOI:10.1002/ange.201604268
Abstract
Herein, we report the palladium-catalyzed direct arylation of unactivated aliphatic C−H bonds in free primary amines. This method takes advantage of an exo-imine-type directing group (DG) that can be generated and removed in situ. A range of unprotected aliphatic amines are suitable substrates, undergoing site-selective arylation at the γ-position. Methyl as well as cyclic and acyclic methylene groups can be activated. Furthermore, when aniline-derived substrates were used, preliminary success with δ-C−H arylation was achieved. The feasibility of using the DG component in a catalytic fashion was also demonstrated.
Co-reporter:Yan Xu;Tianshun Su;Zhongxing Huang;Dr. Guangbin Dong
Angewandte Chemie International Edition 2016 Volume 55( Issue 7) pp:2559-2563
Publication Date(Web):
DOI:10.1002/anie.201510638
Abstract
Direct arylation of cyclopentanones has been a long-standing challenge because of competitive self-aldol condensation and multiple arylations. Reported herein is a direct mono-α-C−H arylation of cyclopentanones with aryl bromides which is enabled by palladium/amine cooperative catalysis. This method is scalable and chemoselective with broad functional-group tolerance. Application to controlled sequential arylation of cyclopentanones has been also demonstrated.
Co-reporter:Zhongxing Huang;Chengpeng Wang ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2016 Volume 55( Issue 17) pp:5299-5303
Publication Date(Web):
DOI:10.1002/anie.201600912
Abstract
Described is a new hydrazone-based exo-directing group (DG) strategy developed for the functionalization of unactivated primary β C−H bonds of aliphatic amines. Conveniently synthesized from protected primary amines, the hydrazone DGs are shown to site-selectively promote the β-acetoxylation and tosyloxylation via five-membered exo-palladacycles. Amines with a wide scope of skeletons and functional groups are tolerated. Moreover, the hydrazone DG can be readily removed, and a one-pot C−H acetoxylation/DG removal protocol was also discovered.
Co-reporter:Yan Xu;Dr. Michael C. Young;Chengpeng Wang;David M. Magness ; Guangbin Dong
Angewandte Chemie International Edition 2016 Volume 55( Issue 31) pp:9084-9087
Publication Date(Web):
DOI:10.1002/anie.201604268
Abstract
Herein, we report the palladium-catalyzed direct arylation of unactivated aliphatic C−H bonds in free primary amines. This method takes advantage of an exo-imine-type directing group (DG) that can be generated and removed in situ. A range of unprotected aliphatic amines are suitable substrates, undergoing site-selective arylation at the γ-position. Methyl as well as cyclic and acyclic methylene groups can be activated. Furthermore, when aniline-derived substrates were used, preliminary success with δ-C−H arylation was achieved. The feasibility of using the DG component in a catalytic fashion was also demonstrated.
Co-reporter:Zhi Ren and Guangbin Dong
Organometallics 2016 Volume 35(Issue 8) pp:1057-1059
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.organomet.6b00185
Here we describe the first preparation of tertiary alkyl palladium complexes via C–H activation. Enabled by an exo-type oxime directing group, cyclopalladation occurred smoothly at the bridgehead position. Treatment of the resulting complex with iodine led to C–H iodination at the methine carbon. This study provides important mechanistic information for palladium-catalyzed functionalization of methine C–H bonds.
Co-reporter:Yan Xu;Tianshun Su;Zhongxing Huang;Dr. Guangbin Dong
Angewandte Chemie 2016 Volume 128( Issue 7) pp:2605-2609
Publication Date(Web):
DOI:10.1002/ange.201510638
Abstract
Direct arylation of cyclopentanones has been a long-standing challenge because of competitive self-aldol condensation and multiple arylations. Reported herein is a direct mono-α-C−H arylation of cyclopentanones with aryl bromides which is enabled by palladium/amine cooperative catalysis. This method is scalable and chemoselective with broad functional-group tolerance. Application to controlled sequential arylation of cyclopentanones has been also demonstrated.
Co-reporter:Zhongxing Huang;Chengpeng Wang ;Dr. Guangbin Dong
Angewandte Chemie 2016 Volume 128( Issue 17) pp:5385-5389
Publication Date(Web):
DOI:10.1002/ange.201600912
Abstract
Described is a new hydrazone-based exo-directing group (DG) strategy developed for the functionalization of unactivated primary β C−H bonds of aliphatic amines. Conveniently synthesized from protected primary amines, the hydrazone DGs are shown to site-selectively promote the β-acetoxylation and tosyloxylation via five-membered exo-palladacycles. Amines with a wide scope of skeletons and functional groups are tolerated. Moreover, the hydrazone DG can be readily removed, and a one-pot C−H acetoxylation/DG removal protocol was also discovered.
Co-reporter:Zhongxing Huang, Hee Nam Lim, Fanyang Mo, Michael C. Young and Guangbin Dong
Chemical Society Reviews 2015 vol. 44(Issue 21) pp:7764-7786
Publication Date(Web):17 Jul 2015
DOI:10.1039/C5CS00272A
Transition metal-catalyzed C–H functionalization has evolved into a prominent and indispensable tool in organic synthesis. While nitrogen, phosphorus and sulfur-based functional groups (FGs) are widely employed as effective directing groups (DGs) to control the site-selectivity of C–H activation, the use of common FGs (e.g. ketone, alcohol and amine) as DGs has been continuously pursued. Ketones are an especially attractive choice of DGs and substrates due to their prevalence in various molecules and versatile reactivity as synthetic intermediates. Over the last two decades, transition metal-catalyzed C–H functionalization that is directed or mediated by ketones has experienced vigorous growth. This review summarizes these advancements into three major categories: use of ketone carbonyls as DGs, direct β-functionalization, and α-alkylation/alkenylation with unactivated olefins and alkynes. Each of these subsections is discussed from the perspective of strategic design and reaction discovery.
Co-reporter:Rong Zeng
Journal of the American Chemical Society 2015 Volume 137(Issue 4) pp:1408-1411
Publication Date(Web):January 8, 2015
DOI:10.1021/ja512306a
Herein we report a [5 + 2 – 1] transformation though catalytic decarbonylative coupling between isatins and alkynes, which provides a unique way to synthesize 2-quinolinone derivatives. A broad range of alkynes can be coupled efficiently with high regioselectivity. This reaction is proposed to go through C–C activation of isatins, followed by decarbonylation and alkyne insertion. Directing group (DG) plays a critical role in this transformation. Assisted by the DG, the C–C cleavage of isatins occurs at room temperature.
Co-reporter:Samuel J. Thompson; Danny Q. Thach
Journal of the American Chemical Society 2015 Volume 137(Issue 36) pp:11586-11589
Publication Date(Web):August 31, 2015
DOI:10.1021/jacs.5b07384
Here, a palladium-catalyzed functionalization of unactivated sp3 C–H bonds with internal alcohol nucleophiles is described. Directed by an oxime-masked alcohol, annulation chemoselectively occurs at the β position, leading to a range of aliphatic cyclic ethers with four- to seven-membered rings. Tethered primary, secondary, and tertiary free hydroxyl groups can all react to give the corresponding cyclized products. In addition, benzyl and silyl protected alcohols can also be directly coupled. An sp3 C–H activation/intramolecular SN2 pathway was proposed.
Co-reporter:Xuan Zhou
Journal of the American Chemical Society 2015 Volume 137(Issue 42) pp:13715-13721
Publication Date(Web):October 6, 2015
DOI:10.1021/jacs.5b09799
Herein we describe a rhodium-catalyzed (4+1) cyclization between cyclobutanones and allenes, which provides a distinct [4.2.1]-bicyclic skeleton containing two quaternary carbon centers. The reaction involves C–C activation of cyclobutanones and employs allenes as a one-carbon unit. A variety of functional groups can be tolerated, and a diverse range of polycyclic scaffolds can be accessed. Excellent enantioselectivity can be obtained, which is enabled by a TADDOL-derived phosphoramidite ligand. The bridged bicyclic products can be further functionalized or derivatized though simple transformations.
Co-reporter:Lin Deng; Tao Xu; Hongbo Li
Journal of the American Chemical Society 2015 Volume 138(Issue 1) pp:369-374
Publication Date(Web):December 16, 2015
DOI:10.1021/jacs.5b11120
Herein we describe the first enantioselective Rh-catalyzed carboacylation of oximes (imines) via C–C activation. In this transformation, the benzocyclobutenone C1–C2 bond is selectively activated by a low valent rhodium catalyst and subsequently the resulting two Rh–C bonds add across a C═N bond, which provides a unique approach to access chiral lactams. A range of polycyclic nitrogen-containing scaffolds were obtained in good yields with excellent enantioselectivity. Further derivatization of the lactam products led to a rapid entry to various novel fused heterocycles.
Co-reporter:Fanyang Mo; Hee Nam Lim
Journal of the American Chemical Society 2015 Volume 137(Issue 49) pp:15518-15527
Publication Date(Web):November 13, 2015
DOI:10.1021/jacs.5b10466
Here, we describe a detailed study of the rhodium(I)-catalyzed, bifunctional ligand-assisted ketone α-C–H alkenylation using internal alkynes. Through controlling the reaction conditions, conjugated enamines, α,β- or β,γ-unsaturated ketones, can be selectively accessed. Both aromatic and aliphatic alkynes can be employed as coupling partners. The reaction conditions also tolerate a broad range of functional groups, including carboxylic esters, malonates, secondary amides, thioethers, and free alcohols. In addition, excellent E-selectivity was observed for the tetra-substituted alkene when forming the α,β-unsaturated ketone products. The mechanism of this transformation was explored through control experiments, kinetic monitoring, synthesizing the rhodium–hydride intermediates and their reactions with alkynes, deuterium-labeling experiments, and identification of the resting states of the catalyst.
Co-reporter:Zhe Dong; Jianchun Wang
Journal of the American Chemical Society 2015 Volume 137(Issue 18) pp:5887-5890
Publication Date(Web):April 24, 2015
DOI:10.1021/jacs.5b02809
Herein we report a highly meta-selective C–H arylation using simple tertiary amines as the directing group. This method takes advantage of Pd/norbornene catalysis, offering a distinct strategy to control the site selectivity. The reaction was promoted by commercially available AsPh3 as the ligand and a unique “acetate cocktail”. Aryl iodides with an ortho electron-withdrawing group were employed as the coupling partner. A wide range of functional groups, including some heteroarenes, are tolerated under the reaction conditions. In addition, the amine directing group can be easily installed and transformed to other common versatile functional groups. We expect this C–H functionalization mode to have broad implications for developing other meta-selective transformations beyond this work.
Co-reporter:Zhi Ren, Jonathan E. Schulz, and Guangbin Dong
Organic Letters 2015 Volume 17(Issue 11) pp:2696-2699
Publication Date(Web):May 12, 2015
DOI:10.1021/acs.orglett.5b01098
A Pd-catalyzed ortho-acetoxylation of masked benzyl alcohols to synthesize various 2-hydroxyalkylphenol derivatives is reported. The 2,6-dimethoxyl benzaldoxime proved to be an efficient exo-type directing group for arene (sp2) C–H functionalization. Two strategies were demonstrated to remove the directing group through N–O and C–O bond cleavages. A high catalyst turnover (>1000) was obtained to illustrate the practicality of this method.
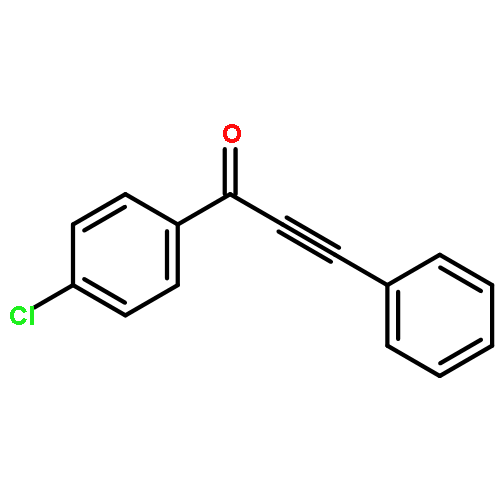
Co-reporter:Rachel E. Whittaker and Guangbin Dong
Organic Letters 2015 Volume 17(Issue 21) pp:5504-5507
Publication Date(Web):October 27, 2015
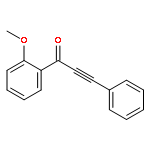
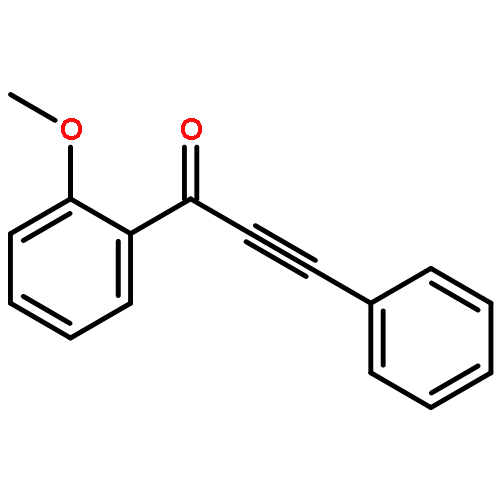
DOI:10.1021/acs.orglett.5b02911
A Rh-catalyzed controlled decarbonylation of alkynyl α-diones is described. By using different ligand and solvent combinations, mono- and double-decarbonylation can be selectively achieved to give conjugated ynones and disubstituted alkynes, respectively. A fundamental study on catalytic activation of unstrained C–C bonds under nonoxidative conditions is presented.
Co-reporter:Xuan Zhou, Imran Zafar, Guangbin Dong
Tetrahedron 2015 Volume 71(26–27) pp:4478-4483
Publication Date(Web):1 July 2015
DOI:10.1016/j.tet.2015.02.087
Here we describe a rhodium-catalyzed intramolecular decarbonylative coupling between 3-aminocyclobutenones and alkenes for synthesis of substituted [3.1.0] bicycles. This transformation represents a formal cyclopropanation reaction, in which the cyclobutenones serve as a one-carbon-unit synthon.
Co-reporter:Zhe Dong;Jianchun Wang;Zhi Ren ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2015 Volume 54( Issue 43) pp:12664-12668
Publication Date(Web):
DOI:10.1002/anie.201506397
Abstract
Reported herein is a palladium/norbornene-catalyzed ortho-arene acylation of aryl iodides by a Catellani-type CH functionalization. This transformation is enabled by isopropyl carbonate anhydrides, which serve as both an acyl cation equivalent and a hydride source.
Co-reporter:Dr. Hee Nam Lim ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2015 Volume 54( Issue 50) pp:15294-15298
Publication Date(Web):
DOI:10.1002/anie.201507741
Abstract
Two complementary methods for catalytic intramolecular ketone alkylation reactions with unactivated olefins, resulting in Conia-ene-type reactions, are reported. The transformations are enabled by dual activation of both the ketone and the olefin and are atom-economical as stoichiometric oxidants or reductants are not required. Assisted by Kool’s aniline catalyst, the reaction conditions can be both pH- and redox-neutral. A broad range of functional groups are thus tolerated. Whereas the rhodium catalysts are effective for the formation of five-membered rings, a ruthenium-based system that affords the six-membered ring products was also developed.
Co-reporter:Zhe Dong;Jianchun Wang;Zhi Ren ;Dr. Guangbin Dong
Angewandte Chemie 2015 Volume 127( Issue 43) pp:12855-12859
Publication Date(Web):
DOI:10.1002/ange.201506397
Abstract
Reported herein is a palladium/norbornene-catalyzed ortho-arene acylation of aryl iodides by a Catellani-type CH functionalization. This transformation is enabled by isopropyl carbonate anhydrides, which serve as both an acyl cation equivalent and a hydride source.
Co-reporter:Dr. Hee Nam Lim ;Dr. Guangbin Dong
Angewandte Chemie 2015 Volume 127( Issue 50) pp:15509-15513
Publication Date(Web):
DOI:10.1002/ange.201507741
Abstract
Two complementary methods for catalytic intramolecular ketone alkylation reactions with unactivated olefins, resulting in Conia-ene-type reactions, are reported. The transformations are enabled by dual activation of both the ketone and the olefin and are atom-economical as stoichiometric oxidants or reductants are not required. Assisted by Kool’s aniline catalyst, the reaction conditions can be both pH- and redox-neutral. A broad range of functional groups are thus tolerated. Whereas the rhodium catalysts are effective for the formation of five-membered rings, a ruthenium-based system that affords the six-membered ring products was also developed.
Co-reporter:Peng-hao Chen;Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie 2014 Volume 126( Issue 6) pp:1700-1704
Publication Date(Web):
DOI:10.1002/ange.201310100
Abstract
A tunable rhodium-catalyzed intramolecular alkyne insertion reaction proceeding through the CC cleavage of benzocyclobutenones is described. Selective formation of either the direct or decarbonylative insertion product can be controlled by using different catalytic systems. A variety of fused β-naphthol and indene scaffolds were obtained in good yields with high functional group tolerance. This work illustrates a divergent approach to synthesize fused-ring systems by CC activation/functionalization.
Co-reporter:Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie 2014 Volume 126( Issue 40) pp:10909-10912
Publication Date(Web):
DOI:10.1002/ange.201404802
Abstract
The first total syntheses of the proposed structure of cycloinumakiol (1) and its C5 epimer (18) are achieved in a concise and efficient fashion. Starting from the known 3-hydroxybenzocyclobutenone, 1 and 18 are obtained in nine and five steps with overall yields of 15 % and 33 %, respectively. The key for the success of this approach is the use of a catalytic CC activation strategy for constructing the tetracyclic core of 1 through carboacylation of a sterically hindered trisubstituted olefin with benzocyclobutenone. In addition, the structure of the natural cycloinumakiol was reassigned to 19-hydroxytotarol (7) through X-ray diffraction analysis. This work demonstrates the potential of CC activation for streamlining complex natural product synthesis.
Co-reporter:Peng-hao Chen;Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2014 Volume 53( Issue 6) pp:1674-1678
Publication Date(Web):
DOI:10.1002/anie.201310100
Abstract
A tunable rhodium-catalyzed intramolecular alkyne insertion reaction proceeding through the CC cleavage of benzocyclobutenones is described. Selective formation of either the direct or decarbonylative insertion product can be controlled by using different catalytic systems. A variety of fused β-naphthol and indene scaffolds were obtained in good yields with high functional group tolerance. This work illustrates a divergent approach to synthesize fused-ring systems by CC activation/functionalization.
Co-reporter:Dr. Tao Xu;Nikolas A. Savage ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2014 Volume 53( Issue 7) pp:1891-1895
Publication Date(Web):
DOI:10.1002/anie.201310149
Abstract
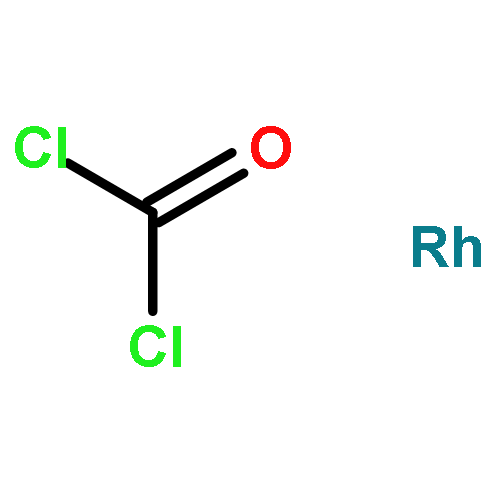
The rhodium-catalyzed formation of all-carbon spirocenters involves a decarbonylative coupling of trisubstituted cyclic olefins and benzocyclobutenones through CC activation. The metal–ligand combination [{Rh(CO)2Cl}2]/P(C6F5)3 catalyzed this transformation most efficiently. A range of diverse spirocycles were synthesized in good to excellent yields and many sensitive functional groups were tolerated. A mechanistic study supports a hydrogen-transfer process that occurs through a β-H elimination/decarbonylation pathway.
Co-reporter:Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:10733-10736
Publication Date(Web):
DOI:10.1002/anie.201404802
Abstract
The first total syntheses of the proposed structure of cycloinumakiol (1) and its C5 epimer (18) are achieved in a concise and efficient fashion. Starting from the known 3-hydroxybenzocyclobutenone, 1 and 18 are obtained in nine and five steps with overall yields of 15 % and 33 %, respectively. The key for the success of this approach is the use of a catalytic CC activation strategy for constructing the tetracyclic core of 1 through carboacylation of a sterically hindered trisubstituted olefin with benzocyclobutenone. In addition, the structure of the natural cycloinumakiol was reassigned to 19-hydroxytotarol (7) through X-ray diffraction analysis. This work demonstrates the potential of CC activation for streamlining complex natural product synthesis.
Co-reporter:P.-H. Chen, Nikolas A. Savage, Guangbin Dong
Tetrahedron 2014 70(27–28) pp: 4135-4146
Publication Date(Web):
DOI:10.1016/j.tet.2014.03.080
Co-reporter:Samuel J. Thompson and Guangbin Dong
Organometallics 2014 Volume 33(Issue 14) pp:3757-3767
Publication Date(Web):July 7, 2014
DOI:10.1021/om500438s
Alkylation of rhodium(III) porphyrins [RhIII(por)] was achieved under relatively mild conditions in up to 98% yields, where readily available ammonium and quinolinium salts were utilized as the alkylating agents. This transformation tolerates air and water, thus serving as a convenient method to prepare a variety of alkyl- and benzyl-RhIII(por) complexes. Preliminary mechanistic studies support an SN2-like reaction pathway involving a RhI(por) anion intermediate.
Co-reporter:Dr. Tao Xu;Nikolas A. Savage ;Dr. Guangbin Dong
Angewandte Chemie 2014 Volume 126( Issue 7) pp:1922-1926
Publication Date(Web):
DOI:10.1002/ange.201310149
Abstract
The rhodium-catalyzed formation of all-carbon spirocenters involves a decarbonylative coupling of trisubstituted cyclic olefins and benzocyclobutenones through CC activation. The metal–ligand combination [{Rh(CO)2Cl}2]/P(C6F5)3 catalyzed this transformation most efficiently. A range of diverse spirocycles were synthesized in good to excellent yields and many sensitive functional groups were tolerated. A mechanistic study supports a hydrogen-transfer process that occurs through a β-H elimination/decarbonylation pathway.
Co-reporter:Fanyang Mo
Science 2014 Volume 345(Issue 6192) pp:68-72
Publication Date(Web):04 Jul 2014
DOI:10.1126/science.1254465
Carbon-carbon bonds without byproducts
Environmental and cost concerns are spurring development of chemical methods that minimize byproduct formation. In this vein, Mo and Dong present a catalyst that inserts olefins such as ethylene directly into the C-H bonds of ketones. Traditional methods to form such products rely on the preliminary reaction of the ketone with a base, followed by subsequent reaction with an alkyl halide. The authors used a ligand that simultaneously activates the ketone and guides the catalytic rhodium to the right location. This approach removes the need for the other reagents and eliminates the associated halide salt byproducts.
Science, this issue p. 68
Co-reporter:Zhongxing Huang
Journal of the American Chemical Society 2013 Volume 135(Issue 47) pp:17747-17750
Publication Date(Web):November 15, 2013
DOI:10.1021/ja410389a
Herein we report a direct β-arylation of simple ketones with widely available aryl iodides, combining palladium-catalyzed ketone oxidation, aryl-halide activation, and conjugate addition through a single catalytic cycle. Simple cyclic ketones with different ring-sizes, as well as acyclic ketones, can be directly arylated at the β-position with complete site-selectivity and excellent functional group tolerance.
Co-reporter:Zhe Dong
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18350-18353
Publication Date(Web):November 20, 2013
DOI:10.1021/ja410823e
A Pd and norbornene-catalyzed ortho-arene amination via Catellani-type C–H functionalization is reported. Aryl halides are used as substrates; N-benzoyloxyamines and isopropanol are employed as the amine source (oxidant) and reductant respectively. Examples are provided in 50–99% yields with high functional group tolerance. This method gives complementary site selectivity at the ortho- instead of ipso-position of aryl halides.
Co-reporter:Alpay Dermenci, Rachel E. Whittaker, and Guangbin Dong
Organic Letters 2013 Volume 15(Issue 9) pp:2242-2245
Publication Date(Web):April 15, 2013
DOI:10.1021/ol400815y
Utilization of C–C bond activation as a unique mode of reactivity for constructing C–C bonds provides new strategies for preparing important organic molecules. Development of a Rh(I)-catalyzed C–C activation of diynones to synthesize symmetrical and unsymmetrical conjugated diynes through decarbonylation is reported. This C–C cleavage strategy takes advantage of the innate reactivity of conjugated ynones without relying on any ring strain or auxiliary directing group. This alkynation method also has orthogonal properties compared to typical cross-coupling reactions.
Co-reporter:Fanyang Mo, Guangbin Dong, Yan Zhang and Jianbo Wang
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 10) pp:1582-1593
Publication Date(Web):11 Jan 2013
DOI:10.1039/C3OB27366K
Arene diazonium salts are common, easily prepared and highly useful intermediates in organic synthesis due to their rich reactivity and diverse transformations. In this review, recent advances involving arene diazonium salts as starting materials or active intermediates for various synthetically useful applications are summarized.
Co-reporter:Alpay Dermenci
Science China Chemistry 2013 Volume 56( Issue 6) pp:685-701
Publication Date(Web):2013 June
DOI:10.1007/s11426-012-4791-7
New methods for carbon-carbon (C-C) forming reactions are constantly emerging in the field of organic synthesis. In this review, a brief history followed by recent developments of decarbonylative C-C forming reactions mediated by transition metals is described. Many different substrates are presented and the review is organized by the different carbonyl precursors, such as acyl chlorides, aldehydes, anhydrides, esters and ketones, used in the respective transformations. Furthermore, the broad scope of these reactions is exhibited by the application to several reaction types (e.g. Heck-type reactions, Suzuki cross-coupling type reactions, C-H activation, etc.) as well as a natural product synthesis (e.g. muscroride A). While several examples are provided, this review marks the beginning of a new field that is still in its infancy and for what might be a new approach to achieve highly efficient reactions that come closer to meeting the standards of chemical economies (e.g. atom, redox, step, etc.) and green chemistry.
Co-reporter:Zhi Ren ; Fanyang Mo
Journal of the American Chemical Society 2012 Volume 134(Issue 41) pp:16991-16994
Publication Date(Web):October 2, 2012
DOI:10.1021/ja3082186
We describe a Pd-catalyzed site-selective functionalization of unactivated aliphatic C–H bonds, providing chemically differentiated 1,2-diols from monoalcohol derivatives. The oxime was employed as both a directing group (DG) and an alcohol surrogate for this transformation. As demonstrated in a range of substrates, the C–H bonds β to the oxime group are selectively oxidized. Besides activation of the methyl groups, methylene groups (CH2) in cyclic substrates and methine groups (CH) at bridge-head positions can also be functionalized. In addition, an intriguing oxidative skeleton rearrangement was observed using the menthol-derived substrate. The use of exo-directing groups in C–H activation, as illustrated in this work, would potentially open doors for the discovery of new transformations and new cleavable DGs.
Co-reporter:Tao Xu ; Haye Min Ko ; Nikolas A. Savage
Journal of the American Chemical Society 2012 Volume 134(Issue 49) pp:20005-20008
Publication Date(Web):November 21, 2012
DOI:10.1021/ja309978c
Here we report the first highly enantioselective Rh-catalyzed carboacylation of olefins via C–C bond activation of benzocyclobutenones. Good yields and excellent enantioselectivities (92–99% ee, 14 examples) were obtained for substrates with various steric and electronic properties. In addition, fully saturated poly-fused rings were prepared from the carboacylation products through a challenging catalytic reductive dearomatization approach. These investigations provide a distinct way to prepare chiral carbon frameworks that are nontrivial to access with conventional methods.
Co-reporter:Dr. Fanyang Mo;Louis J. Trzepkowski ;Dr. Guangbin Dong
Angewandte Chemie 2012 Volume 124( Issue 52) pp:13252-13256
Publication Date(Web):
DOI:10.1002/ange.201207479
Co-reporter:Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2012 Volume 51( Issue 30) pp:7567-7571
Publication Date(Web):
DOI:10.1002/anie.201202771
Co-reporter:Dr. Fanyang Mo;Louis J. Trzepkowski ;Dr. Guangbin Dong
Angewandte Chemie International Edition 2012 Volume 51( Issue 52) pp:13075-13079
Publication Date(Web):
DOI:10.1002/anie.201207479
Co-reporter:Dr. Tao Xu ;Dr. Guangbin Dong
Angewandte Chemie 2012 Volume 124( Issue 30) pp:7685-7689
Publication Date(Web):
DOI:10.1002/ange.201202771
Co-reporter:Zhiqian Wang ; Brandon J. Reinus
Journal of the American Chemical Society () pp:
Publication Date(Web):2017-2-22
DOI:10.1021/ja306123m
We describe the first example of Rh-catalyzed intermolecular C-alkylation of cyclic 1,2-diketones using simple terminal olefins as alkylating agents. Aminopyridine is employed as a recyclable directing group. First, it reacts with ketones to give enamines and delivers Rh to activate the vinyl C–H bonds in the same pot; second, it can be cleaved off and recovered via hydrolysis. A broad range of olefins can be utilized as substrates, including aliphatic, aromatic olefins and vinyl esters. The efficiency of this method is also demonstrated in the synthesis of a natural flavoring compound, 3-ethyl-5-methyl-1,2-cyclopentadione (one-pot 53% yield vs a previous four-step route 16% yield from the same starting material). This work is expected to serve as a seminal study toward catalytic ketone α-alkylation with unactivated olefins.
Co-reporter:Fanyang Mo, Guangbin Dong, Yan Zhang and Jianbo Wang
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 10) pp:NaN1593-1593
Publication Date(Web):2013/01/11
DOI:10.1039/C3OB27366K
Arene diazonium salts are common, easily prepared and highly useful intermediates in organic synthesis due to their rich reactivity and diverse transformations. In this review, recent advances involving arene diazonium salts as starting materials or active intermediates for various synthetically useful applications are summarized.
Co-reporter:Zhongxing Huang, Hee Nam Lim, Fanyang Mo, Michael C. Young and Guangbin Dong
Chemical Society Reviews 2015 - vol. 44(Issue 21) pp:NaN7786-7786
Publication Date(Web):2015/07/17
DOI:10.1039/C5CS00272A
Transition metal-catalyzed C–H functionalization has evolved into a prominent and indispensable tool in organic synthesis. While nitrogen, phosphorus and sulfur-based functional groups (FGs) are widely employed as effective directing groups (DGs) to control the site-selectivity of C–H activation, the use of common FGs (e.g. ketone, alcohol and amine) as DGs has been continuously pursued. Ketones are an especially attractive choice of DGs and substrates due to their prevalence in various molecules and versatile reactivity as synthetic intermediates. Over the last two decades, transition metal-catalyzed C–H functionalization that is directed or mediated by ketones has experienced vigorous growth. This review summarizes these advancements into three major categories: use of ketone carbonyls as DGs, direct β-functionalization, and α-alkylation/alkenylation with unactivated olefins and alkynes. Each of these subsections is discussed from the perspective of strategic design and reaction discovery.
Co-reporter:Alpay Dermenci, Jotham W. Coe and Guangbin Dong
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 5) pp:NaN581-581
Publication Date(Web):2014/03/27
DOI:10.1039/C4QO00053F
New modes of chemical reactivity are of high value to synthetic organic chemistry. In this vein, carbon–carbon (C–C) activation is an emerging field that offers new possibilities for synthesizing valuable complex molecules. This review discusses the pioneering stoichiometric discoveries in this field up to the most recent synthetic applications that apply catalytic transformations. Specifically, the review focuses on C–C activation in relatively unstrained systems, including stoichiometric reactions, chelation-directed and chelation-free catalytic reactions. While the field of C–C activation of relatively unstrained systems is underdeveloped, we expect that this review will provide insight into new developments and pave the path for robust, practical applications.









![Cyclobutanone, 3-[2-(1-methylethenyl)phenyl]-](http://img.cochemist.com/ccimg/918300/918299-10-2.png)
![Cyclobutanone, 3-[2-(1-methylethenyl)phenyl]-](http://img.cochemist.com/ccimg/918300/918299-10-2_b.png)







![PYRIDINE, 3-[(4-FLUOROPHENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/866700/866684-98-2.png)
![PYRIDINE, 3-[(4-FLUOROPHENYL)ETHYNYL]-](http://img.cochemist.com/ccimg/866700/866684-98-2_b.png)






![2-Propyn-1-one, 3-phenyl-1-[4-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/850000/849941-88-4.png)
![2-Propyn-1-one, 3-phenyl-1-[4-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/850000/849941-88-4_b.png)

![Pyridine, 3-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/817600/817574-43-9.png)
![Pyridine, 3-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/817600/817574-43-9_b.png)


![2-Propyn-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/742700/742699-28-1.png)
![2-Propyn-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/742700/742699-28-1_b.png)
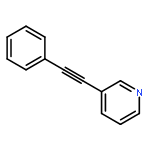
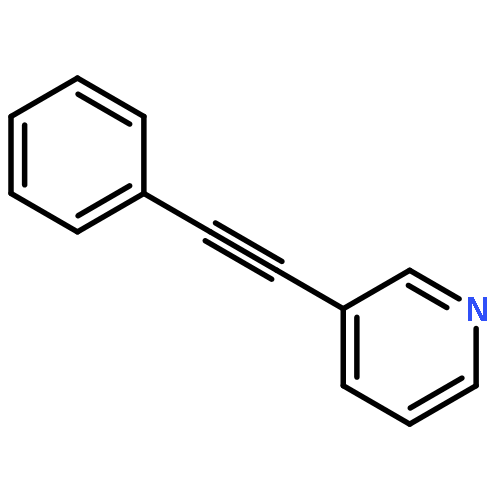
![Pyridine, 3-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/733100/733035-88-6.png)
![Pyridine, 3-[(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/733100/733035-88-6_b.png)




![Pyridine, 3-[(4-chlorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/676600/676562-55-3.png)
![Pyridine, 3-[(4-chlorophenyl)ethynyl]-](http://img.cochemist.com/ccimg/676600/676562-55-3_b.png)

















![2-BROMO-5-[TERT-BUTYL(DIMETHYL)SILYL]OXYBENZALDEHYDE](http://img.cochemist.com/ccimg/351500/351418-50-3.png)
![2-BROMO-5-[TERT-BUTYL(DIMETHYL)SILYL]OXYBENZALDEHYDE](http://img.cochemist.com/ccimg/351500/351418-50-3_b.png)

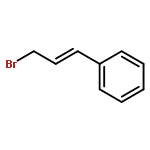
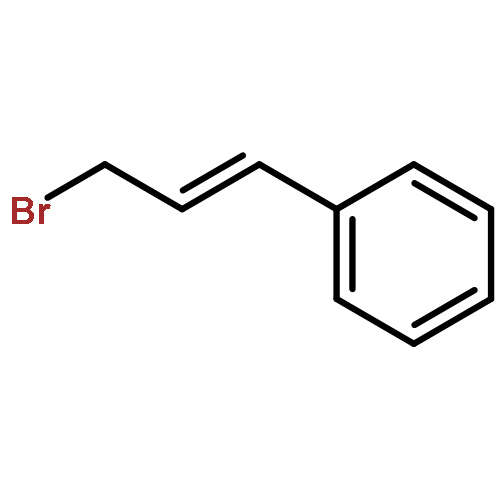
![Benzene, [[[2-(bromomethyl)-2-propenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/347900/347886-00-4.png)
![Benzene, [[[2-(bromomethyl)-2-propenyl]oxy]methyl]-](http://img.cochemist.com/ccimg/347900/347886-00-4_b.png)





![[1,1'-Biphenyl]-4-amine,3-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/275800/275795-17-0.png)
![[1,1'-Biphenyl]-4-amine,3-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/275800/275795-17-0_b.png)


















![Benzenesulfonamide, N-[(4-methoxyphenyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/171500/171414-16-7.png)
![Benzenesulfonamide, N-[(4-methoxyphenyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/171500/171414-16-7_b.png)







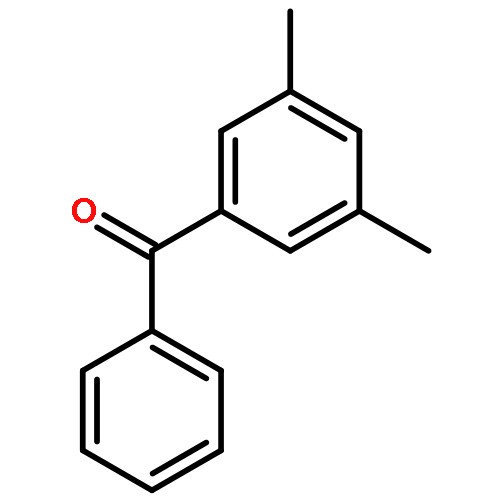
![Benzene, 1,3,5-tris[[3,5-bis(1,1-dimethylethyl)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/155100/155064-29-2.png)
![Benzene, 1,3,5-tris[[3,5-bis(1,1-dimethylethyl)phenyl]ethynyl]-](http://img.cochemist.com/ccimg/155100/155064-29-2_b.png)








![Methyl 4'-formyl-[1,1'-biphenyl]-2-carboxylate](http://img.cochemist.com/ccimg/144300/144291-47-4.png)
![Methyl 4'-formyl-[1,1'-biphenyl]-2-carboxylate](http://img.cochemist.com/ccimg/144300/144291-47-4_b.png)



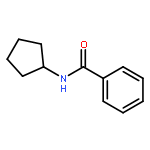
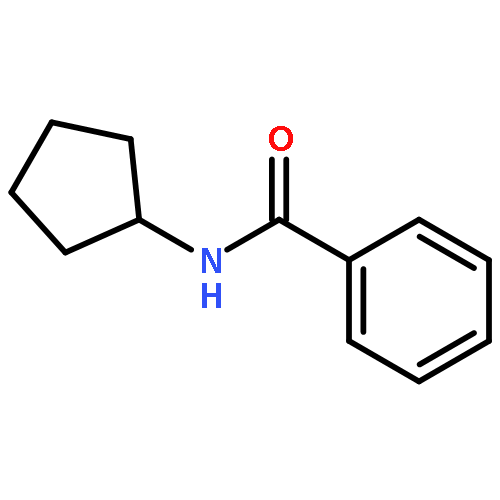
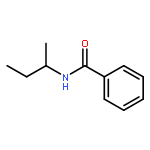
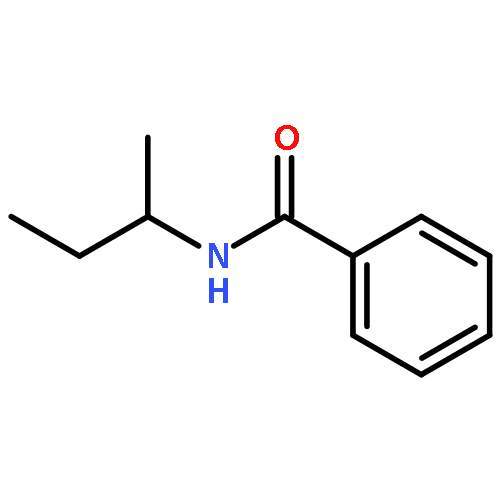
![Benzamide, N-[3-(4-methoxyphenyl)-1-methylpropyl]-](http://img.cochemist.com/ccimg/139000/138963-89-0.png)
![Benzamide, N-[3-(4-methoxyphenyl)-1-methylpropyl]-](http://img.cochemist.com/ccimg/139000/138963-89-0_b.png)
![Benzamide, N-[1-methyl-3-(4-methylphenyl)propyl]-](http://img.cochemist.com/ccimg/139000/138963-87-8.png)
![Benzamide, N-[1-methyl-3-(4-methylphenyl)propyl]-](http://img.cochemist.com/ccimg/139000/138963-87-8_b.png)


![2-Hexanol, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/138700/138611-32-2.png)
![2-Hexanol, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/138700/138611-32-2_b.png)


![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-phenyl-](http://img.cochemist.com/ccimg/134900/134810-65-4.png)
![2-Butanone, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4-phenyl-](http://img.cochemist.com/ccimg/134900/134810-65-4_b.png)
![2-Tosyl-2,3,3a,4-tetrahydrocyclopenta[c]pyrrol-5(1H)-one](http://img.cochemist.com/ccimg/133900/133886-43-8.png)
![2-Tosyl-2,3,3a,4-tetrahydrocyclopenta[c]pyrrol-5(1H)-one](http://img.cochemist.com/ccimg/133900/133886-43-8_b.png)
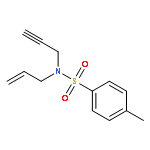
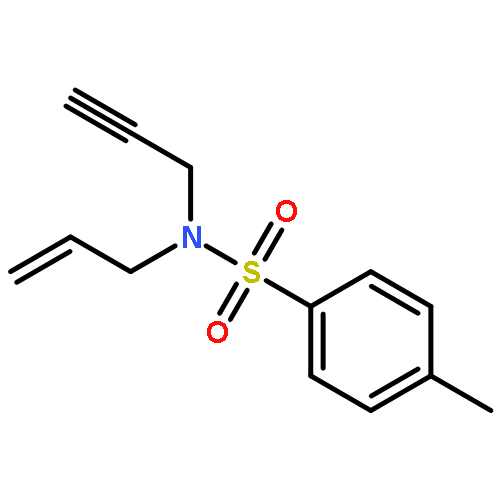


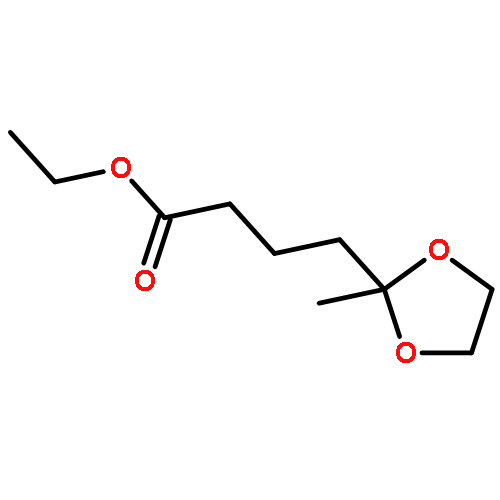
![1-Oxa-8-azaspiro[4.5]decane-8-carboxylic acid, 3-hydroxy-, ethyl ester](http://img.cochemist.com/ccimg/133400/133382-30-6.png)
![1-Oxa-8-azaspiro[4.5]decane-8-carboxylic acid, 3-hydroxy-, ethyl ester](http://img.cochemist.com/ccimg/133400/133382-30-6_b.png)


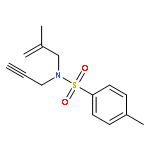
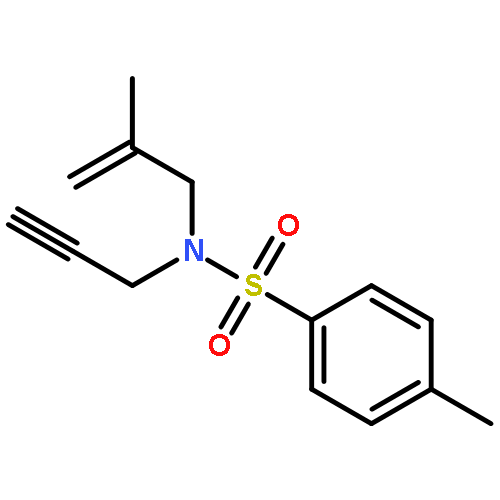



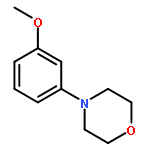
![Pyrrolidine, 1-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/122500/122439-13-8.png)
![Pyrrolidine, 1-[(3-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/122500/122439-13-8_b.png)










![Bicyclo[2.2.1]hepta-2,5-diene, 7-octyl-](http://img.cochemist.com/ccimg/108400/108380-53-6.png)
![Bicyclo[2.2.1]hepta-2,5-diene, 7-octyl-](http://img.cochemist.com/ccimg/108400/108380-53-6_b.png)




















![Benzoic acid, 3-[(dimethylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/90000/89999-70-2.png)
![Benzoic acid, 3-[(dimethylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/90000/89999-70-2_b.png)


![Silane, (1,1-dimethylethyl)[(1-methoxy-1-propenyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/89400/89337-61-1.png)
![Silane, (1,1-dimethylethyl)[(1-methoxy-1-propenyl)oxy]dimethyl-](http://img.cochemist.com/ccimg/89400/89337-61-1_b.png)


![3-Azabicyclo[3.2.1]octane, 1-methyl-](http://img.cochemist.com/ccimg/88800/88799-00-2.png)
![3-Azabicyclo[3.2.1]octane, 1-methyl-](http://img.cochemist.com/ccimg/88800/88799-00-2_b.png)
























![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 8-methyl-](http://img.cochemist.com/ccimg/69000/68913-17-7.png)
![Bicyclo[4.2.0]octa-1,3,5-trien-7-one, 8-methyl-](http://img.cochemist.com/ccimg/69000/68913-17-7_b.png)







![BICYCLO[2.2.1]HEPTA-2,5-DIENE-7-METHANOL](http://img.cochemist.com/ccimg/63700/63603-20-3.png)
![BICYCLO[2.2.1]HEPTA-2,5-DIENE-7-METHANOL](http://img.cochemist.com/ccimg/63700/63603-20-3_b.png)




![1-(Benzo[d][1,3]dioxol-5-yl)-N,N-dimethylmethanamine](http://img.cochemist.com/ccimg/59000/58995-64-5.png)
![1-(Benzo[d][1,3]dioxol-5-yl)-N,N-dimethylmethanamine](http://img.cochemist.com/ccimg/59000/58995-64-5_b.png)
























![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0.png)
![Benzaldehyde, 2-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/52100/52095-44-0_b.png)









![Bicyclo[2.2.1]hepta-2,5-diene,7-phenyl-](http://img.cochemist.com/ccimg/40200/40156-12-5.png)
![Bicyclo[2.2.1]hepta-2,5-diene,7-phenyl-](http://img.cochemist.com/ccimg/40200/40156-12-5_b.png)


















































![heptacyclo[6.6.0.0~2,6~.0~3,13~.0~4,11~.0~5,9~.0~10,14~]tetradecane (non-preferred name)](http://img.cochemist.com/ccimg/17900/17872-39-8.png)
![heptacyclo[6.6.0.0~2,6~.0~3,13~.0~4,11~.0~5,9~.0~10,14~]tetradecane (non-preferred name)](http://img.cochemist.com/ccimg/17900/17872-39-8_b.png)






![[1,1'-Biphenyl]-2-carboxylicacid, 2'-formyl-, methyl ester](http://img.cochemist.com/ccimg/16300/16231-67-7.png)
![[1,1'-Biphenyl]-2-carboxylicacid, 2'-formyl-, methyl ester](http://img.cochemist.com/ccimg/16300/16231-67-7_b.png)




![Benzene, 1-methoxy-3-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14064-41-6.png)
![Benzene, 1-methoxy-3-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14064-41-6_b.png)









![Bicyclo[2.2.1]hept-5-ene-2-carboxamide, N-methyl-, endo- (9CI)](http://img.cochemist.com/ccimg/13300/13295-40-4.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxamide, N-methyl-, endo- (9CI)](http://img.cochemist.com/ccimg/13300/13295-40-4_b.png)




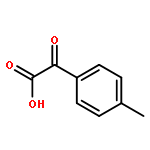
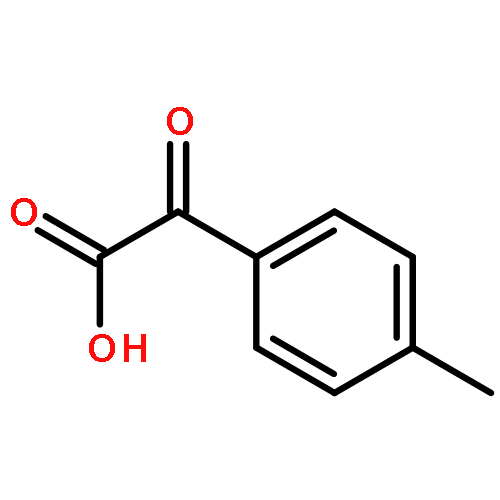
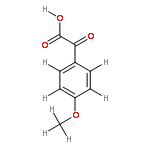
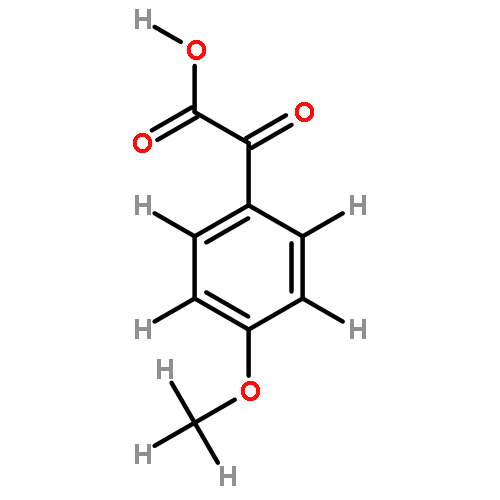










![BICYCLO[2.2.1]HEPTA-2,5-DIENE-7-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/5600/5597-66-0.png)
![BICYCLO[2.2.1]HEPTA-2,5-DIENE-7-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/5600/5597-66-0_b.png)



























![5-(Bromomethyl)benzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/2700/2606-51-1.png)
![5-(Bromomethyl)benzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/2700/2606-51-1_b.png)

















![(1R,2R,4R)-Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/1200/1195-12-6.png)
![(1R,2R,4R)-Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/1200/1195-12-6_b.png)





![Bicyclo[2.2.1]hepta-2,5-dien-7-ol](http://img.cochemist.com/ccimg/900/822-80-0.png)
![Bicyclo[2.2.1]hepta-2,5-dien-7-ol](http://img.cochemist.com/ccimg/900/822-80-0_b.png)
![N,N-DIMETHYL-1-[3-(TRIFLUOROMETHYL)PHENYL]METHANAMINE](http://img.cochemist.com/ccimg/800/713-93-9.png)
![N,N-DIMETHYL-1-[3-(TRIFLUOROMETHYL)PHENYL]METHANAMINE](http://img.cochemist.com/ccimg/800/713-93-9_b.png)




![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8.png)
![Tetracyclo[3.2.0.02,7.04,6]heptane](http://img.cochemist.com/ccimg/300/278-06-8_b.png)


![Cyclobutanone, 3-methyl-3-[2-(3-methyl-1,2-butadien-1-yl)phenyl]-](/data/chemimg/3722300/1817683-28-5.png)
![Cyclobutanone, 3-methyl-3-[2-(3-methyl-1,2-butadien-1-yl)phenyl]-](/data/chemimg/3722300/1817683-28-5_b.png)



![Tris[3,5-bis(trifluoromethyl)phenyl]phosphine](http://img.cochemist.com/ccimg/175200/175136-62-6.png)
![Tris[3,5-bis(trifluoromethyl)phenyl]phosphine](http://img.cochemist.com/ccimg/175200/175136-62-6_b.png)

![2-Butanol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/105000/104925-50-0.png)
![2-Butanol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/105000/104925-50-0_b.png)









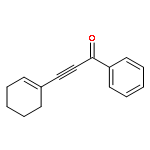

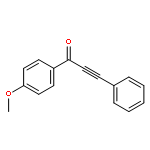
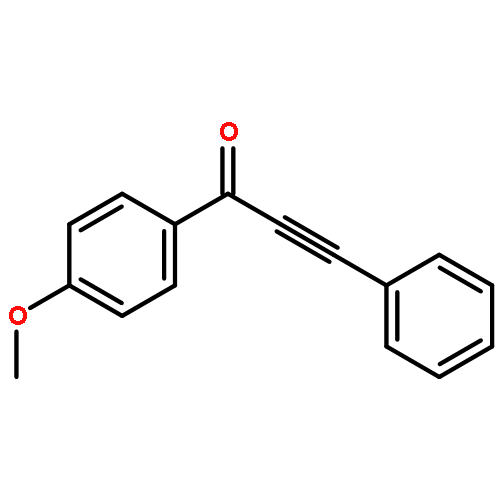
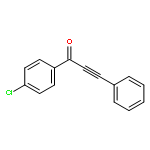









![Benzenesulfonamide, N-[(4-methoxyphenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/54900/54879-64-0.png)
![Benzenesulfonamide, N-[(4-methoxyphenyl)methyl]-4-methyl-](http://img.cochemist.com/ccimg/54900/54879-64-0_b.png)













































































































































































































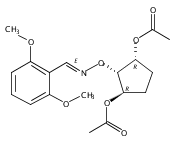







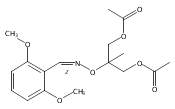











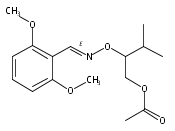
































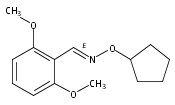




























































































































































![Silane, (1,1-dimethylethyl)[(2-iodophenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/271800/271769-81-4.png)
![Silane, (1,1-dimethylethyl)[(2-iodophenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/271800/271769-81-4_b.png)




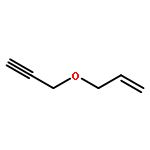
![Benzene, 1-[(3-butynyloxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/218500/218434-90-3.png)
![Benzene, 1-[(3-butynyloxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/218500/218434-90-3_b.png)































![Benzene, [4-(1-cyclohexen-1-yl)-1,3-butadiynyl]-](http://img.cochemist.com/ccimg/105900/105850-88-2.png)
![Benzene, [4-(1-cyclohexen-1-yl)-1,3-butadiynyl]-](http://img.cochemist.com/ccimg/105900/105850-88-2_b.png)




















![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5.png)
![1-FLUORO-4-[4-(4-FLUOROPHENYL)BUTA-1,3-DIYNYL]BENZENE](http://img.cochemist.com/ccimg/55700/55606-94-5_b.png)































![Morpholine, 4-[(4-iodophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/23000/22950-14-7.png)
![Morpholine, 4-[(4-iodophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/23000/22950-14-7_b.png)















![3-BROMO-5-[2-(TRIFLUOROMETHYL)PHENYL]PYRIDINE](http://img.cochemist.com/ccimg/7100/7025-91-4.png)
![3-BROMO-5-[2-(TRIFLUOROMETHYL)PHENYL]PYRIDINE](http://img.cochemist.com/ccimg/7100/7025-91-4_b.png)














![Bicyclo[2.2.1]heptan-1-ol](http://img.cochemist.com/ccimg/500/497-36-9.png)
![Bicyclo[2.2.1]heptan-1-ol](http://img.cochemist.com/ccimg/500/497-36-9_b.png)
































![1H-Indole, 5-iodo-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/110600/174715-24-3.png)
![1H-Indole, 5-iodo-1-[(4-methylphenyl)sulfonyl]-](/data/chemimg/110600/174715-24-3_b.png)