Co-reporter:Cameron L. Brown, Meredith H. Barbee, Jeong Hoon Ko, Heather D. Maynard, and Stephen L. Craig
Journal of Chemical Education November 14, 2017 Volume 94(Issue 11) pp:1752-1752
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.jchemed.6b00806
An easy-to-implement science outreach demonstration featuring a mechanically and photochemically color-changing polymer is described. The active polymeric material is a filled poly(dimethylsiloxane) (PDMS) elastomer that is covalently functionalized with spiropyran (SP), which is both a photochemical and mechanochemical switch. The material can be reversibly changed from colorless to dark purple by exposing it to light from a blue laser pointer or providing a mechanical stimulus such as hitting the polymer with a hammer or dragging a blunt object across the surface. The keynote demonstration is a PDMS chemical-drawing board that allows children literally to “write without ink” using a laser pointer or a blunt stylus. Collectively, these demonstrations are suitable for various student groups and encompass concepts in polymer and materials chemistry, photochemistry, and mechanochemistry. This demonstration has been successfully employed dozens of times in multiple universities across North America.Keywords: Demonstrations; Elementary/Middle School Science; Hands-On-Learning/Manipulatives; Materials Science; Photochemistry; Polymer Chemistry; Public Understanding/Outreach;
Co-reporter:Yang Liu, Juneyoung Lee, Kathryn M. Mansfield, Jeong Hoon Ko, Sahar Sallam, Chrys Wesdemiotis, and Heather D. Maynard
Bioconjugate Chemistry March 15, 2017 Volume 28(Issue 3) pp:836-836
Publication Date(Web):January 3, 2017
DOI:10.1021/acs.bioconjchem.6b00659
Biocompatible polymers such as poly(ethylene glycol) (PEG) have been successfully conjugated to therapeutic proteins to enhance their pharmacokinetics. However, many of these polymers, including PEG, only improve the in vivo lifetimes and do not protect proteins against inactivation during storage and transportation. Herein, we report a polymer with trehalose side chains (PolyProtek) that is capable of improving both the external stability and the in vivo plasma half-life of a therapeutic protein. Insulin was employed as a model biologic, and high performance liquid chromatography and dynamic light scattering confirmed that addition of trehalose glycopolymer as an excipient or covalent conjugation prevented thermal or agitation-induced aggregation of insulin. The insulin–trehalose glycopolymer conjugate also showed significantly prolonged plasma circulation time in mice, similar to the analogous insulin–PEG conjugate. The insulin–trehalose glycopolymer conjugate was active as tested by insulin tolerance tests in mice and retained bioactivity even after exposure to high temperatures. The trehalose glycopolymer was shown to be nontoxic to mice up to at least 1.6 mg/kg dosage. These results together suggest that the trehalose glycopolymer should be further explored as an alternative to PEG for long circulating protein therapeutics.
Co-reporter:Jeong Hoon Ko, Takaya Terashima, Mitsuo Sawamoto, and Heather D. Maynard
Macromolecules December 12, 2017 Volume 50(Issue 23) pp:9222-9222
Publication Date(Web):November 21, 2017
DOI:10.1021/acs.macromol.7b01973
Fluorine-containing polymers have potential for use in medicine and other applications, but the synthesis of degradable fluorous polymers is underexplored. In this report, we present a facile route to degradable fluorinated polymers and characterize the effect of fluorous comonomer identity on the polymerization as well as the degradation kinetics of the resulting polymer. Copolymers of poly(ethylene glycol methyl ether methacrylate) (PEGMA), fluorous methacrylate (1H,1H,2H,2H-perfluorooctyl or 1H,1H,2H,2H,3H,3H-perfluoropentyl methacrylate), and cyclic ketene acetal 5,6-benzo-2-methylene-1,3-dioxepane (BMDO) were synthesized via ruthenium-catalyzed living radical polymerization. It was observed that increasing the fluorous monomer content led to enhanced BMDO incorporation in the resulting polymer. Density functional theory calculations suggest that this is due to the decreased energy gap between the singly occupied molecular orbital (SOMO) of the methacrylate radical and the highest occupied molecular orbital (HOMO) of BMDO. Moreover, polymers with higher fluorous monomer content were more hydrolytically stable, with a degradation rate constant 100-fold smaller for the polymer with highest fluorous content compared to the nonfluorous polymer. This work provides easy access to degradable fluorous polymers using vinyl monomers. In addition, the insights gained into modulation of reactivity of cyclic ketene acetals and polymer degradation will be useful in applying fluorous polymers for a variety of biomedical applications.
Co-reporter:Emma M. Pelegri-O’Day, Samantha J. Paluck, and Heather D. Maynard
Journal of the American Chemical Society 2017 Volume 139(Issue 3) pp:1145-1154
Publication Date(Web):January 12, 2017
DOI:10.1021/jacs.6b10776
Many proteins, especially those used as therapeutics, are unstable to storage and shipping temperatures, leading to increased costs in research and industry. Therefore, the design and synthesis of novel stabilizers is an important area of investigation. Herein we report new degradable polymers that stabilize proteins to environmental stressors such as refrigeration and elevated temperature. Specifically, polycaprolactones with different pendant groups were synthesized and surveyed for their ability to stabilize an important therapeutic protein to storage and shipping conditions. Ring-opening polymerization (ROP) of an allyl-substituted caprolactone monomer was carried out using the organocatalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) to yield a well-defined, alkene-substituted degradable polymer, which was used as a common backbone to control for the degree of polymerization. Relevant side chains such as trehalose, lactose, glucose, carboxybetaine, and oligo(ethylene glycol) were installed via postpolymerization thiol–ene reactions. These degradable polymers were then employed as excipients for the stabilization of the therapeutic protein granulocyte colony-stimulating factor (G-CSF) against storage at 4 °C and shipping temperatures of 60 °C. The best stabilization was observed using the trehalose- and zwitterion- substituted polyesters. Both the trehalose- and carboxybetaine-substituted pCL were further investigated with regard to molecular weight dependence, and it was found that the molecular weight was minimally important for stabilization to refrigeration, but critical for G-CSF stabilization at elevated temperatures. Both high performing zwitterionic and trehalose polyesters were also degraded, and the polymers and degradation products were shown to be noncytotoxic. This work provides potential biocompatible polymers for stabilization of the important therapeutic G-CSF, as well as a general platform for the future discovery of new polymeric protein stabilizers.
Co-reporter:Jose M. Medina, Jeong Hoon Ko, Heather D. MaynardNeil K. Garg
Macromolecules 2017 Volume 50(Issue 2) pp:
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.macromol.6b02376
Benzonorbornadiene polymers synthesized by ring-opening metathesis polymerization (ROMP) are typically prone to oxidation at the benzylic/allylic position under ambient conditions. Accordingly, the use of benzonorbornadiene polymers in practical applications has remained limited. In this manuscript, we report the synthesis of poly(benzonorbornadiene) polymers using a strategic blend of benzyne chemistry and ROMP. Through a comparative study, we show that substitution at the benzylic/allylic position prevents oxidative deformation, yet does not inhibit polymerization by common ruthenium catalysts with good control over molecular weight dispersity. We expect the benzyne/ROMP reaction sequence will allow easy access to air-stable benzonorbornadiene polymers for various applications.
Co-reporter:Samantha J. Paluck
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 31) pp:4548-4556
Publication Date(Web):2017/08/08
DOI:10.1039/C7PY00861A
Fibroblast growth factor-2 (FGF2) is a heparin binding protein that plays a role in a range of biological functions such as wound healing and bone regeneration. Heparin, a highly sulfated glycosaminoglycan, is required for FGF2 to bind to its receptor. Therefore, polymeric mimics of heparin are widely studied for their ability to manipulate FGF2-induced biological interactions. It is known that altering the degree of sulfonated monomer incorporation and size of heparin-mimicking polymers can affect protein-receptor binding. To elucidate the relationship between degree of sulfonation and receptor binding for the heparin-mimicking polymer, poly(styrene sulfonate-co-poly(ethylene glycol) methyl ether methacrylate) (p(SS-co-PEGMA)) a library was synthesized to contain nine polymers with degrees of sulfonation ranging from 0–100%. Kinetics of the polymerization were evaluated and reactivity ratios compared to literature results. These polymers were then tested for their ability to enhance FGF2 binding with its receptor as both covalent conjugates and as excipients. In a receptor based enzyme-linked immunosorbant assay (ELISA), as well as a cell-based study, the polymer with 81% SS incorporation enhanced receptor binding compared to FGF2 alone, and to a greater extent than the other polymers. Therefore, another library of polymers was prepared maintaining the degree of sulfonation at 81% and changing the size from 41 to 390 monomer repeat units. The polymers were again tested in receptor based ELISA and cell studies, and all of the different sizes performed similarly, except for degree of polymerization 295 and 390, which had reduced response in the cellular assay. These results provide important information for the use of pSS-co-PEGMA as a potential heparin-mimicking therapeutic.
Co-reporter:Marco S. Messina;Jeong Hoon Ko;Zhongyue Yang;M. Jane Strouse;K. N. Houk
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 33) pp:4781-4788
Publication Date(Web):2017/08/22
DOI:10.1039/C7PY00700K
There is considerable interest in the use of proteins as therapeutics and as chemical and biochemical reagents. However, many proteins are unstable and aggregate when exposed to stressors, including increased temperature, pH change, agitation, and desiccation. Polymers with side chain trehalose units were shown to be effective protein stabilizers, preventing aggregation and prolonging activity. Herein, we report the synthesis and characterization of four trehalose regioisomers containing a vinylbenzyl ether moiety at either the 2-O, 3-O, 4-O, or 6-O position. Computational analysis of these regioisomers suggested that they differ in their conformational flexibility, but all retained the native clam shell conformation of trehalose. Polymers were synthesized from the monomers separately via free radical polymerization and one polymer was prepared containing all of the regioisomers. The polymers were tested for their ability to stabilize insulin, and were found to prevent agitation-induced aggregation comparably. The results show that for insulin the effect of trehalose positional modification is minimal and suggest that the clam shell conformation itself may be more important than the polymer backbone attachment site for stabilization of proteins.
Co-reporter:Emma M. Pelegri-O’Day and Heather D. Maynard
Accounts of Chemical Research 2016 Volume 49(Issue 9) pp:1777
Publication Date(Web):September 2, 2016
DOI:10.1021/acs.accounts.6b00258
Protein–polymer conjugates are unique constructs that combine the chemical properties of a synthetic polymer chain with the biological properties of a biomacromolecule. This often leads to improved stabilities, solubilities, and in vivo half-lives of the resulting conjugates, and expands the range of applications for the proteins. However, early chemical methods for protein–polymer conjugation often required multiple polymer modifications, which were tedious and low yielding. To solve these issues, work in our laboratory has focused on the development of controlled radical polymerization (CRP) techniques to improve synthesis of protein−polymer conjugates. Initial efforts focused on the one-step syntheses of protein-reactive polymers through the use of functionalized initiators and chain transfer agents. A variety of functional groups such as maleimide and pyridyl disulfide could be installed with high end-group retention, which could then react with protein functional groups through mild and biocompatible chemistries. While this grafting to method represented a significant advance in conjugation technique, purification and steric hindrance between large biomacromolecules and polymer chains often led to low conjugation yields. Therefore, a grafting from approach was developed, wherein a polymer chain is grown from an initiating site on a functionalized protein. These conjugates have demonstrated improved homogeneity, characterization, and easier purification, while maintaining protein activity.Much of this early work utilizing CRP techniques focused on polymers made up of biocompatible but nonfunctional monomer units, often containing oligoethylene glycol meth(acrylate) or N-isopropylacrylamide. These branched polymers have significant advantages compared to the historically used linear poly(ethylene glycols) including decreased viscosities and thermally responsive behavior, respectively. Recently, we were motivated to use CRP techniques to develop polymers with rationally designed and functional biological properties for conjugate preparation. Specifically, two families of saccharide-inspired polymers were developed for stabilization and activation of therapeutic biomolecules. A series of polymers with trehalose side-chains and vinyl backbones were prepared and used to stabilize proteins against heat and lyophilization stress as both conjugates and additives. These materials, which combine properties of osmolytes with nonionic surfactants, have significant potential for in vivo therapeutic use. Additionally, polymers that mimic the structure of the naturally occurring polysaccharide heparin were prepared. These polymers contained negatively charged sulfonate groups and imparted stabilization to a heparin-binding growth factor after conjugation. A screen of other sulfonated polymers led to the development of a polymer with improved heparin mimesis, enhancing both stability and activity of the protein to which it was attached.Chemical improvements over the past decade have enabled the preparation of a diverse set of protein–polymer conjugates by controlled polymerization techniques. Now, the field should thoroughly explore and expand both the range of polymer structures and also the applications available to protein–polymer conjugates. As we move beyond medicine toward broader applications, increased collaboration and interdisciplinary work will result in the further development of this exciting field.
Co-reporter:Peter C. Nauka, Juneyoung Lee and Heather D. Maynard
Polymer Chemistry 2016 vol. 7(Issue 13) pp:2352-2357
Publication Date(Web):14 Mar 2016
DOI:10.1039/C6PY00080K
Polymers with oligoethylene glycol side chains are promising in therapeutic protein–polymer conjugates as replacements for linear polyethylene glycol (PEG). Branched PEG polymers can confer additional stability and advantageous properties compared to linear PEGs. However, branched PEG polymers suffer from low conjugation yields to proteins, likely due to steric interactions between the bulky side chains of the polymer and the protein. In an effort to increase the yield, the linker length between the protein-reactive functional end-group of the polymer chain and the branched PEG side chain was systematically increased. This was accomplished by synthesizing four well-defined poly(poly(ethylene glycol methyl ether) acrylates) (pPEGAs) with pyridyl disulfide end-groups by reversible addition-fragmentation chain transfer (RAFT) polymerization mediated by chain transfer agents (CTAs) with different linker lengths. These, along with linear PEG and poly(N-isopropylacrylamide) (pNIPAAm), were conjugated to two model proteins, bovine serum albumin (BSA) and beta-lactoglobulin (βLG). The conjugation yields were determined by gel electrophoresis. The length of the linker affected the conjugation yield for both proteins. For BSA, the conjugation yield step increased from 10% to 24% when the linker was altered from 1 ethylene glycol (EG) unit to 3, with no additional increase for 4 and 6 EG units. In the case of βLG, the yield gradually increased from 9% to 33% when the linker length was increased from 1 to 6. PEG and pNIPAAm reacted with yields as high as 75%, further emphasizing the effect of steric hindrance in lowering conjugation yields.
Co-reporter:Rock J. Mancini, Samantha J. Paluck, Erhan Bat, and Heather D. Maynard
Langmuir 2016 Volume 32(Issue 16) pp:4043-4051
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.langmuir.6b00560
Electron beam (e-beam) lithography was employed to prepare one protein immobilized hydrogel encapsulated inside another by first fabricating protein-reactive hydrogels of orthogonal reactivity and subsequently conjugating the biomolecules. Exposure of thin films of eight arm star poly(ethylene glycol) (PEG) functionalized with biotin (Biotin-PEG), alkyne (Alkyne-PEG) or aminooxy (AO-PEG) end-groups to e-beam radiation resulted in cross-linked hydrogels with the respective functionality. It was determined via confocal microscopy that a nominal size exclusion effect exists for streptavidin immobilized on Biotin-PEG hydrogels of feature sizes ranging from 5 to 40 μm. AO-PEG was subsequently patterned as an encapsulated core inside a contiguous outer shell of Biotin-PEG. Similarly, Alkyne-PEG was patterned as a core inside an AO-PEG shell. The hydrogel reactive end-groups were conjugated to dyes or proteins of complementary reactivity, and the three-dimensional (3-D) spatial orientation was determined for both configurations using confocal microscopy. The enzyme glucose oxidase (GOX) was immobilized in the core of the encapsulated Alkyne-PEG core/ AO-PEG shell architecture, and horseradish peroxidase (HRP) was conjugated to the shell periphery. Bioactivity for the HRP-GOX enzyme pair was observed in this encapsulated configuration by demonstrating that the enzyme pair was capable of enzyme cascade reactions.
Co-reporter:Maltish M. Lorenzo, Caitlin G. Decker, Muhammet U. Kahveci, Samantha J. Paluck, and Heather D. Maynard
Macromolecules 2016 Volume 49(Issue 1) pp:30-37
Publication Date(Web):December 23, 2015
DOI:10.1021/acs.macromol.5b02323
Tetrazine end-functionalized telechelic polymers were synthesized by controlled radical polymerization (CRP) and employed to generate T4 lysozyme homodimers. Mutant T4 lysozyme (V131C), containing a single surface-exposed cysteine, was modified with a protein-reactive trans-cyclooctene (T4L-TCO). Reversible addition–fragmentation chain transfer (RAFT) polymerization yielded poly(N-isopropylacrylamide) (pNIPAAm) with a number-average molecular weight (Mn by 1H NMR) of 2.0 kDa and a dispersity (Đ by GPC) of 1.05. pNIPAAm was then modified at both ends by postpolymerization with 6-methyltetrazine. For comparison, 2.0 kDa bis-tetrazine poly(ethylene glycol) (PEG) and 2.0 kDa bis-maleimide pNIPAAm were synthesized. Ligation of T4L-TCO to bis-tetrazine pNIPAAm or bis-tetrazine PEG resulted in protein homodimer in 38% yield and 37% yield, respectively, after only 1 h, whereas bis-maleimide pNIPAAm resulted in only 5% yield of dimer after 24 h. This work illustrates the advantage of employing tetrazine ligation over maleimide thiol–ene chemistry for the synthesis of protein homodimer conjugates.
Co-reporter:Samantha J. Paluck, Thi H. Nguyen, Jonghan P. Lee, and Heather D. Maynard
Biomacromolecules 2016 Volume 17(Issue 10) pp:3386
Publication Date(Web):August 31, 2016
DOI:10.1021/acs.biomac.6b01182
Fibroblast growth factor 2 (FGF2) is a protein involved in cellular functions in applications such as wound healing and tissue regeneration. Stabilization of this protein is important for its use as a therapeutic since the native protein is unstable during storage and delivery. Additionally, the ability to increase the activity of FGF2 is important for its application, particularly in chronic wound healing and the treatment of various ischemic conditions. Here we report a heparin mimicking block copolymer, poly(styrenesulfonate-co-poly(ethylene glycol) methyl ether methacrylate)-b-vinyl sulfonate) (p(SS-co-PEGMA)-b-VS, that contains a segment that enhances the stability of FGF2 and one that binds to the FGF2 receptor. The FGF2 conjugate retained activity after exposure to refrigeration (4 °C) and room temperature (23 °C) for 7 days, while unmodified FGF2 was inactive after these standard storage conditions. A cell study performed with a cell line lacking native heparan sulfate proteoglycans indicated that the conjugated block copolymer facilitated binding of FGF2 to its receptor similar to the addition of heparin to FGF2. A receptor-based enzyme-linked immunosorbant assay (ELISA) confirmed the results. The conjugate also increased the migration of endothelial cells by 80% compared to FGF2 alone. Additionally, the FGF2-p(SS-co-PEGMA)-b-VS stimulated endothelial cell sprouting 250% better than FGF2 at low concentration. These data verify that this rationally designed protein-block copolymer conjugate enhances receptor binding, cellular processes such as migration and tube-like formation, and stability, and suggest that it may be useful for applications in biomaterials, tissue regeneration, and wound healing.
Co-reporter:Samantha J. Paluck, Thi H. Nguyen, and Heather D. Maynard
Biomacromolecules 2016 Volume 17(Issue 11) pp:3417
Publication Date(Web):October 14, 2016
DOI:10.1021/acs.biomac.6b01147
Heparin is a naturally occurring, highly sulfated polysaccharide that plays a critical role in a range of different biological processes. Therapeutically, it is mostly commonly used as an injectable solution as an anticoagulant for a variety of indications, although it has also been employed in other forms such as coatings on various biomedical devices. Due to the diverse functions of this polysaccharide in the body, including anticoagulation, tissue regeneration, anti-inflammation, and protein stabilization, and drawbacks of its use, analogous heparin-mimicking materials are also widely studied for therapeutic applications. This review focuses on one type of these materials, namely, synthetic heparin-mimicking polymers. Utilization of these polymers provides significant benefits compared to heparin, including enhancing therapeutic efficacy and reducing side effects as a result of fine-tuning heparin-binding motifs and other molecular characteristics. The major types of the various polymers are summarized, as well as their applications. Because development of a broader range of heparin-mimicking materials would further expand the impact of these polymers in the treatment of various diseases, future directions are also discussed.
Co-reporter:Uland Y. Lau, Sina S. Saxer, Juneyoung Lee, Erhan Bat, and Heather D. Maynard
ACS Nano 2016 Volume 10(Issue 1) pp:723
Publication Date(Web):December 17, 2015
DOI:10.1021/acsnano.5b05781
Simultaneous detection of multiple biomarkers, such as extracellular signaling molecules, is a critical aspect in disease profiling and diagnostics. Precise positioning of antibodies on surfaces, especially at the micro- and nanoscale, is important for the improvement of assays, biosensors, and diagnostics on the molecular level, and therefore, the pursuit of device miniaturization for parallel, fast, low-volume assays is a continuing challenge. Here, we describe a multiplexed cytokine immunoassay utilizing electron beam lithography and a trehalose glycopolymer as a resist for the direct writing of antibodies on silicon substrates, allowing for micro- and nanoscale precision of protein immobilization. Specifically, anti-interleukin 6 (IL-6) and antitumor necrosis factor alpha (TNFα) antibodies were directly patterned. Retention of the specific binding properties of the patterned antibodies was shown by the capture of secreted cytokines from stimulated RAW 264.7 macrophages. A sandwich immunoassay was employed using gold nanoparticles and enhancement with silver for the detection and visualization of bound cytokines to the patterns by localized surface plasmon resonance detected with dark-field microscopy. Multiplexing with both IL-6 and TNFα on a single chip was also successfully demonstrated with high specificity and in relevant cell culture conditions and at different times after cell stimulation. The direct fabrication of capture antibody patterns for cytokine detection described here could be useful for biosensing applications.Keywords: antibody patterning; biosensor; dark-field microscopy; electron beam lithography; localized surface plasmon resonance; trehalose glycopolymer;
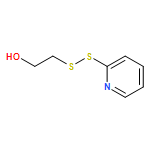
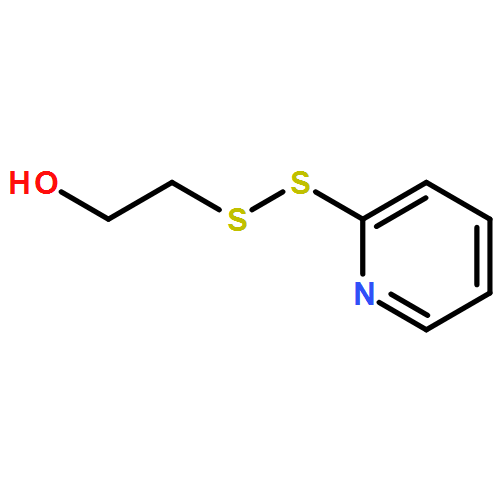
Co-reporter:Juneyoung Lee, Jeong Hoon Ko, En-Wei Lin, Peter Wallace, Frank Ruch and Heather D. Maynard
Polymer Chemistry 2015 vol. 6(Issue 18) pp:3443-3448
Publication Date(Web):07 Apr 2015
DOI:10.1039/C5PY00121H
Enzymes can catalyze various reactions with high selectivity and are involved in many important biological processes. However, the general instability of enzymes against high temperature often limits their application. To address this, we synthesized a trehalose-based hydrogel in two steps from commercial starting materials with minimal purification procedures. Mono- and multi-functional trehalose monomers were cross-linked by redox-initiated radical polymerization to form a hydrogel. Phytase, an important enzyme utilized in animal feedstock, was employed to study the effectiveness of the trehalose hydrogel to stabilize proteins against heat. Addition of the phytase solution to the hydrogel resulted in enzyme internalization as confirmed by confocal microscopy. The phytase in the hydrogel retained 100% activity upon heating at 90 °C compared to 39% when the hydrogel was absent. The enzyme could also be recovered from the hydrogel. The trehalose hydrogel synthesis reported herein should be readily scalable for thermal stabilization of a wide variety of enzymes.
Co-reporter:Yuta Koda, Takaya Terashima, Mitsuo Sawamoto and Heather D. Maynard
Polymer Chemistry 2015 vol. 6(Issue 2) pp:240-247
Publication Date(Web):23 Oct 2014
DOI:10.1039/C4PY01346H
Herein, amphiphilic/fluorous random copolymers bearing poly(ethylene glycol) (PEG) chains and perfluorinated alkane pendants were developed as novel non-cytotoxic polymers for protein conjugation. Three kinds of random copolymers with different initiating terminals (carboxylic acid, pyridyl disulfide, and N-hydroxysuccinimide ester) were prepared by reversible addition–fragmentation chain transfer (RAFT) copolymerization of a PEG methyl ether methacrylate and a perfluorinated alkane methacrylate with the corresponding functional chain transfer agents. All of the polymers were soluble in water to form nanostructures with perfluorinated compartments via fluorous interaction: large aggregates from the intermolecular multi-chain association and compact unimer micelles from the intramolecular single-chain folding. Such a PEGylated and perfluorinated random copolymer was non-cytotoxic to NIH 3T3 mouse embryonic fibroblast cells and human umbilical vein endothelial cells (HUVECs). Additionally, a random copolymer with a pyridyl disulfide terminal was also successfully conjugated with a thiolated lysozyme.
Co-reporter:Nicholas M. Matsumoto, George W. Buchman, Leonard H. Rome, Heather D. Maynard
European Polymer Journal 2015 Volume 69() pp:532-539
Publication Date(Web):August 2015
DOI:10.1016/j.eurpolymj.2015.01.043
•We describe dually-responsive vault-polymer biohybrid nanoparticles.•Poly(N-isopropylacrylamide-co-acrylic acid) was conjugated to human major vault protein.•The smart vault reversibly aggregates depending on pH and temperature.Multiply responsive protein nanoparticles are interesting for a variety of applications. Herein, we describe the synthesis of a vault nanoparticle that responds to both temperature and pH. Specifically, poly(N-isopropylacrylamide-co-acrylic acid) with a pyridyl disulfide end group was prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization. The polymer had a lower critical solution temperature (LCST) of 31.9 °C at pH 5, 44.0 °C at pH 6 and above 60 °C at pH 7. The polymer was conjugated to human major vault protein (hMVP), and the resulting nanoparticle was analyzed by UV–Vis, dynamic light scattering (DLS) and electron microscopy. The data demonstrated that the poly(N-isopropylacrylamide-co-acrylic acid)-vault conjugate did not respond to temperatures below 60 °C at pH 7, while the nanoparticles reversibly aggregated at pH 6. Furthermore, it was shown that the vault nanoparticle structure remained intact for at least three heat and cooling cycles. Thus, these dually responsive nanoparticles may serve as a platform for drug delivery and other applications.Graphical abstract
Co-reporter:Caitlin G. Decker, Heather D. Maynard
European Polymer Journal 2015 Volume 65() pp:305-312
Publication Date(Web):April 2015
DOI:10.1016/j.eurpolymj.2015.01.025
•Degradable PEG-like, protein-reactive polymers were synthesized by RAFT polymerization.•p(PEGMA-co-BMDO)-lysozyme conjugates were prepared with or without masked activity.•Degradation of the polymers alone and from lysozyme was assessed.•The activity of lysozyme reductively released from the polymer(s) was analyzed.Poly(ethylene glycol) (PEG)–protein therapeutics exhibit enhanced pharmacokinetics, but have drawbacks including decreased protein activities and polymer accumulation in the body. Therefore a major aim for second-generation polymer therapeutics is to introduce degradability into the backbone. Herein we describe the synthesis of poly(poly(ethylene glycol methyl ether methacrylate)) (pPEGMA) degradable polymers with protein-reactive end-groups via reversible addition–fragmentation chain transfer (RAFT) polymerization, and the subsequent covalent attachment to lysozyme through a reducible disulfide linkage. RAFT copolymerization of cyclic ketene acetal (CKA) monomer 5,6-benzo-2-methylene-1,3-dioxepane (BMDO) with PEGMA yielded two polymers with number-average molecular weight (Mn) (GPC) of 10.9 and 20.9 kDa and molecular weight dispersities (Đ) of 1.34 and 1.71, respectively. Hydrolytic degradation of the polymers was analyzed by 1H NMR and GPC under basic and acidic conditions. The reversible covalent attachment of these polymers to lysozyme, as well as the hydrolytic and reductive cleavage of the polymer from the protein, was analyzed by gel electrophoresis and mass spectrometry. Following reductive cleavage of the polymer, an increase in activity was observed for both conjugates, with the released protein having full activity. This represents a method to prepare PEGylated proteins, where the polymer is readily cleaved from the protein and the main chain of the polymer is degradable.
Co-reporter:En-Wei Lin and Heather D. Maynard
Macromolecules 2015 Volume 48(Issue 16) pp:5640-5647
Publication Date(Web):August 14, 2015
DOI:10.1021/acs.macromol.5b00846
Small interfering ribonucleic acids (siRNAs) are important therapeutic agents and are challenging to deliver efficiently. To address this, covalent attachment of synthetic polymers to siRNA has become of great interest. In this report, we present a synthetic route to siRNA–polymer conjugates by the grafting from method, meaning that the polymerization of monomers occurs from an initiating site that is attached to siRNA. Specifically, a siRNA macroinitiator (siRNA-I) was prepared through disulfide exchange of 5′-thiol-modified siRNA with a pyridyl disulfide initiator. Activator generated by electron transfer atom transfer radical polymerization (AGET ATRP) of two monomers, poly(ethylene glycol) methyl ether methacrylate (PEGMA; Mn = 300) and di(ethylene glycol) methyl ether methacrylate (DEGMA), was undertaken to synthesize a series of siRNA–polymer conjugates. The resulting conjugates were characterized by mass spectrometry and 1H nuclear magnetic resonance spectroscopy and compared to siRNA–pDEGMA conjugates prepared by the grafting to method in gel electrophoresis to estimate polymer molecular weight.
Co-reporter:Natalie Boehnke, Cynthia Cam, Erhan Bat, Tatiana Segura, and Heather D. Maynard
Biomacromolecules 2015 Volume 16(Issue 7) pp:
Publication Date(Web):June 10, 2015
DOI:10.1021/acs.biomac.5b00519
A shortage of available organ donors has created a need for engineered tissues. In this context, polymer-based hydrogels that break down inside the body are often used as constructs for growth factors and cells. Herein, we report imine cross-linked gels where degradation is controllable by the introduction of mixed imine cross-links. Specifically, hydrazide-functionalized poly(ethylene glycol) (PEG) reacts with aldehyde-functionalized PEG (PEG-CHO) to form hydrazone linked hydrogels that degrade quickly in media. The time to degradation can be controlled by changing the structure of the hydrazide group or by introducing hydroxylamines to form nonreversible oxime linkages. Hydrogels containing adipohydrazide-functionalized PEG (PEG-ADH) and PEG-CHO were found to degrade more rapidly than gels formed from carbodihydrazide-functionalized PEG (PEG-CDH). Incorporating oxime linkages via aminooxy-functionalized PEG (PEG-AO) into the hydrazone cross-linked gels further stabilized the hydrogels. This imine cross-linking approach should be useful for modulating the degradation characteristics of 3D cell culture supports for controlled cell release.
Co-reporter:Emma M. Pelegri-O’Day ; En-Wei Lin
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14323-14332
Publication Date(Web):September 12, 2014
DOI:10.1021/ja504390x
Protein–polymer conjugates are widely used as therapeutics. All Food and Drug Administration (FDA)-approved protein conjugates are covalently linked to poly(ethylene glycol) (PEG). These PEGylated drugs have longer half-lives in the bloodstream, leading to less frequent dosing, which is a significant advantage for patients. However, there are some potential drawbacks to PEG that are driving the development of alternatives. Polymers that display enhanced pharmacokinetic properties along with additional advantages such as improved stability or degradability will be important to advance the field of protein therapeutics. This perspective presents a summary of protein–PEG conjugates for therapeutic use and alternative technologies in various stages of development as well as suggestions for future directions. Established methods of producing protein–PEG conjugates and new approaches utilizing controlled radical polymerization are also covered.
Co-reporter:En-Wei Lin, Natalie Boehnke, and Heather D. Maynard
Bioconjugate Chemistry 2014 Volume 25(Issue 10) pp:1902
Publication Date(Web):September 27, 2014
DOI:10.1021/bc500380r
A photoactivated, site-selective conjugation of poly(ethylene glycol) (PEG) to the glutathione (GSH) binding pocket of glutathione S-transferase (GST) is described. To achieve this, a GSH analogue (GSH-BP) was designed and chemically synthesized with three functionalities: (1) the binding affinity of GSH to GST, (2) a free thiol for polymer functionalization, and (3) a photoreactive benzophenone (BP) component. Different molecular weights (2 kDa, 5 kDa, and 20 kDa) of GSH-BP modified PEGs (GSBP-PEGs) were synthesized and showed conjugation efficiencies between 52% and 76% to GST. Diazirine (DA) PEG were also prepared but gave conjugation yields lower than for GSBP-PEGs. PEGs with different end-groups were also synthesized to validate the importance of each component in the end-group design. End-groups included glutathione (GS-PEG) and benzophenone (BP-PEG). Results showed that both GSH and BP were crucial for successful conjugation to GST. In addition, conjugations of 5 kDa GSBP-PEG to different proteins were investigated, including bovine serum albumin (BSA), lysozyme (Lyz), ubiquitin (Ubq), and GST-fused ubiquitin (GST-Ubq) to ensure specific binding to GST. By combining noncovalent and covalent interactions, we have developed a new phototriggered protein–polymer conjugation method that is generally applicable to GST-fusion proteins.
Co-reporter:Erhan Bat;En-Wei Lin;Sina Saxer
Macromolecular Rapid Communications 2014 Volume 35( Issue 14) pp:1260-1265
Publication Date(Web):
DOI:10.1002/marc.201400160
Co-reporter:Steevens N. S. Alconcel;Sung Hye Kim;Lei Tao
Macromolecular Rapid Communications 2013 Volume 34( Issue 12) pp:983-989
Publication Date(Web):
DOI:10.1002/marc.201300205
Co-reporter:Nicholas M. Matsumoto, Panchami Prabhakaran, Leonard H. Rome, and Heather D. Maynard
ACS Nano 2013 Volume 7(Issue 1) pp:867
Publication Date(Web):December 21, 2012
DOI:10.1021/nn3053457
Synthetic modification of a recombinant protein cage called a vault with stimuli-responsive smart polymers provides access to a new class of biohybrid materials; the polymer nanocapsules retain the structure of the protein cage and exhibit the responsive nature of the polymer. Vaults are naturally occurring ubiquitous ribonucleoprotein particles 41 × 41 × 72.5 nm composed of a protein shell enclosing multiple copies of two proteins and multiple copies of one or more small untranslated RNAs. Recombinant vaults are structurally identical but lack the vault content. Poly(N-isopropylacrylamide) (pNIPAAm), a polymer responsive to heat, was conjugated to recombinant vaults that were composed of ∼78 copies of the major vault protein (MVP) modified to contain a cysteine rich region at the N-terminus (CP-MVP). The polymer was synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization to have a dansyl group at the alpha end and modified to have a thiol-reactive pyridyl disulfide at the omega end, which readily coupled to CP-MVP vaults. The resulting vault nanocapsules underwent reversible aggregation upon heating above the lower critical solution temperature (LCST) of the polymer as determined by electron microscopy (EM), dynamic light scattering experiments, and UV–vis turbidity analysis. The vault structure remained entirely intact throughout the phase transition; suggesting its use in a myriad of biomedical and biotechnology applications.Keywords: poly(N-isopropylacrylamide); protein cage; protein−polymer conjugate; thermo-responsive; vault
Co-reporter:Jingquan Liu, Ronald C. Li, Gregory J. Sand, Volga Bulmus, Thomas P. Davis, and Heather D. Maynard
Macromolecules 2013 Volume 46(Issue 1) pp:
Publication Date(Web):December 13, 2012
DOI:10.1021/ma302183g
A new methacrylate monomer with a reactive ketone side chain, 2-(4-oxopentanoate)ethyl methacrylate (PAEMA), was synthesized and subsequently polymerized by reversible addition–fragmentation chain transfer (RAFT) polymerization to give a polymer with a narrow molecular weight distribution (PDI = 1.25). The polymer was chain extended with poly(ethylene glycol methyl ether methacrylate) (PEGMA) to yield a block copolymer. Aminooxy-containing small molecules and oligoethylene glycol were conjugated to the ketone functionality of the side chains in high yields. Cytotoxicity of the oxime-linked tetra(ethylene glycol) polymer to mouse fibroblast cells was investigated; the polymer was found to be noncytotoxic up to 1 mg/mL. The ease with which this polymer is functionalized suggests that it may be useful in forming tailored polymeric medicines.
Co-reporter:Juneyoung Lee, En-Wei Lin, Uland Y. Lau, James L. Hedrick, Erhan Bat, and Heather D. Maynard
Biomacromolecules 2013 Volume 14(Issue 8) pp:
Publication Date(Web):June 18, 2013
DOI:10.1021/bm4003046
Herein, the synthesis of four different trehalose glycopolymers and investigation of their ability to stabilize proteins to heat and lyophilization stress are described. The disaccharide, α,α-trehalose, was modified with a styrenyl acetal, methacrylate acetal, styrenyl ether, or methacrylate moiety resulting in four different monomers. These monomers were then separately polymerized using free radical polymerization with azobisisobutyronitrile (AIBN) as an initiator to synthesize the glycopolymers. Horseradish peroxidase and glucose oxidase were incubated at 70 and 50 °C, respectively, and β-galactosidase was lyophilized multiple times in the presence of various ratios of the polymers or trehalose. The protein activities were subsequently tested and found to be significantly higher when the polymers were present during the stress compared to no additive and to equivalent amounts of trehalose. Different molecular weights (10 kDa, 20 kDa, and 40 kDa) were tested, and all were equivalent in their stabilization ability. However, some subtle differences were observed regarding stabilization ability between the different polymer samples, depending on the stress. Small molecules such as benzyl ether trehalose were not better stabilizers than trehalose, and the trehalose monomer decreased protein activity, suggesting that hydrophobized trehalose was not sufficient and that the polymeric structure was required. In addition, cytotoxicity studies with NIH 3T3 mouse embryonic fibroblast cells, RAW 264.7 murine macrophages, human dermal fibroblasts (HDFs), and human umbilical vein endothelial cells (HUVECs) were conducted with polymer concentrations up to 8 mg/mL. The data showed that all four polymers were noncytotoxic for all tested concentrations. The results together suggest that trehalose glycopolymers are promising as additives to protect proteins from a variety of stressors.
Co-reporter:Dr. Zachary P. Tolstyka;Wade Richardson;Dr. Erhan Bat;Caitlin J. Stevens;Dayanara P. Parra;Jonathan K. Dozier; Dr. Mark D. Distefano; Dr. Bruce Dunn; Dr. Heather D. Maynard
ChemBioChem 2013 Volume 14( Issue 18) pp:2464-2471
Publication Date(Web):
DOI:10.1002/cbic.201300478
Abstract
Herein, a combination of microcontact printing of functionalized alkanethiols and site-specific modification of proteins is utilized to chemoselectively immobilize proteins onto gold surfaces, either by oxime- or copper-catalyzed alkyne–azide click chemistry. Two molecules capable of click reactions were synthesized, an aminooxy-functionalized alkanethiol and an azide-functionalized alkanethiol, and self-assembled monolayer (SAM) formation on gold was confirmed by IR spectroscopy. The alkanethiols were then individually patterned onto gold surfaces by microcontact printing. Site-specifically modified proteins—horse heart myoglobin (HHMb) containing an N-terminal α-oxoamide and a red fluorescent protein (mCherry-CVIA) with a C-terminal alkyne—were immobilized by incubation onto respective stamped functionalized alkanethiol patterns. Pattern formation was confirmed by fluorescence microscopy.
Co-reporter:Rock J. Mancini ; Juneyoung Lee
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8474-8479
Publication Date(Web):April 20, 2012
DOI:10.1021/ja2120234
Herein, we report the synthesis of trehalose side chain polymers for stabilization of protein conjugates to environmental stressors. The glycomonomer 4,6-O-(4-vinylbenzylidene)-α,α-trehalose was synthesized in 40% yield over two steps without the use of protecting group chemistry. Polymers containing the trehalose pendent groups were prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization using two different thiol-reactive chain transfer agents (CTAs) for subsequent conjugation to proteins through disulfide linkages. The resulting glycopolymers were well-defined, and a range of molecular weights from 4200 to 49 500 Da was obtained. The polymers were conjugated to thiolated hen egg white lysozyme and purified. The glycopolymers when added or covalently attached to protein significantly increased stability toward lyophilization and heat relative to wild-type protein. Up to 100% retention of activity was observed when lysozyme was stressed ten times with lyophilization and 81% activity when the protein was heated at 90 °C for 1 h; this is in contrast to 16% and 18% retention of activity, respectively, for the wild-type protein alone. The glycopolymers were compared to equivalent concentrations of trehalose and poly(ethylene glycol) (PEG) and found to be superior at stabilizing the protein to lyophilization and heat. In addition, the protein–glycopolymer conjugates exhibited significant increases in lyophilization stability when compared to adding the same concentration of unconjugated polymer to the protein.
Co-reporter:Christopher M. Kolodziej
Journal of the American Chemical Society 2012 Volume 134(Issue 30) pp:12386-12389
Publication Date(Web):July 17, 2012
DOI:10.1021/ja304860q
Herein, features that alter their shape to form a different pattern upon an external trigger are described. Electron-beam lithography was used to fabricate micrometer- and nanometer-sized surface immobilized poly(triethylene glycol methacrylate) (pTEGMA) that exhibits significant thermal responsivity; the resulting hydrogels collapsed by up to 95% of their height upon addition of heat. Multicomponent features composed of both the thermoresponsive polymer and nonresponsive poly(ethylene glycol) (PEG) were then prepared. Upon increase in temperature, only the thermally responsive component of the pattern collapsed, causing a significant and predictable alteration in the overall pattern. Reversible micrometer- and nanometer-sized square-to-triangles, squares-to-checkerboards, smiles-to-neutral face, and zeros-to-ones shapes were shown.
Co-reporter:Christopher M. Kolodziej and Heather D. Maynard
Chemistry of Materials 2012 Volume 24(Issue 5) pp:774
Publication Date(Web):December 21, 2011
DOI:10.1021/cm202669f
This review summarizes the use of electron beam (e-beam) lithography to pattern biomolecules on surfaces. The focus is on approaches that employ poly(ethylene glycol) (PEG) resists. Overview of the different strategies used, including ablation of self-assembled monolayers and cross-linking of PEG, is provided. Subsequent use of surfaces to immobilize cells for tissue engineering applications is summarized.Keywords: cell adhesion; e-beam lithography; microarrays; nanoarrays; protein patterning;
Co-reporter:Gregory N. Grover, Jonathan Lam, Thi H. Nguyen, Tatiana Segura, and Heather D. Maynard
Biomacromolecules 2012 Volume 13(Issue 10) pp:
Publication Date(Web):September 12, 2012
DOI:10.1021/bm301346e
Oxime Click chemistry was used to form hydrogels that support cell adhesion. Eight-armed aminooxy poly(ethylene glycol) (PEG) was mixed with glutaraldehyde to form oxime-linked hydrogels. The mechanical properties, gelation kinetics, and water swelling ratios were studied and found to be tunable. It was also shown that gels containing the integrin ligand arginine-glycine-aspartic acid (RGD) supported mesenchymal stem cell (MSC) incorporation. High cell viability and proliferation of the encapsulated cells demonstrated biocompatibility of the material.
Co-reporter:Dr. Vimary Vázquez-Dorbatt;Juneyoung Lee;En-Wei Lin ; Heather D. Maynard
ChemBioChem 2012 Volume 13( Issue 17) pp:2478-2487
Publication Date(Web):
DOI:10.1002/cbic.201200480
Abstract
Natural saccharides are involved in numerous biological processes. It has been shown that these carbohydrates play a role in cell adhesion and proliferation, as well as protein stabilization, organization, and recognition. Certain carbohydrates also serve as receptors for viruses and bacteria. They are over expressed in diseases such as cancer. Hence, a lot of effort has been focused on mimicking these sugars. Polymers with pendent saccharide groups, also known as glycopolymers, are studied as oligo- and polysaccharide mimics. Controlled radical polymerization (CRP) techniques such as atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT) polymerization, and nitroxide-mediated polymerization (NMP), as well as cyanoxyl-mediated free radical polymerization have allowed chemists to synthesize well-defined glycopolymers that, in some cases, have particular end-group functionalities. This review focuses on the synthesis of glycopolymers by these methods and the applications of glycopolymers as natural saccharide mimics.
Co-reporter:Christopher M. Kolodziej ; Sung Hye Kim ; Rebecca M. Broyer ; Sina S. Saxer ; Caitlin G. Decker
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:247-255
Publication Date(Web):November 29, 2011
DOI:10.1021/ja205524x
Understanding and controlling cell adhesion on engineered scaffolds is important in biomaterials and tissue engineering. In this report we used an electron-beam (e-beam) lithography technique to fabricate patterns of a cell adhesive integrin ligand combined with a growth factor. Specifically, micron-sized poly(ethylene glycol) (PEG) hydrogels with aminooxy- and styrene sulfonate-functional groups were fabricated. Cell adhesion moieties were introduced using a ketone-functionalized arginine-glycine-aspartic acid (RGD) peptide to modify the O-hydroxylamines by oxime bond formation. Basic fibroblast growth factor (bFGF) was immobilized by electrostatic interaction with the sulfonate groups. Human umbilical vein endothelial cells (HUVECs) formed focal adhesion complexes on RGD- and RGD and bFGF-immobilized patterns as shown by immunostaining of vinculin and actin. In the presence of both bFGF and RGD, cell areas were larger. The data demonstrate confinement of cellular focal adhesions to chemically and physically well-controlled microenvironments created by a combination of e-beam lithography and “click” chemistry techniques. The results also suggest positive implications for addition of growth factors into adhesive patterns for cell-material interactions.
Co-reporter:Christopher M. Kolodziej, Chien-Wen Chang and Heather D. Maynard
Journal of Materials Chemistry A 2011 vol. 21(Issue 5) pp:1457-1461
Publication Date(Web):27 Oct 2010
DOI:10.1039/C0JM02370A
The ability to immobilize biomolecules on nanopatterned substrates is important for diverse applications including tissue engineering and proteomics. The design of general linkers can allow a single substrate to be used for multiple purposes, as well as reconfiguration of devices if the linkage is reversible. In this work, the glutathione S-transferase (GST) tag was used to reversibly immobilize green fluorescent protein (GFP) and basic fibroblast growth factor (FGF2) on glutathione (GSH)-functionalized nanopatterns fabricated by electron-beam lithography.
Co-reporter:Rebecca M. Broyer, Gregory N. Grover and Heather D. Maynard
Chemical Communications 2011 vol. 47(Issue 8) pp:2212-2226
Publication Date(Web):12 Jan 2011
DOI:10.1039/C0CC04062B
Protein–polymer conjugates are important in diverse fields including drug delivery, biotechnology, and nanotechnology. This feature article highlights recent advances in the synthesis and application of protein–polymer conjugates by controlled radical polymerization techniques. Special emphasis on new applications of the materials, particularly in biomedicine, is provided.
Co-reporter:Steevens N. S. Alconcel, Arnold S. Baas and Heather D. Maynard
Polymer Chemistry 2011 vol. 2(Issue 7) pp:1442-1448
Publication Date(Web):27 Apr 2011
DOI:10.1039/C1PY00034A
PEGylation or covalent attachment of poly(ethylene glycol) improves the pharmacokinetic properties of protein drugs. In vivo circulation lifetimes are increased and dosages are decreased, resulting in improved patient quality of life. PEG may be attached to proteins using a variety of different chemical reactions. This review discusses currently available FDA-approved PEGylated protein drugs, their intended use and target, and the PEG attachment chemistry utilized.
Co-reporter:Rebecca M. Broyer, Eric Schopf, Christopher M. Kolodziej, Yong Chen and Heather D. Maynard
Soft Matter 2011 vol. 7(Issue 21) pp:9972-9977
Publication Date(Web):30 Aug 2011
DOI:10.1039/C1SM05991B
In the study described in this report orthogonal Click reactions were utilized to immobilize two different proteins on surfaces side-by-side and in multilayer constructs. Alkyne- and azide-functionalized poly(ethylene glycol) hydrogel features were fabricated. Copper-catalyzed Huisgen 1,3 dipolar cycloaddition and oxime chemistry were employed to conjugate an azide-functionalized ubiquitin and oxoamide-modified myoglobin, respectively. Multicomponent patterning was verified by fluorescence imaging.
Co-reporter:Karen L. Christman, Rebecca M. Broyer, Eric Schopf, Christopher M. Kolodziej, Yong Chen, and Heather D. Maynard
Langmuir 2011 Volume 27(Issue 4) pp:1415-1418
Publication Date(Web):December 30, 2010
DOI:10.1021/la103978x
Patterning proteins on the nanoscale is important for applications in biology and medicine. As feature sizes are reduced, it is critical that immobilization strategies provide site-specific attachment of the biomolecules. In this study, oxime chemistry was exploited to conjugate proteins onto nanometer-sized features. Poly(Boc-aminooxy tetra(ethylene glycol) methacrylate) was synthesized by free radical polymerization. The polymer was patterned onto silicon wafers using an electron beam writer. Trifluoroacetic acid removal of the Boc groups provided the desired aminooxy functionality. In this manner, patterns of concentric squares and contiguous bowtie shapes were fabricated with 150−170-nm wide features. Ubiquitin modified at the N-terminus with an α-ketoamide group and Nε-levulinyl lysine-modified bovine serum albumin were subsequently conjugated to the polymer nanopatterns. Protein immobilization was confirmed by fluorescence microscopy. Control studies on protected surfaces and using proteins presaturated with O-methoxyamine indicated that attachment occurred via oxime bond formation.
Co-reporter:Karina L. Heredia, Lei Tao, Gregory N. Grover and Heather D. Maynard
Polymer Chemistry 2010 vol. 1(Issue 2) pp:168-170
Publication Date(Web):18 Jan 2010
DOI:10.1039/B9PY00369J
A heterotelechelic biotin–maleimide polymer containing a cleavable disulfide bond was synthesized by RAFT polymerization and used to reversibly modify surfaces with proteins.
Co-reporter:Chien-Wen Chang;Thi H. Nguyen
Macromolecular Rapid Communications 2010 Volume 31( Issue 19) pp:1691-1695
Publication Date(Web):
DOI:10.1002/marc.201000333
Co-reporter:Eric Schopf, Rebecca Broyer, Lei Tao, Yong Chen and Heather D. Maynard
Chemical Communications 2009 (Issue 32) pp:4818-4820
Publication Date(Web):17 Jul 2009
DOI:10.1039/B908282D
A pyrene-functionalized polymer was patterned viaelectron beam lithography onto a silicon wafer and shown to selectively bind with carbon nanotubes.
Co-reporter:Lei Tao, Catherine S. Kaddis, Rachel R. Ogorzalek Loo, Gregory N. Grover, Joseph A. Loo and Heather D. Maynard
Chemical Communications 2009 (Issue 16) pp:2148-2150
Publication Date(Web):18 Mar 2009
DOI:10.1039/B822799C
Synthesis of well-defined homodimeric protein–polymer conjugates using RAFT polymerization is described.
Co-reporter:Rock J. Mancini, Ronald C. Li, Zachary P. Tolstyka and Heather D. Maynard
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 23) pp:4954-4959
Publication Date(Web):06 Oct 2009
DOI:10.1039/B904195H
A photo-caged aminooxy alkane thiol synthesized in 7 steps and 15% overall yield was used to form a self-assembled monolayer (SAM). Photo-deprotection on the surface was confirmed by FT-IR spectroscopy and contact angle goniometry. Conjugation of a small molecule ketone, ethyl levulinate, further confirmed the presence of aminooxy groups on the surface.
Co-reporter:Karina L. Heredia, Gregory N. Grover, Lei Tao and Heather D. Maynard
Macromolecules 2009 Volume 42(Issue 7) pp:2360-2367
Publication Date(Web):January 30, 2009
DOI:10.1021/ma8022712
We describe a straightforward approach to synthesize polymers with end-groups that bind site-specifically to two different proteins. Telechelic biotin−maleimide poly(N-isopropylacrylamide) (pNIPAAm) was synthesized for the formation of streptavidin (SAv)−bovine serum albumin (BSA) polymer conjugates. Reversible addition−fragmentation chain transfer (RAFT) polymerization of NIPAAm was conducted in the presence of biotinylated chain transfer agents (CTAs) with either ester or amide linkages, and the resultant α-biotinylated pNIPAAms were formed with low polydispersity indices (PDI ≤ 1.09). UV−vis analysis of the trithiocarbonate chain-ends indicated 88% or greater retention of the group. A maleimide was introduced to the ω chain-end via a radical cross-coupling reaction with a functionalized azo-initiator. The polymer structures were characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC). The resultant biotin−maleimide heterotelechelic polymer was used to form a SAv−BSA heterodimer conjugate. Bioconjugate formation was confirmed by gel electrophoresis.
Co-reporter:Gregory N. Grover, Steevens N. S. Alconcel, Nicholas M. Matsumoto and Heather D. Maynard
Macromolecules 2009 Volume 42(Issue 20) pp:7657-7663
Publication Date(Web):October 1, 2009
DOI:10.1021/ma901036x
Herein we report the synthesis of vinyl sulfone end-functionalized PEGylated polymers by reversible addition−fragmentation chain transfer (RAFT) polymerization for conjugation to proteins. Poly(ethylene glycol) methyl ether acrylate (PEGA) was polymerized in the presence of 1-phenylethyl dithiobenzoate with 2,2′-azobis(2-methylpropionitrile) as the initiator to generate well-defined polyPEGAs with number-average molecular weights (Mn) by gel permeation chromatography (GPC) of 6.7, 11.8, and 16.1 kDa. Postpolymerization, the majority of polymer chains contained the dithioester functional group at the omega chain end, and the polydispersity indexes (PDI) of the polymers ranged from 1.08 to 1.24. The dithioesters were subsequently reduced via aminolysis, and the resulting thiols were trapped with divinyl sulfone in situ to produce semitelechelic, vinyl sulfone polyPEGAs with efficiencies ranging between 85% and 99%. It was determined that the retention of vinyl sulfone was directly related to reaction time, with the maximum dithioester being transformed into a vinyl sulfone within 30 min. Longer reaction times resulted in slow decomposition of the vinyl sulfone end group. The resulting semitelechelic vinyl sulfone polymers were then conjugated to a protein containing a free cysteine, bovine serum albumin (BSA). Gel electrophoresis demonstrated that the reaction was highly efficient and that conjugates of increasing size were readily prepared. After polymer attachment, the activity of the BSA was 92% of the unmodified biomolecule.
Co-reporter:Lei Tao, Catherine S. Kaddis, Rachel R. Ogorzalek Loo, Gregory N. Grover, Joseph A. Loo and Heather D. Maynard
Macromolecules 2009 Volume 42(Issue 21) pp:8028-8033
Publication Date(Web):July 29, 2009
DOI:10.1021/ma901540p
Protein−polymer conjugates exhibit superior properties to unmodified proteins, generating a high demand for these materials in the fields of medicine, biotechnology, and nanotechnology. Multimeric conjugates are predicted to surpass the activity of monomeric conjugates. Herein, we report a straightforward method to synthesize multimeric polymer conjugates. Four-armed poly(N-isopropylacrylamide) (pNIPAAm) was synthesized by reversible addition−fragmentation chain transfer (RAFT) polymerization in the presence of a tetrafunctionalized trithiocarbonate chain transfer agent (CTA). The polymer molecular weight, architecture, and polydispersity index (PDI) were verified by gel permeation chromatography (GPC), dynamic light scattering gel permeation chromatography (DLS-GPC), and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. This approach afforded well-defined polymers (PDI’s < 1.06) and the ability to target various molecular weights. Maleimide functional groups were introduced at the chain ends by heating the polymers in the presence of a furan-protected azo-initiator. This allowed for site-specific conjugation of V131C T4 lysozyme to the polymers to generate multimeric protein−polymer conjugates. MALDI-TOF mass spectrometry, electrospray ionization gas-phase electrophoretic mobility macromolecule analysis (ESI-GEMMA), gel electrophoresis, and liquid chromatography tandem mass spectrometry (LC-MS/MS) of the trypsin digests demonstrated that multimeric protein−polymer conjugates had formed. This simple strategy provides ready access to star protein−polymer conjugates for application in the fields of drug discovery, drug delivery, and nanotechnology.
Co-reporter:Vimary Vázquez-Dorbatt, Zachary P. Tolstyka and Heather D. Maynard
Macromolecules 2009 Volume 42(Issue 20) pp:7650-7656
Publication Date(Web):September 30, 2009
DOI:10.1021/ma9013803
A Boc-protected aminooxy end-functionalized poly(N-isopropylacrylamide) (pNIPAAm) was synthesized by reversible addition−fragmentation chain transfer (RAFT) polymerization. The monomer was polymerized in the presence of a Boc-protected aminooxy trithiocarbonate chain transfer agent (CTA) utilizing 2,2′-azobis(2-isobutyronitrile) (AIBN) as the initiator in dimethylformamide (DMF) at 70 °C. The final polymer had a number-average molecular weight (Mn) of 4200 Da as determined by 1H NMR spectroscopy and a narrow polydispersity index (1.14) by gel permeation chromatography (GPC). The Boc group was removed, and the polymer was then incubated with Nε-levulinyllysine-modified bovine serum albumin (BSA). Gel electrophoresis confirmed that the conjugation was successful. The aminooxy end-functionalized pNIPAAm was also immobilized on a gold surface after reduction of the trithiocarbonate end-group. The pNIPAAm surface was then incubated with an aldehyde-modified heparin to yield the polysaccharide-functionalized surface. All surface modifications were monitored by FT-IR spectroscopy.
Co-reporter:Emmanuelle Bays, Lei Tao, Chien-Wen Chang and Heather D. Maynard
Biomacromolecules 2009 Volume 10(Issue 7) pp:
Publication Date(Web):June 9, 2009
DOI:10.1021/bm9001987
Maleimide end functionalized polymers for site-selective conjugation to free cysteines of proteins were synthesized using reversible addition−fragmentation chain transfer (RAFT) polymerization. A furan-protected maleimide chain transfer agent (CTA) was employed in the RAFT polymerization of poly(ethylene glycol) methyl ether acrylate (PEGA). Gel permeation chromatography (GPC) with laser light scattering detection indicated that 20000 and 39000 Da polyPEGA had been made with polydispersity indices of 1.25 and 1.36, respectively. The maleimide group on the polymer chain end was exposed by heating the poly(PEGA)s for 4 h. The deprotection efficiency was estimated to be 80 and 60% for poly(PEGA)20 kDa and poly(PEGA)39 kDa, respectively. Maleimide-poly(PEGA)s were conjugated to V131C T4 lysozyme (T4L), and the resultant polymer−protein conjugates were characterized by size exclusion chromatography and gel electrophoresis.
Co-reporter:Vimary Vázquez-Dorbatt, Zachary P. Tolstyka, Chien-Wen Chang and Heather D. Maynard
Biomacromolecules 2009 Volume 10(Issue 8) pp:
Publication Date(Web):July 16, 2009
DOI:10.1021/bm900395h
A pyridyl disulfide end-functionalized polymer with N-acetyl-d-glucosamine pendant side-chains was synthesized by atom transfer radical polymerization (ATRP). The glycopolymer was prepared from a pyridyl disulfide initiator catalyzed by a Cu(I)/Cu(II)/2,2′-bipyridine system in a mixture of methanol and water at 30 °C. The final polymer had a number-average molecular weight (Mn) of 13.0 kDa determined by 1H NMR spectroscopy and a narrow polydispersity index (1.12) determined by gel permeation chromatography (GPC). The pyridyl disulfide end-group was then utilized to conjugate the glycopolymer to a double-stranded short interfering RNA (siRNA). Characterization of the glycopolymer-siRNA by polyacrylamide gel electrophoresis (PAGE) showed 97% conjugation. The activated disulfide polymer was also patterned on gold via microcontact printing. The pyridyl disulfide allowed for ready immobilization of the glycopolymer into 200 μm sized features on the surface.
Co-reporter:Karina L. Heredia, Thi H. Nguyen, Chien-Wen Chang, Volga Bulmus, Thomas P. Davis and Heather D. Maynard
Chemical Communications 2008 (Issue 28) pp:3245-3247
Publication Date(Web):06 Jun 2008
DOI:10.1039/B804812F
A straightforward synthetic method to prepare pyridyl disulfide end functionalized poly(PEG acrylate) by RAFT polymerization for efficient and reversible conjugation to siRNA is described.
Co-reporter:Heather D. Maynard, Karina L. Heredia, Ronald C. Li, Dayanara P. Parra and Vimary Vázquez-Dorbatt
Journal of Materials Chemistry A 2007 vol. 17(Issue 38) pp:4015-4017
Publication Date(Web):05 Sep 2007
DOI:10.1039/B710513D
The aqueous phase behaviour of poly(N-isopropylacrylamide)–protein conjugates prepared by atom transfer radical polymerization (ATRP) is reported.
Co-reporter:Karen L. Christman, Rebecca M. Broyer, Zachary P. Tolstyka and Heather D. Maynard
Journal of Materials Chemistry A 2007 vol. 17(Issue 19) pp:2021-2027
Publication Date(Web):14 Mar 2007
DOI:10.1039/B618002G
Immobilizing proteins in specific orientations is important for diagnostic protein arrays, biomaterials, and other applications where retention of bioactivity is essential. We report an approach for protein micropatterning that exploits a chemoselective reaction to conjugate proteins at the N-terminus to polymer films. A copolymer from 2-hydroxyethyl methacrylate and a Boc-protected aminooxy tetra(ethylene glycol) methacrylate was synthesized by radical polymerization. Boc groups were locally deprotected using photoacid generator-based photolithography. Micropatterns were verified by fluorescence microscopy utilizing green fluorescent aldehyde microspheres. Streptavidin that was subjected to a transamination reaction to install an α-ketoamide group at the N-terminus was conjugated to the patterns by oxime bond formation. Since the majority of proteins may be modified to contain a reactive carbonyl group, this methodology should be applicable to pattern a wide variety of proteins specifically through the N-terminus.
Co-reporter:Jungyeon Hwang, Ronald C. Li, Heather D. Maynard
Journal of Controlled Release 2007 Volume 122(Issue 3) pp:279-286
Publication Date(Web):8 October 2007
DOI:10.1016/j.jconrel.2007.04.010
Polymers with reactive side chains and narrow molecular weight distributions were synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization, and the potential to utilize these polymers to prepare drug carriers was demonstrated. p-Nitrophenyl methacrylate (NPMA) and diethoxypropyl methacrylate (DEPMA) were polymerized utilizing cumyl dithiobenzoate (CDB) as the chain transfer agent and azobisisobutyronitrile (AIBN) as the initiator to high conversions (≥ 86%). The resulting pNPMA and pDEPMA had narrow molecular weight distributions (polydispersity indices < 1.3). The ability to functionalize these polymers was confirmed. For pNPMA, up to 86% of the side chains were substituted with the amino acid, glycine methyl ester. The side chains of pDEPMA were hydrolyzed to aldehydes and reaction with O-benzylhydroxylamine and O-methylhydroxylamine to form stable oxime bond conjugates was demonstrated. The percent substitution depended on the feed ratios. Conjugation of an aminooxy-functionalized RGD peptide was also demonstrated.
Co-reporter:Ronald C. Li, Jungyeon Hwang and Heather D. Maynard
Chemical Communications 2007 (Issue 35) pp:3631-3633
Publication Date(Web):02 Aug 2007
DOI:10.1039/B709304G
Block copolymers with sequences of differential reactivity were synthesized, and the step-wise and selective derivatization to form a new block copolymer was demonstrated.
Co-reporter:Karina L. Heredia and Heather D. Maynard
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 1) pp:45-53
Publication Date(Web):08 Nov 2006
DOI:10.1039/B612355D
Protein–polymer conjugates are widely employed for applications in medicine, biotechnology and nanotechnology. Covalent attachment of synthetic polymers to proteins improves protein stability, solubility, and biocompatibility. Furthermore, synthetic polymers impart new properties such as self assembly and phase behavior. Polymer attachment at amino acid side-chains and at ligand binding sites is typically exploited. This Emerging Area focuses on synthetic methods to prepare protein-reactive polymers and also employing the protein itself as an initiator for polymerization.
Co-reporter:Karen L. Christman, Vanessa D. Enriquez-Rios and Heather D. Maynard
Soft Matter 2006 vol. 2(Issue 11) pp:928-939
Publication Date(Web):29 Sep 2006
DOI:10.1039/B611000B
A variety of techniques have been developed to site-specifically immobilize biomolecules onto surfaces with resolutions below one micron. The ability to pattern proteins and peptides in particular has great potential for applications in biosensors, biomaterials, and tissue engineering. For example, immobilizing proteins at the nanoscale could lead to the development of diagnostic protein nanoarrays, while patterning peptides could lead to greater control over the cell/biomaterial interface. This review discusses the methods that have been reported for patterning proteins and peptides with submicron and nanometer resolutions.
Co-reporter:Ronald C. Li;Rebecca M. Broyer
Journal of Polymer Science Part A: Polymer Chemistry 2006 Volume 44(Issue 17) pp:5004-5013
Publication Date(Web):21 JUL 2006
DOI:10.1002/pola.21609
The synthesis of a polymer with acetal functionalized side chains, deprotection to aldehydes, and conjugation of aminooxy-modified molecules is reported. Poly(3,3′-diethoxypropyl methacrylate) (PDEPMA) was prepared by atom transfer radical polymerization (ATRP). Kinetic investigation of the ATRP of DEPMA from ethyl 2-bromoisobutyrate in methanol with copper (I) bromide and 2,2′-bipyridine (BIPY) at ambient temperature revealed a controlled polymerization. Altering the initial monomer to initiator ratios resulted in 75–93% conversion to polymers with different molecular weights and narrow molecular weight distributions (PDIs < 1.3). Reactive aldehyde groups were produced by hydrolysis of the acetals in dilute acid. Aminooxy-functionalized oligo(ethylene glycol) and O-(carboxymethyl)hydroxylamine were conjugated to the side chains via oxime linkages. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 5004–5013, 2006
Co-reporter:Debora Bontempo, Ronald C. Li, Tiffany Ly, Carrie E. Brubaker and Heather D. Maynard
Chemical Communications 2005 (Issue 37) pp:4702-4704
Publication Date(Web):22 Aug 2005
DOI:10.1039/B507912H
Low polydispersity poly(N-isopropylacrylamide) with a biotin end-group was obtained in one step from a biotinylated initiator for atom transfer radical polymerization and interacted with streptavidin to generate the thermosensitive polymer–protein conjugate.
Co-reporter:Caitlin G. Decker, Yu Wang, Samantha J. Paluck, Lu Shen, Joseph A. Loo, Alex J. Levine, Lloyd S. Miller, Heather D. Maynard
Biomaterials (March 2016) Volume 81() pp:157-168
Publication Date(Web):March 2016
DOI:10.1016/j.biomaterials.2015.12.003
Site-specific chemical dimerization of fibroblast growth factor 2 (FGF2) with the optimal linker length resulted in a FGF2 homodimer with improved granulation tissue formation and blood vessel formation at exceptionally low concentrations. Homodimers of FGF2 were synthesized through site-specific linkages to both ends of different molecular weight poly(ethylene glycols) (PEGs). The optimal linker length was determined by screening dimer-induced metabolic activity of human dermal fibroblasts and found to be that closest to the inter-cysteine distance, 70 Å, corresponding to 2 kDa PEG. A straightforward analysis of the kinetics of second ligand binding as a function of tether length showed that, as the polymerization index (the number of monomer repeat units in the polymer, N) of the tether decreases, the mean time for second ligand capture decreases as ∼N3/2, leading to an enhancement of the number of doubly bound ligands in steady-state for a given (tethered) ligand concentration. FGF2-PEG2k-FGF2 induced greater fibroblast metabolic activity than FGF2 alone, all other dimers, and all monoconjugates, at each concentration tested, with the greatest difference observed at low (0.1 ng/mL) concentration. FGF2-PEG2k-FGF2 further exhibited superior activity compared to FGF2 for both metabolic activity and migration in human umbilical vein endothelial cells, as well as improved angiogenesis in a coculture model in vitro. Efficacy in an in vivo wound healing model was assessed in diabetic mice. FGF2-PEG2k-FGF2 increased granulation tissue and blood vessel density in the wound bed compared to FGF2. The results suggest that this rationally designed construct may be useful for improving the fibroblast matrix formation and angiogenesis in chronic wound healing.
Co-reporter:Caitlin G. Decker, Yu Wang, Samantha J. Paluck, Lu Shen, Joseph A. Loo, Alex J. Levine, Lloyd S. Miller, Heather D. Maynard
Biomaterials (March 2016) Volume 81() pp:
Publication Date(Web):March 2016
DOI:10.1016/j.biomaterials.2015.12.003
Site-specific chemical dimerization of fibroblast growth factor 2 (FGF2) with the optimal linker length resulted in a FGF2 homodimer with improved granulation tissue formation and blood vessel formation at exceptionally low concentrations. Homodimers of FGF2 were synthesized through site-specific linkages to both ends of different molecular weight poly(ethylene glycols) (PEGs). The optimal linker length was determined by screening dimer-induced metabolic activity of human dermal fibroblasts and found to be that closest to the inter-cysteine distance, 70 Å, corresponding to 2 kDa PEG. A straightforward analysis of the kinetics of second ligand binding as a function of tether length showed that, as the polymerization index (the number of monomer repeat units in the polymer, N) of the tether decreases, the mean time for second ligand capture decreases as ∼N3/2, leading to an enhancement of the number of doubly bound ligands in steady-state for a given (tethered) ligand concentration. FGF2-PEG2k-FGF2 induced greater fibroblast metabolic activity than FGF2 alone, all other dimers, and all monoconjugates, at each concentration tested, with the greatest difference observed at low (0.1 ng/mL) concentration. FGF2-PEG2k-FGF2 further exhibited superior activity compared to FGF2 for both metabolic activity and migration in human umbilical vein endothelial cells, as well as improved angiogenesis in a coculture model in vitro. Efficacy in an in vivo wound healing model was assessed in diabetic mice. FGF2-PEG2k-FGF2 increased granulation tissue and blood vessel density in the wound bed compared to FGF2. The results suggest that this rationally designed construct may be useful for improving the fibroblast matrix formation and angiogenesis in chronic wound healing.
Co-reporter:Heather D. Maynard, Karina L. Heredia, Ronald C. Li, Dayanara P. Parra and Vimary Vázquez-Dorbatt
Journal of Materials Chemistry A 2007 - vol. 17(Issue 38) pp:NaN4017-4017
Publication Date(Web):2007/09/05
DOI:10.1039/B710513D
The aqueous phase behaviour of poly(N-isopropylacrylamide)–protein conjugates prepared by atom transfer radical polymerization (ATRP) is reported.
Co-reporter:Lei Tao, Catherine S. Kaddis, Rachel R. Ogorzalek Loo, Gregory N. Grover, Joseph A. Loo and Heather D. Maynard
Chemical Communications 2009(Issue 16) pp:NaN2150-2150
Publication Date(Web):2009/03/18
DOI:10.1039/B822799C
Synthesis of well-defined homodimeric protein–polymer conjugates using RAFT polymerization is described.
Co-reporter:Ronald C. Li, Jungyeon Hwang and Heather D. Maynard
Chemical Communications 2007(Issue 35) pp:NaN3633-3633
Publication Date(Web):2007/08/02
DOI:10.1039/B709304G
Block copolymers with sequences of differential reactivity were synthesized, and the step-wise and selective derivatization to form a new block copolymer was demonstrated.
Co-reporter:Karina L. Heredia and Heather D. Maynard
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 1) pp:NaN53-53
Publication Date(Web):2006/11/08
DOI:10.1039/B612355D
Protein–polymer conjugates are widely employed for applications in medicine, biotechnology and nanotechnology. Covalent attachment of synthetic polymers to proteins improves protein stability, solubility, and biocompatibility. Furthermore, synthetic polymers impart new properties such as self assembly and phase behavior. Polymer attachment at amino acid side-chains and at ligand binding sites is typically exploited. This Emerging Area focuses on synthetic methods to prepare protein-reactive polymers and also employing the protein itself as an initiator for polymerization.
Co-reporter:Rock J. Mancini, Ronald C. Li, Zachary P. Tolstyka and Heather D. Maynard
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 23) pp:NaN4959-4959
Publication Date(Web):2009/10/06
DOI:10.1039/B904195H
A photo-caged aminooxy alkane thiol synthesized in 7 steps and 15% overall yield was used to form a self-assembled monolayer (SAM). Photo-deprotection on the surface was confirmed by FT-IR spectroscopy and contact angle goniometry. Conjugation of a small molecule ketone, ethyl levulinate, further confirmed the presence of aminooxy groups on the surface.
Co-reporter:Karen L. Christman, Rebecca M. Broyer, Zachary P. Tolstyka and Heather D. Maynard
Journal of Materials Chemistry A 2007 - vol. 17(Issue 19) pp:NaN2027-2027
Publication Date(Web):2007/03/14
DOI:10.1039/B618002G
Immobilizing proteins in specific orientations is important for diagnostic protein arrays, biomaterials, and other applications where retention of bioactivity is essential. We report an approach for protein micropatterning that exploits a chemoselective reaction to conjugate proteins at the N-terminus to polymer films. A copolymer from 2-hydroxyethyl methacrylate and a Boc-protected aminooxy tetra(ethylene glycol) methacrylate was synthesized by radical polymerization. Boc groups were locally deprotected using photoacid generator-based photolithography. Micropatterns were verified by fluorescence microscopy utilizing green fluorescent aldehyde microspheres. Streptavidin that was subjected to a transamination reaction to install an α-ketoamide group at the N-terminus was conjugated to the patterns by oxime bond formation. Since the majority of proteins may be modified to contain a reactive carbonyl group, this methodology should be applicable to pattern a wide variety of proteins specifically through the N-terminus.
Co-reporter:Christopher M. Kolodziej, Chien-Wen Chang and Heather D. Maynard
Journal of Materials Chemistry A 2011 - vol. 21(Issue 5) pp:NaN1461-1461
Publication Date(Web):2010/10/27
DOI:10.1039/C0JM02370A
The ability to immobilize biomolecules on nanopatterned substrates is important for diverse applications including tissue engineering and proteomics. The design of general linkers can allow a single substrate to be used for multiple purposes, as well as reconfiguration of devices if the linkage is reversible. In this work, the glutathione S-transferase (GST) tag was used to reversibly immobilize green fluorescent protein (GFP) and basic fibroblast growth factor (FGF2) on glutathione (GSH)-functionalized nanopatterns fabricated by electron-beam lithography.
Co-reporter:Karina L. Heredia, Thi H. Nguyen, Chien-Wen Chang, Volga Bulmus, Thomas P. Davis and Heather D. Maynard
Chemical Communications 2008(Issue 28) pp:NaN3247-3247
Publication Date(Web):2008/06/06
DOI:10.1039/B804812F
A straightforward synthetic method to prepare pyridyl disulfide end functionalized poly(PEG acrylate) by RAFT polymerization for efficient and reversible conjugation to siRNA is described.
Co-reporter:Rebecca M. Broyer, Gregory N. Grover and Heather D. Maynard
Chemical Communications 2011 - vol. 47(Issue 8) pp:NaN2226-2226
Publication Date(Web):2011/01/12
DOI:10.1039/C0CC04062B
Protein–polymer conjugates are important in diverse fields including drug delivery, biotechnology, and nanotechnology. This feature article highlights recent advances in the synthesis and application of protein–polymer conjugates by controlled radical polymerization techniques. Special emphasis on new applications of the materials, particularly in biomedicine, is provided.
Co-reporter:Eric Schopf, Rebecca Broyer, Lei Tao, Yong Chen and Heather D. Maynard
Chemical Communications 2009(Issue 32) pp:NaN4820-4820
Publication Date(Web):2009/07/17
DOI:10.1039/B908282D
A pyrene-functionalized polymer was patterned viaelectron beam lithography onto a silicon wafer and shown to selectively bind with carbon nanotubes.
.jpg)




![PENTANOIC ACID, 4-OXO-, 2-[2-(2-HYDROXYETHOXY)ETHOXY]ETHYL ESTER](http://img.cochemist.com/ccimg/879900/879871-77-9.png)
![PENTANOIC ACID, 4-OXO-, 2-[2-(2-HYDROXYETHOXY)ETHOXY]ETHYL ESTER](http://img.cochemist.com/ccimg/879900/879871-77-9_b.png)






![Poly[(2,3-dihydro-1H-indene-1,3-diyl)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/401000/400900-20-1.png)
![Poly[(2,3-dihydro-1H-indene-1,3-diyl)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/401000/400900-20-1_b.png)
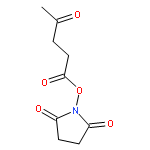
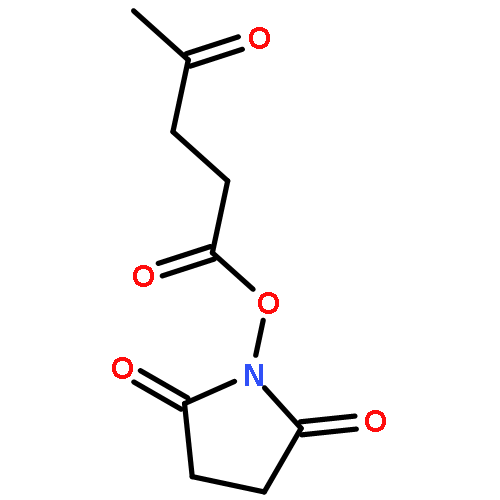







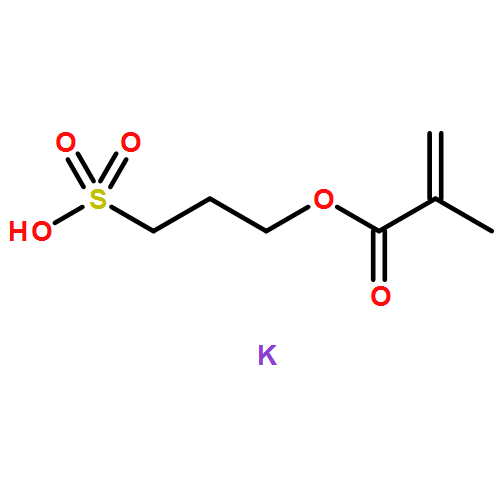

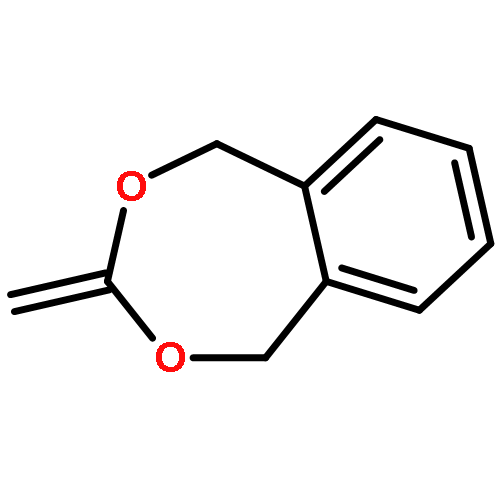


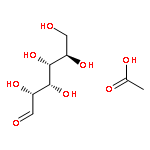








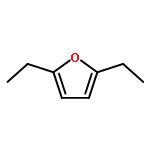







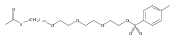
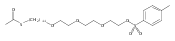
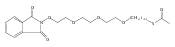





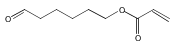
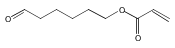
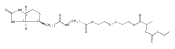
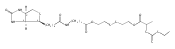







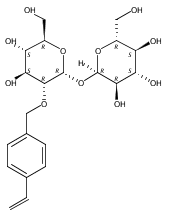


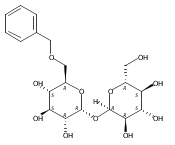
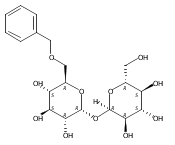
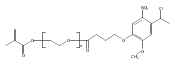
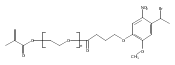



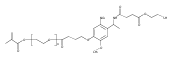

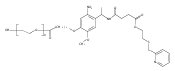

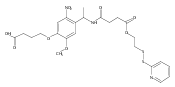









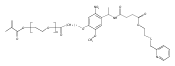





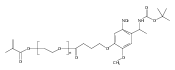
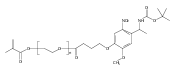

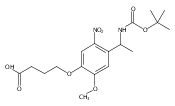




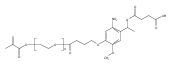
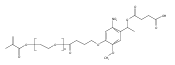

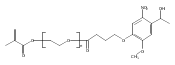
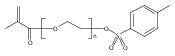
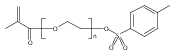













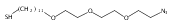
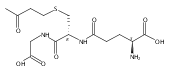

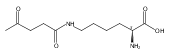
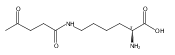










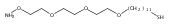
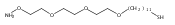

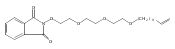

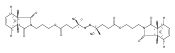


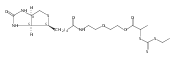













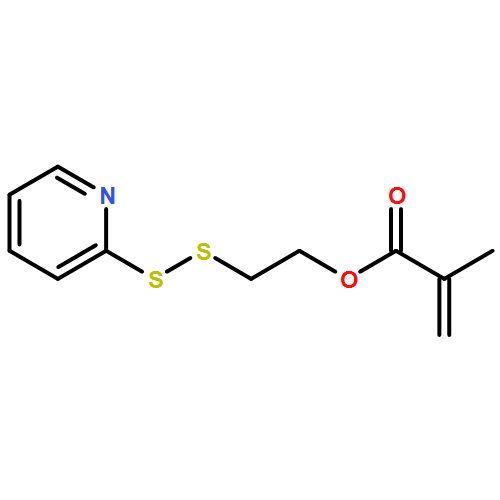















![Pentanoic acid,5-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-oxo-](http://img.cochemist.com/ccimg/160200/160111-41-1.png)
![Pentanoic acid,5-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-oxo-](http://img.cochemist.com/ccimg/160200/160111-41-1_b.png)


![Poly(oxy-1,2-ethanediyl), α-[(4-ethenylphenyl)methyl]-ω-methoxy-, homopolymer](http://img.cochemist.com/ccimg/138500/138433-30-4.png)
![Poly(oxy-1,2-ethanediyl), α-[(4-ethenylphenyl)methyl]-ω-methoxy-, homopolymer](http://img.cochemist.com/ccimg/138500/138433-30-4_b.png)



![Ethanol,2-[2-[2-(10-undecen-1-yloxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/130800/130727-45-6.png)
![Ethanol,2-[2-[2-(10-undecen-1-yloxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/130800/130727-45-6_b.png)










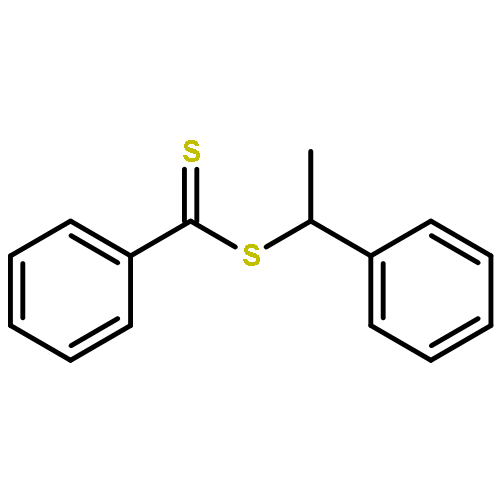



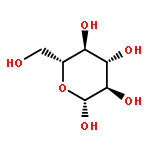
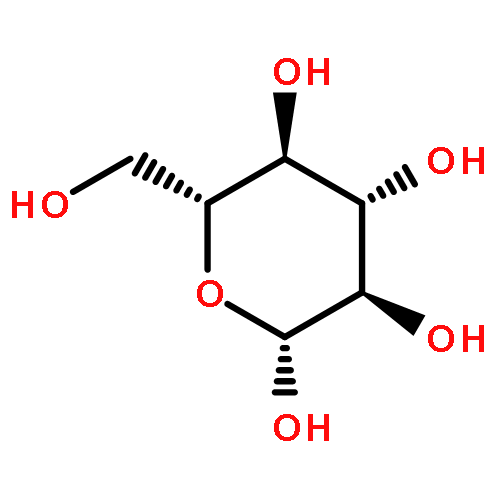




![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)




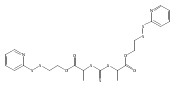
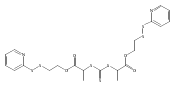


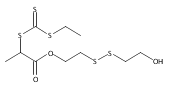



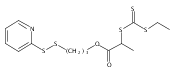

![Ethanol, 2-[2-[2-[2-(aminooxy)ethoxy]ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/106500/106492-60-8.png)
![Ethanol, 2-[2-[2-[2-(aminooxy)ethoxy]ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/106500/106492-60-8_b.png)
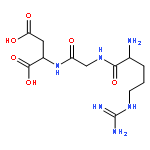

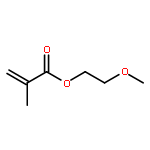
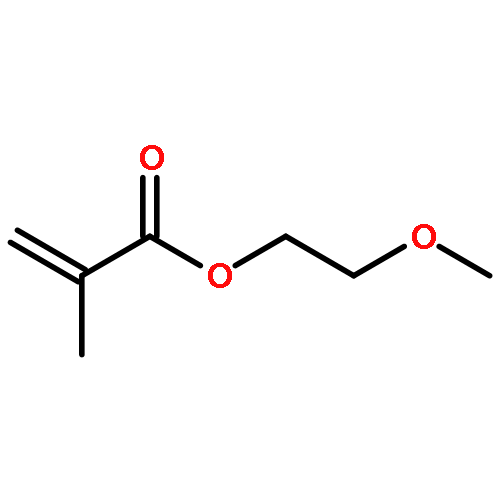




![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis-](/data/chemimg/630600/6332-91-8.png)
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis-](/data/chemimg/630600/6332-91-8_b.png)


![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-](/data/chemimg/178400/1035996-09-8.png)
![Propanoic acid, 2-[[(ethylthio)thioxomethyl]thio]-](/data/chemimg/178400/1035996-09-8_b.png)