Co-reporter:Qiu Li, Hai-Kui Yang, Qi Sun, Wen-Wei You, Pei-Liang Zhao
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmcl.2017.07.076
Based on our previous work, a series of novel 2-amino-7,8-dihydropteridin-6(5H)-one derivatives were designed and synthesized via a ring-closing strategy. Biological evaluation with four human cancer cell lines (BT549, T47D, MDA-MB-468, and MDA-MB-231) showed that most of these compounds possessed moderate to potent antiproliferative activities. The most promising compound 8-benzyl-2-(phenethylamino)-7,8-dihydropteridin-6(5H)-one (6q) possessing IC50 values of 7.75, 6.37, and 10.73 μM against MDA-MB-468, T47D, and BT549, respectively, which were 49, 11, and 8 folds more active than the positive control fluorouracil. Moreover, fluorescence-activated cell sorting analysis revealed that compound 6q displayed a significant effect on G1 cell-cycle arrest in a concentration-dependent manner in T47D cells. The initial structure–activity relationship studies indicated that linker-length of amine chain in C-2 position of pyrimidine ring played a crucial role in modulating the antitumor activity, which could be of help in the rational design of dihydropteridin-6(5H)-ones as novel anticancer drugs.Download high-res image (104KB)Download full-size image
Co-reporter:Peng-Cheng Diao, Qiu Li, Meng-Jin Hu, Yu-Feng Ma, Wen-Wei You, Kwon Ho Hong, Pei-Liang Zhao
European Journal of Medicinal Chemistry 2017 Volume 134(Volume 134) pp:
Publication Date(Web):7 July 2017
DOI:10.1016/j.ejmech.2017.04.011
•22 novel indole-pyrimidine hybrids were designed and synthesized.•Antiproliferative activities of these compounds were evaluated.•Compound 14 arrested HeLa cells in G2/M phase of cell cycle.•Hybrid 15 exhibited powerful tubulin inhibitory activity.•Molecular modeling suggested that 15 binds well in the colchicine binding site of α,β-tubulin.Based on our previous screening hit compound 1, a series of novel indole-pyrimidine hybrids possessing morpholine or thiomorpholine moiety were synthesized via an efficient one-pot multistep synthetic method. The antiproliferative activities of the synthesized compounds were evaluated in vitro against four cancer cell lines including HeLa, MDA-MB-231, MCF-7, and HCT116. The results revealed that most compounds possessed moderate to excellent potency. The IC50 values of the most promising compound 15 are 0.29, 4.04, and 9.48 μM against MCF-7, HeLa, and HCT116 cell lines, respectively, which are 48.0, 4.9, and 1.8 folds more active than the lead compound 1. Moreover, fluorescence-activated cell sorting analysis revealed that compound 14 showing the highest activity against HeLa (IC50 = 2.51 μM) displayed a significant effect on G2/M cell-cycle arrest in a concentration-dependent manner in HeLa cell line. In addition, representative nine active hybrids were evaluated for tubulin polymerization inhibitory activities, and compound 15 exhibited the most potent anti-tubulin activity showing 42% inhibition at 10 μM. These preliminary results encourage a further investigation on indole-pyrimidine hybrids for the development of potent anticancer agents that inhibit tubulin polymerization.Download high-res image (239KB)Download full-size image
Co-reporter:Pei-Liang Zhao, Yan-Hong Li, Hai-Kui Yang, Peng Chen, Bei Zhang, Qi Sun, Qiu Li, Wen-Wei You
European Journal of Medicinal Chemistry 2016 Volume 118() pp:161-169
Publication Date(Web):8 August 2016
DOI:10.1016/j.ejmech.2016.04.038
•23 novel pyrimidine derivatives were designed and synthesized via a ring-opening strategy.•Antiproliferative activity of these compounds was evaluated.•Compound 7w exhibited much stronger antitumor activity than fluorouracil.•Flow-activated cell sorting analysis suggested that compound 7w mainly arrested HepG2 cells in G2/M stage.In order to discover new anticancer drug leads, a series of novel alkylamino pyrimidine derivatives were designed and synthesized based on our previous work via a ring-opening strategy. Biological evaluation with four human cancer cell lines (MDA-MB-231, A549, HepG2, and MCF-7) showed that most of these compounds possessed moderate to potent antiproliferative activities. The most promising compound 7w displayed a three-fold improvement compared with commercial anticancer drug fluorouracil in inhibiting HepG2 cell proliferation with IC50 value of 10.37 μM. Moreover, flow-activated cell sorting analysis suggested that compound 7w mainly arrested HepG2 cells in G2/M stage. Hence, it could serve as a promising lead for the design of novel anticancer small-molecule drugs.A series of novel alkylamino pyrimidine derivatives were designed and synthesized based on our previous work via a ring-opening strategy. Biological evaluation showed that some compounds possessed potent antiproliferative activities.
Co-reporter:Pei-Liang Zhao, Peng Chen, Qiu Li, Meng-Jin Hu, Peng-Cheng Diao, En-Shan Pan, Wen-Wei You
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3679-3683
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.05.086
Based on our previous work, a series of novel 3-alkylsulfanyl-4-amino-1,2,4-triazole derivatives were designed, synthesized and evaluated for their antiproliferative activities. The results indicated that some compounds possessed significant antiproliferative activities against four cancer cell lines, HepG2, HCT116, PC-3, and Hela. Particularly, the most promising compound 8d displayed 184-, 18-, and 17-fold improvement compared to fluorouracil in inhibiting HCT116, Hela and PC-3 cell proliferation with IC50 values of 0.37, 2.94, and 31.31 μM, respectively. Most interestingly, the compound did not affect the normal human embryonic kidney cells, HEK-293. Moreover, mechanistic investigation showed that the representative compound 8d induced apoptosis and blocked cell cycle in G2/M phase in Hela cells in a dose-dependent manner. These findings suggest that compound 8d may have potential to be developed as a promising lead for the design of novel anticancer small-molecule drugs.
Co-reporter:Bei Zhang, Yan-Hong Li, Yang Liu, Yu-Rong Chen, En-Shan Pan, Wen-Wei You, Pei-Liang Zhao
European Journal of Medicinal Chemistry 2015 Volume 103() pp:335-342
Publication Date(Web):20 October 2015
DOI:10.1016/j.ejmech.2015.08.053
•26 novel 1,2,4-triazolo [3,4-b][1,3,4] thiadiazines bearing furan and thiophene nucleus were synthesized.•Antiproliferative activity of these compounds was evaluated.•The most promising compound 32 displayed much stronger antitumor activity than fluorouracil.•Compound 32 displayed a significant effect on G2/M cell-cycle arrest in a dose-dependent manner in PC-3 cells.Twenty-six novel 1,2,4-triazolo [3,4-b][1,3,4] thiadiazines containing furan and thiophene nucleus were designed, synthesized and evaluated for their antiproliferative activities. The results indicated that most of the compounds showed moderate to potent antiproliferative activities against four cancer cell lines, PC-3, HepG2, A549, and MCF-7. Particularly, compound 32 showed eleven-, three-, and two-fold improvement compared to positive control fluorouracil in inhibiting HepG2, PC-3, and A549 cell proliferation with IC50 values of 5.09, 3.70 and 12.74 μM, respectively. Further flow-activated cell sorting analysis revealed that the most promising compound 32 displayed a significant effect on G2/M cell-cycle arrest in a dose-dependent manner in PC-3 cells. These encouraging results should provide important information for the development of new anticancer agents.Compound 32 exhibited eleven-, three-, and two-fold improvement compared to fluorouracil in inhibiting HepG2, PC-3, and A549 cell proliferation with IC50 values of 5.09, 3.70 and 12.74 μM, respectively.
Co-reporter:Meng-Jin Hu;Bei Zhang;Hai-Kui Yang;Yang Liu;Yu-Rong Chen;Tian-Zhu Ma;Ling Lu;Wen-Wei You
Chemical Biology & Drug Design 2015 Volume 86( Issue 6) pp:1491-1500
Publication Date(Web):
DOI:10.1111/cbdd.12616
A series of novel hybrids of indole–pyrimidine-containing piperazine moiety were designed, synthesized and evaluated for their antiproliferative and tubulin polymerization inhibitory activities. The results indicated that most of these compounds possessed significant cytotoxic potency against four cancer cell lines, HT-29, A549, MDA-MB-231 and MCF-7. Particularly, the most promising compound 34 showed more potent and broad-spectrum cytotoxic activities with the IC50 values ranged from 5.01 to 14.36 μm against A549, MDA-MB-231 and MCF-7 cell lines. Meanwhile, 34 also displayed the most potent tubulin polymerization inhibitory activity with IC50 value of 11.2 μm. Furthermore, molecular docking analysis demonstrated 34 interacts and binds efficiently with the tubulin protein at the colchicine-binding site. It was worth noting that the compound did not affect the normal human embryonic kidney cells, HEK-293. These results suggest that this novel class of indole–pyrimidine hybrids may have potential to be developed as new a class of tubulin polymerization inhibitors.
Co-reporter:Wei-Feng Ma, Hai-Kui Yang, Meng-Jin Hu, Qian Li, Tian-Zhu Ma, Zhong-Zhen Zhou, Rui-Yuan Liu, Wen-Wei You, Pei-Liang Zhao
European Journal of Medicinal Chemistry 2014 Volume 84() pp:127-134
Publication Date(Web):12 September 2014
DOI:10.1016/j.ejmech.2014.07.017
•24 novel 2,4-diaminopyrimidines were synthesized via a one-pot reaction.•Antiproliferative activity of these compounds was evaluated.•Two compounds displayed much stronger antitumor activity than Fluorouracil.•Compound 28 displayed a significant effect on G2/M cell-cycle arrest in MDA-MB-231.A series of novel 2,4-diaminopyrimidines containing piperidine and piperazine moieties were synthesized via an efficient one-pot methodology. The bioassay tests demonstrated that compounds 27 and 28 displayed much stronger antitumor activities against four human cancer cell lines (HepG2, A549, MDA-MB-231 and MCF-7) than positive control fluorouracil. Particularly, compound 28 showed a two-fold improvement compared to fluorouracil in inhibiting MDA-MB-231 and A549 cell proliferation with IC50 values of 7.46 and 12.78 μM, respectively. Further flow-activated cell sorting analysis revealed that the most promising compound 28 displayed a significant effect on G2/M cell-cycle arrest in a dose-dependent manner in MDA-MB-231 cells.

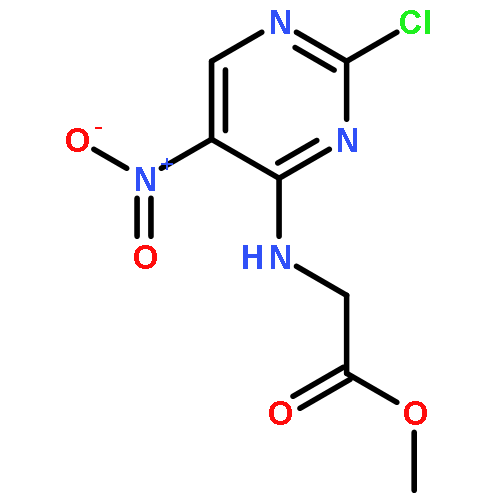
![Glycine, N-[2-[(4-chlorophenyl)amino]-5-nitro-4-pyrimidinyl]-, ethyl ester](http://img.cochemist.com/ccimg/514900/514836-83-0.png)
![Glycine, N-[2-[(4-chlorophenyl)amino]-5-nitro-4-pyrimidinyl]-, ethyl ester](http://img.cochemist.com/ccimg/514900/514836-83-0_b.png)
![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3.png)
![Acetamide,2-chloro-N-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/99600/99585-88-3_b.png)
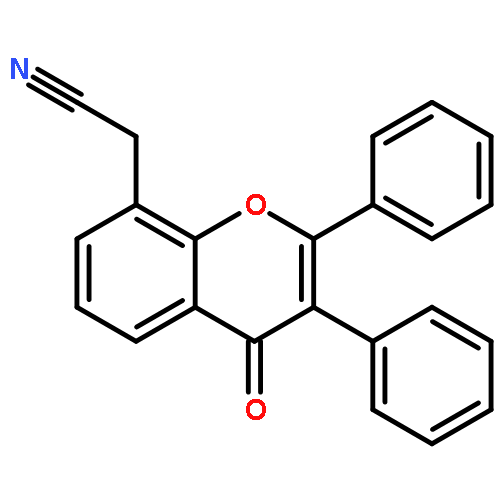
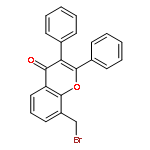
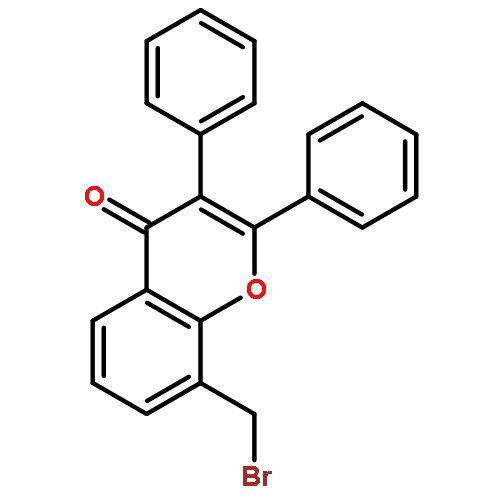
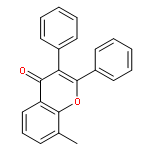
![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2.png)
![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2_b.png)
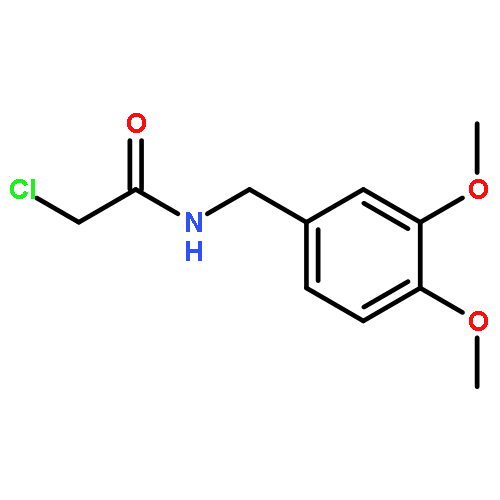
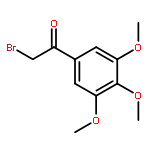

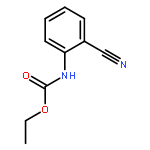
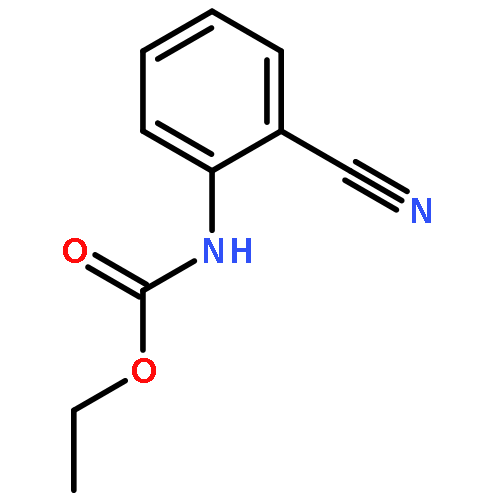



![2-CHLORO-N-[2-(4-CHLORO-PHENYL)-ETHYL]-ACETAMIDE](http://img.cochemist.com/ccimg/3900/3840-66-2.png)
![2-CHLORO-N-[2-(4-CHLORO-PHENYL)-ETHYL]-ACETAMIDE](http://img.cochemist.com/ccimg/3900/3840-66-2_b.png)