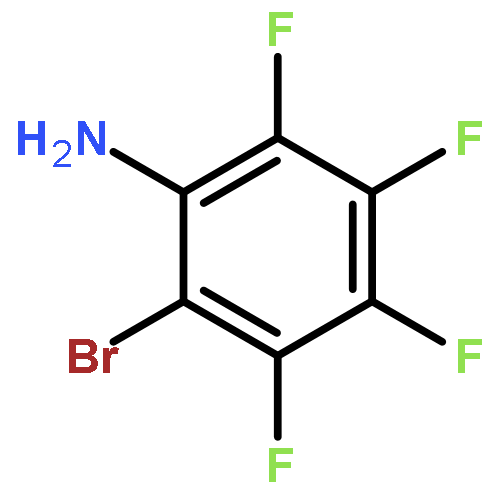
Co-reporter:Qianxiang Ai, Yulia A. Getmanenko, Karol Jarolimek, Raúl Castañeda, Tatiana V. Timofeeva, and Chad Risko
The Journal of Physical Chemistry Letters September 21, 2017 Volume 8(Issue 18) pp:4510-4510
Publication Date(Web):September 1, 2017
DOI:10.1021/acs.jpclett.7b01816
Mixed cocrystals derived from electron-rich donor (D) and electron-deficient acceptor (A) molecules showcase electronic, optical, and magnetic properties of interest for a wide range of applications. We explore the structural and electronic properties of a cocrystal synthesized from dithieno[3,2-a:2′,3′-c]phenazine (DTPhz) and 7,7,8,8-tetracyanoquinodimethane (TCNQ), which has a mixed-stack packing arrangement of the (π-electronic) face-to-face stacks in a 2:1 D:A stoichiometry. Density functional theory investigations reveal that the primary electronic characteristics of the cocrystal are not determined by electronic interactions along the face-to-face stacks, but rather they are characterized by stronger electronic interactions orthogonal to these stacks that follow the edge-to-edge donor–donor or acceptor–acceptor contacts. These distinctive electronic characteristics portend semiconducting properties that are unusual for semiconducting mixed cocrystals and suggest further potential to design organic semiconductors with orthogonal transport characteristics for different charge carriers.
Co-reporter:Karl J. Thorley, Tristan W. Finn, Karol Jarolimek, John E. Anthony, and Chad Risko
Chemistry of Materials March 28, 2017 Volume 29(Issue 6) pp:2502-2502
Publication Date(Web):November 21, 2016
DOI:10.1021/acs.chemmater.6b04211
The functionalization of oligoacenes and similar π-conjugated chromophores with trialkylsilylethynyl groups has proven to be a versatile means to enhance solubility and solution processability and engineer solid-state packing arrangements to produce organic semiconductors that demonstrate outstanding charge-carrier transport characteristics. While a general, empirical-based geometric model has been developed and implemented to direct the solid-state packing arrangements of these molecular materials, there exist numerous examples where the model falters. Here, we employ electronic structure methods to probe the noncovalent, intermolecular interactions of two closely related systems that result in two very different crystal packing configurations: triisopropylsilylethynyl (TIPS) pentacene and its triethylsilylethynyl (TES) analog. The quantum-chemical evaluation details how the slightly larger electron density contained within the volume of the TIPS moiety with respect to TES is in part responsible for the solid-state packing variations. We also make use of periodic density functional theory (DFT) methods to develop in silico polymorphs of these systems and explore the electronic characteristics of varied packing arrangements. The results suggest that TES pentacene, if processed correctly, could be developed into a material with improved charge-carrier transport characteristics when compared to its native form. Overall, the theory-driven insight developed in this work lays an important foundation to build a more robust crystal engineering paradigm for these technologically relevant organic semiconductors.
Co-reporter:Abby-Jo Payne;Shi Li;Sergey V. Dayneko;Gregory C. Welch
Chemical Communications 2017 vol. 53(Issue 76) pp:10608-10608
Publication Date(Web):2017/09/21
DOI:10.1039/C7CC90360J
Correction for ‘An unsymmetrical non-fullerene acceptor: synthesis via direct heteroarylation, self-assembly, and utility as a low energy absorber in organic photovoltaic cells’ by Abby-Jo Payne et al., Chem. Commun., 2017, 53, 10168–10171.
Co-reporter:Abby-Jo Payne;Shi Li;Sergey V. Dayneko;Gregory C. Welch
Chemical Communications 2017 vol. 53(Issue 73) pp:10168-10171
Publication Date(Web):2017/09/12
DOI:10.1039/C7CC05836E
This study reports on the design and synthesis of an unsymmetrical π-conjugated organic molecule composed of perylene diimide, thienyl diketopyrrolopyrrole, and indoloquinoxaline pieced together using direct heteroarylation. This material demonstrates unprecedented response in the thin-film upon post-deposition solvent vapor annealing, resulting in dramatic red-shifts in optical absorption. Such changes were utilized to enhance photocurrent generation in P3HT based organic solar cells.
Co-reporter:Christopher Sutton, Chad Risko, and Jean-Luc Brédas
Chemistry of Materials 2016 Volume 28(Issue 1) pp:3
Publication Date(Web):October 30, 2015
DOI:10.1021/acs.chemmater.5b03266
Noncovalent intermolecular interactions, which can be tuned through the toolbox of synthetic chemistry, determine not only the molecular packing but also the resulting electronic, optical, and mechanical properties of materials derived from π-conjugated molecules, oligomers, and polymers. Here, we provide an overview of the theoretical underpinnings of noncovalent intermolecular interactions and briefly discuss the computational chemistry approaches used to understand the magnitude of these interactions. These methodologies are then exploited to illustrate how noncovalent intermolecular interactions impact important electronic properties—such as the electronic coupling between adjacent molecules, a key parameter for charge-carrier transport—through a comparison between the prototype organic semiconductor pentacene with a series of N-substituted heteropentacenes. Incorporating an understanding of these interactions into the design of organic semiconductors can assist in developing novel materials systems from this fascinating molecular class.
Co-reporter:Sean M. Ryno, Chad Risko, and Jean-Luc Brédas
Chemistry of Materials 2016 Volume 28(Issue 11) pp:3990
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.chemmater.6b01340
Noncovalent interactions determine in large part the thermodynamic aspects of molecular packing in organic crystals. Using a combination of symmetry-adapted perturbation theory (SAPT) and classical multipole electrostatics, we describe the interaction potential energy surfaces for dimers of the oligoacene family, from benzene to hexacene, including up to 5000 configurations for each system. An analysis of these surfaces and a thorough assessment of dimers extracted from the reported crystal structures underline that high-order interactions (i.e., three-body nonadditive interactions) must be considered in order to rationalize the details of the crystal structures. A comparison of the SAPT electrostatic energy with the multipole interaction energy demonstrates the importance of the contribution of charge penetration, which is shown to account for up to 50% of the total interaction energy in dimers extracted from the experimental single crystals; in the case of the most stable cofacial model dimers, this contribution is even larger than the total interaction energy. Our results highlight the importance of taking account of charge penetration in studies of the larger oligoacenes.
Co-reporter:Karl J. Thorley and Chad Risko
Journal of Materials Chemistry A 2016 vol. 4(Issue 18) pp:4040-4048
Publication Date(Web):11 Mar 2016
DOI:10.1039/C5TC03900B
Many high performing organic semiconductor materials contain heteroaromatic rings in order to control the molecular packing and material electronic properties. Here we use a combination of density functional theory and symmetry-adapted perturbation theory calculations to explore the intermolecular noncovalent interactions, which guide solid-state molecular packing, and electronic couplings in a series of benzodithiophene-based dimer models. A novel concept, termed the disordermer, is introduced to delineate how the reduced molecular symmetry of benzodithiophene, when compared to the more highly symmetric anthracene molecule, can present intermolecular isomerism in the solid state that results in a wide range of available molecular packing arrangements that in turn influence the magnitudes of the electronic couplings. The insight developed through the investigation of these disordermers is demonstrated to hold important implications in the design of new generations of organic semiconductor materials.
Co-reporter:Karl J. Thorley and Chad Risko
Journal of Materials Chemistry A 2016 vol. 4(Issue 17) pp:3825-3832
Publication Date(Web):22 Dec 2015
DOI:10.1039/C5TC03765D
The ability to effectively transport charge carriers is often a key determinant concerning the deployment of materials derived from π-conjugated molecules and polymers in (opto)electronic applications. Theoretical models to evaluate charge-carrier transport parameters across a range of organic materials often work under the approximation of evaluating the intermolecular electronic couplings for supermolecular complexes (i.e. dimers) in the neutral state. Here, we investigate how the explicit inclusion of the nature of the charged state (i.e. both the neutral and radical-cation states) impacts the assessment of the intermolecular electronic couplings, and how considerations of the density functionals often used to determine these couplings effect the computed magnitude. From a materials perspective, we explore the role that the dimer configuration plays in determining the magnitudes of the electronic couplings for oligoacenes. The results suggest that appropriate consideration of translational alignment along the long and short acene axes, even in configurations with near perpendicular edge-to-face interactions, can lead to molecular packing arrangements in the solid state with large electronic couplings. These results give insight into ways to fine tune solid-state molecular packing to ensure the highest possible electronic couplings.
Co-reporter:Sean M. Ryno, Yao-Tsung Fu, Chad Risko, and Jean-Luc Brédas
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 24) pp:15524-15534
Publication Date(Web):May 31, 2016
DOI:10.1021/acsami.6b02851
We probe the energetic landscape at a model pentacene/fullerene (C60) interface to investigate the interactions between positive and negative charges, which are critical to the processes of charge separation and recombination in organic solar cells. Using a polarizable force field, we find that polarization energy, i.e., the stabilization a charge feels due to its environment, is larger at the interface than in the bulk for both a positive and a negative charge. The combination of the charge being more stabilized at the interface and the Coulomb attraction between the charges results in a barrier to charge separation at the pentacene/C60 interface that can be in excess of 0.7 eV for static configurations of the donor and acceptor locations. However, the impact of molecular motions, i.e., the dynamics, at the interface at room temperature results in a distribution of polarization energies and in charge separation barriers that can be significantly reduced. The dynamic nature of the interface is thus critical, with the polarization energy distributions indicating that sites along the interface shift in time between favorable and unfavorable configurations for charge separation.
Co-reporter:Sean M. Ryno, Chad Risko, and Jean-Luc Brédas
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 22) pp:14053-14062
Publication Date(Web):May 16, 2016
DOI:10.1021/acsami.6b02579
The polarizable environment surrounding charge carriers in organic semiconductors impacts the efficiency of the charge transport process. Here, we consider two representative organic semiconductors, tetracene and rubrene, and evaluate their polarization energies in the bulk and at the organic–vacuum interface using a polarizable force field that accounts for induced-dipole and quadrupole interactions. Though both oligoacenes pack in a herringbone motif, the tetraphenyl substituents on the tetracene backbone of rubrene alter greatly the nature of the packing. The resulting change in relative orientations of neighboring molecules is found to reduce the bulk polarization energy of holes in rubrene by some 0.3 eV when compared to tetracene. The consideration of model organic-vacuum interfaces highlights the significant variation in the electrostatic environment for a charge carrier at a surface although the net change in polarization energy is small; interestingly, the environment of a charge even just one layer removed from the surface can be viewed already as representative of the bulk. Overall, it is found that in these herringbone-type layered crystals the polarization energy has a much stronger dependence on the intralayer packing density than interlayer packing density.
Co-reporter:Christopher Sutton; Michael S. Marshall; C. David Sherrill; Chad Risko;Jean-Luc Brédas
Journal of the American Chemical Society 2015 Volume 137(Issue 27) pp:8775-8782
Publication Date(Web):June 15, 2015
DOI:10.1021/jacs.5b04066
Rubrene is one of the most studied molecular semiconductors; its chemical structure consists of a tetracene backbone with four phenyl rings appended to the two central fused rings. Derivatization of these phenyl rings can lead to two very different solid-state molecular conformations and packings: One in which the tetracene core is planar and there exists substantive overlap among neighboring π-conjugated backbones; and another where the tetracene core is twisted and the overlap of neighboring π-conjugated backbones is completely disrupted. State-of-the-art electronic structure calculations show for all isolated rubrene derivatives that the twisted conformation is more favorable (by −1.7 to −4.1 kcal mol–1), which is a consequence of energetically unfavorable exchange–repulsion interactions among the phenyl side groups. Calculations based on available crystallographic structures reveal that planar conformations of the tetracene core in the solid state result from intermolecular interactions that can be tuned through well-chosen functionalization of the phenyl side groups and lead to improved intermolecular electronic couplings. Understanding the interplay of these intramolecular and intermolecular interactions provides insight into how to chemically modify rubrene and similar molecular semiconductors to improve the intrinsic materials electronic properties.
Co-reporter:Khanh Do, Chad Risko, John E. Anthony, Aram Amassian, and Jean-Luc Brédas
Chemistry of Materials 2015 Volume 27(Issue 22) pp:7643
Publication Date(Web):October 22, 2015
DOI:10.1021/acs.chemmater.5b02983
In the quest to improve the performance of organic bulk heterojunction solar cells, many recent efforts have focused on developing molecular and polymer alternatives to commonly used fullerene acceptors. Here, molecular dynamics simulations are used to investigate polymer:molecule blends comprised of the polymer donor poly(3-hexylthiophene) (P3HT) with a series of acceptors based on trialkylsilylethynyl-substituted pentacene. A matrix of nine pentacene derivatives, consisting of systematic chemical variation both in the nature of the alkyl groups and electron-withdrawing moieties appended to the acene, is used to draw connections between the chemical structure of the acene acceptor and the nanoscale properties of the polymer:molecule blend, which include polymer and molecular diffusivity, donor–acceptor packing and interfacial (contact) area, and miscibility. The results point to the very significant role that seemingly modest changes in chemical structure play during the formation of polymer:molecule blend morphologies.
Co-reporter:Naga Rajesh Tummala, Christopher Sutton, Saadullah G. Aziz, Michael F. Toney, Chad Risko, and Jean-Luc Bredas
Chemistry of Materials 2015 Volume 27(Issue 24) pp:8261
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.chemmater.5b03254
High-boiling-point solvent additives, employed during the solution processing of active-layer formulations, impact the efficiency of bulk heterojunction (BHJ) organic solar cells by influencing the morphological/topological features of the multicomponent thin film. Here, we aim at a better understanding of how these additives change the aggregation landscape in the casting solution prior to film deposition via a multiscale computational study of the aggregation phenomena of phenyl-C61-butyric-acid methyl ester (PCBM) in various solutions. The energetic landscape of PCBM-solvent/solvent-additive intermolecular interactions is evaluated at the electronic-structure level through symmetry-adapted perturbation theory to determine the nature and strength of noncovalent forces important to aggregation. Molecular dynamics simulations highlight how the choice of solvent and solvent additives control the formation of molecular aggregates. Our results indicate that high-boiling point solvent additives change the effective interactions among the PCBM and casting-solvent molecules and alter equilibrium PCBM aggregate size in solution.
Co-reporter:Naga Rajesh Tummala, Christopher Bruner, Chad Risko, Jean-Luc Brédas, and Reinhold H. Dauskardt
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 18) pp:9957
Publication Date(Web):April 21, 2015
DOI:10.1021/acsami.5b02202
Quantifying cohesion and understanding fracture phenomena in thin-film electronic devices are necessary for improved materials design and processing criteria. For organic photovoltaics (OPVs), the cohesion of the photoactive layer portends its mechanical flexibility, reliability, and lifetime. Here, the molecular mechanism for the initiation of cohesive failure in bulk heterojunction (BHJ) OPV active layers derived from the semiconducting polymer poly(3-hexylthiophene) [P3HT] and two monosubstituted fullerenes is examined experimentally and through molecular-dynamics simulations. The results detail how, under identical conditions, cohesion significantly changes due to minor variations in the fullerene adduct functionality, an important materials consideration that needs to be taken into account across fields where soluble fullerene derivatives are used.Keywords: cohesion and fracture; molecular dynamics; P3HT; solar cells; substituted fullerenes; thin films;
Co-reporter:Matthew D. Casselman, Aman Preet Kaur, Kishore Anand Narayana, Corrine F. Elliott, Chad Risko and Susan A. Odom
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 10) pp:6905-6912
Publication Date(Web):22 Jan 2015
DOI:10.1039/C5CP00199D
The stability and reactivity of the multiple oxidation states of aromatic compounds are critical to the performance of these species as additives and electrolytes in energy-storage applications. Both for the overcharge mitigation in ion-intercalation batteries and as electroactive species in redox flow batteries, neutral, radical-cation, and radical-anion species may be present during charging and discharging processes. Despite the wide range of compounds evaluated for both applications, the progress identifying stable materials has been slow, limited perhaps by the overall lack of analysis of the failure mechanism when a material is utilized in an energy-storage device. In this study, we examined the reactivity of phenothiazine derivatives, which have found interest as redox shuttles in lithium-ion battery applications. We explored the products of the reactions of neutral compounds in battery electrolytes and the products of radical cation formation using bulk electrolysis and coin cell cycling. Following the failure of each cell, the electrolytes were characterized to identify redox shuttle decomposition products. Based on these results, a set of decomposition mechanisms is proposed and further explored using experimental and theoretical approaches. The results highlight the necessity to fully characterize and understand the chemical degradation mechanisms of the redox species in order to develop new generations of electroactive materials.
Co-reporter:Rebecca L. Gieseking; Chad Risko;Jean-Luc Brédas
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 12) pp:2158-2162
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.jpclett.5b00812
Understanding the relationships between the molecular nonlinear optical (NLO) properties and the bond-length alternation (BLA) or π-bond-order alternation (BOA) along the molecular backbone of linear π-conjugated systems has proven widely useful in the development of NLO organic chromophores and materials. Here, we examine model polymethines to elucidate the reliability of these relationships. While BLA is solely a measure of molecular geometric structure, BOA includes information pertaining to the electronic structure. As a result, BLA is found to be a good predictor of NLO properties only when optimized geometries are considered, whereas BOA is more broadly applicable. Proper understanding of the distinction between BLA and BOA is critical when designing computational studies of NLO properties, especially for molecules in complex environments or in nonequilibrium geometries.
Co-reporter:Naga Rajesh Tummala;Christopher Bruner;Reinhold H. Dauskardt;Jean-Luc Brédas
Journal of Polymer Science Part B: Polymer Physics 2015 Volume 53( Issue 13) pp:934-942
Publication Date(Web):
DOI:10.1002/polb.23722
ABSTRACT
Due to their inherent mechanical flexibility and stretchability, organic-based electronic devices have garnered a great deal of academic and industrial interest. Here, molecular-dynamics simulations are used to examine the molecular-scale details that govern the relationships among molecular weight, chain entanglement, persistence length, and the elastic characteristics of the widely studied π-conjugated polymer poly-(3-hexyl thiophene), P3HT. Oligomers containing at least 50 monomer units are required in the simulations to observe elastic behavior in P3HT, while much longer chains are required to ensure description of appropriate levels of entanglement: only when the molecular weight is greater than 50 kDa, that is, oligomers with approximately 400 monomer units, is truly entangled behavior observed. Interestingly, results from primitive path analysis of amorphous P3HT matches well with the observed onsets of inter-chain excitonic coherence with increased molecular weight. The simulations also indicate that the P3HT modulus saturates at 1.6 GPa for chain lengths of 50–100 monomers, a result that compares well with experimental results. This work highlights the care that needs to be taken to accurately model P3HT morphologies in relation to experimental measurements. © 2015 Wiley Periodicals, Inc. J. Polym. Sci., Part B: Polym. Phys. 2015, 53, 934–942
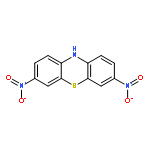
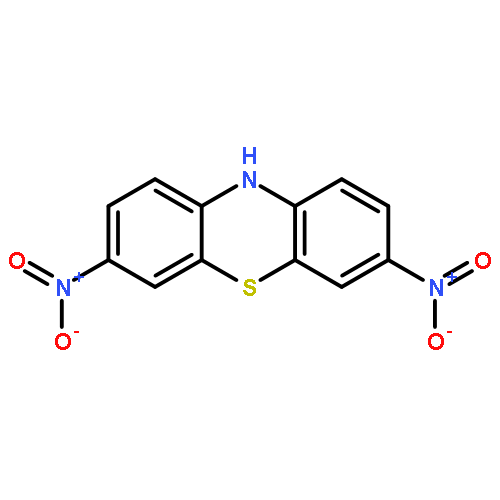
Co-reporter:Kishore An Narayana;Dr. Matthew D. Casselman;Corrine F. Elliott;Dr. Selin Ergun;Dr. Sean R. Parkin; Chad Risko; Susan A. Odom
ChemPhysChem 2015 Volume 16( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/cphc.201590032
Co-reporter:Kishore An Narayana;Dr. Matthew D. Casselman;Corrine F. Elliott;Dr. Selin Ergun;Dr. Sean R. Parkin; Chad Risko; Susan A. Odom
ChemPhysChem 2015 Volume 16( Issue 6) pp:1179-1189
Publication Date(Web):
DOI:10.1002/cphc.201402674
Abstract
Phenothiazine and five N-substituted derivatives were evaluated as electrolyte additives for overcharge protection in LiFePO4/synthetic graphite lithium-ion batteries. We report on the stability and reactivity of both the neutral and radical-cation forms of these six compounds. While three of the compounds show extensive overcharge protection, the remaining three last for only one to a few cycles. UV/Vis studies of redox shuttle stability in the radical cation form are consistent with the overcharge performance: redox shuttles with spectra that show little change over time exhibit extensive overcharge performance, whereas those with changing spectra have limited overcharge protection. In one case, we determined that a CN bond cleaves upon oxidation, forming the phenothiazine radical cation and leading to premature overcharge protection failure; in another case, poor solubility appears to limit protection.
Co-reporter:Igo T. Lima, Chad Risko, Saadullah G. Aziz, Demétrio A. da Silva Filho and Jean-Luc Brédas
Journal of Materials Chemistry A 2014 vol. 2(Issue 42) pp:8873-8879
Publication Date(Web):22 Aug 2014
DOI:10.1039/C4TC01264J
Donor–acceptor π-conjugated copolymers are of interest for a wide range of electronic applications, including field-effect transistors and solar cells. Here, we present a density functional theory (DFT) study of the impact of varying the conjugation pathway on the geometric, electronic, and optical properties of donor–acceptor systems. We consider both linear (“in series”), traditional conjugation among the donor–acceptor moieties versus structures where the acceptor units are appended orthogonally to the linear, donor-only conjugated backbone. Long-range-corrected hybrid functionals are used in the investigation with the values of the tuned long-range separation parameters providing an estimate of the extent of conjugation as a function of the oligomer architecture. Considerable differences in the electronic and optical properties are determined as a function of the nature of the conjugation pathway, features that should be taken into account in the design of donor–acceptor copolymers.
Co-reporter:Monika Williams, Naga Rajesh Tummala, Saadullah G. Aziz, Chad Risko, and Jean-Luc Brédas
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 19) pp:3427-3433
Publication Date(Web):September 17, 2014
DOI:10.1021/jz501559q
Co-reporter:Karl J. Thorley and Chad Risko
Journal of Materials Chemistry A 2016 - vol. 4(Issue 17) pp:NaN3832-3832
Publication Date(Web):2015/12/22
DOI:10.1039/C5TC03765D
The ability to effectively transport charge carriers is often a key determinant concerning the deployment of materials derived from π-conjugated molecules and polymers in (opto)electronic applications. Theoretical models to evaluate charge-carrier transport parameters across a range of organic materials often work under the approximation of evaluating the intermolecular electronic couplings for supermolecular complexes (i.e. dimers) in the neutral state. Here, we investigate how the explicit inclusion of the nature of the charged state (i.e. both the neutral and radical-cation states) impacts the assessment of the intermolecular electronic couplings, and how considerations of the density functionals often used to determine these couplings effect the computed magnitude. From a materials perspective, we explore the role that the dimer configuration plays in determining the magnitudes of the electronic couplings for oligoacenes. The results suggest that appropriate consideration of translational alignment along the long and short acene axes, even in configurations with near perpendicular edge-to-face interactions, can lead to molecular packing arrangements in the solid state with large electronic couplings. These results give insight into ways to fine tune solid-state molecular packing to ensure the highest possible electronic couplings.
Co-reporter:Matthew D. Casselman, Aman Preet Kaur, Kishore Anand Narayana, Corrine F. Elliott, Chad Risko and Susan A. Odom
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 10) pp:NaN6912-6912
Publication Date(Web):2015/01/22
DOI:10.1039/C5CP00199D
The stability and reactivity of the multiple oxidation states of aromatic compounds are critical to the performance of these species as additives and electrolytes in energy-storage applications. Both for the overcharge mitigation in ion-intercalation batteries and as electroactive species in redox flow batteries, neutral, radical-cation, and radical-anion species may be present during charging and discharging processes. Despite the wide range of compounds evaluated for both applications, the progress identifying stable materials has been slow, limited perhaps by the overall lack of analysis of the failure mechanism when a material is utilized in an energy-storage device. In this study, we examined the reactivity of phenothiazine derivatives, which have found interest as redox shuttles in lithium-ion battery applications. We explored the products of the reactions of neutral compounds in battery electrolytes and the products of radical cation formation using bulk electrolysis and coin cell cycling. Following the failure of each cell, the electrolytes were characterized to identify redox shuttle decomposition products. Based on these results, a set of decomposition mechanisms is proposed and further explored using experimental and theoretical approaches. The results highlight the necessity to fully characterize and understand the chemical degradation mechanisms of the redox species in order to develop new generations of electroactive materials.
Co-reporter:Igo T. Lima, Chad Risko, Saadullah G. Aziz, Demétrio A. da Silva Filho and Jean-Luc Brédas
Journal of Materials Chemistry A 2014 - vol. 2(Issue 42) pp:NaN8879-8879
Publication Date(Web):2014/08/22
DOI:10.1039/C4TC01264J
Donor–acceptor π-conjugated copolymers are of interest for a wide range of electronic applications, including field-effect transistors and solar cells. Here, we present a density functional theory (DFT) study of the impact of varying the conjugation pathway on the geometric, electronic, and optical properties of donor–acceptor systems. We consider both linear (“in series”), traditional conjugation among the donor–acceptor moieties versus structures where the acceptor units are appended orthogonally to the linear, donor-only conjugated backbone. Long-range-corrected hybrid functionals are used in the investigation with the values of the tuned long-range separation parameters providing an estimate of the extent of conjugation as a function of the oligomer architecture. Considerable differences in the electronic and optical properties are determined as a function of the nature of the conjugation pathway, features that should be taken into account in the design of donor–acceptor copolymers.
Co-reporter:Karl J. Thorley and Chad Risko
Journal of Materials Chemistry A 2016 - vol. 4(Issue 18) pp:NaN4048-4048
Publication Date(Web):2016/03/11
DOI:10.1039/C5TC03900B
Many high performing organic semiconductor materials contain heteroaromatic rings in order to control the molecular packing and material electronic properties. Here we use a combination of density functional theory and symmetry-adapted perturbation theory calculations to explore the intermolecular noncovalent interactions, which guide solid-state molecular packing, and electronic couplings in a series of benzodithiophene-based dimer models. A novel concept, termed the disordermer, is introduced to delineate how the reduced molecular symmetry of benzodithiophene, when compared to the more highly symmetric anthracene molecule, can present intermolecular isomerism in the solid state that results in a wide range of available molecular packing arrangements that in turn influence the magnitudes of the electronic couplings. The insight developed through the investigation of these disordermers is demonstrated to hold important implications in the design of new generations of organic semiconductor materials.
.jpg)

![Anthra[2,3-b:7,6-b']dithiophene](http://img.cochemist.com/ccimg/144500/144413-59-2.png)
![Anthra[2,3-b:7,6-b']dithiophene](http://img.cochemist.com/ccimg/144500/144413-59-2_b.png)
![Benzo[1,2-b:4,5-b']dithiophene](/data/chemimg/477000/267-65-2.png)
![Benzo[1,2-b:4,5-b']dithiophene](/data/chemimg/477000/267-65-2_b.png)