Co-reporter:Bumki Kim, Ranjala Ratnayake, Hyunji Lee, Guqin Shi, Sabrina L. Zeller, Chenglong Li, Hendrik Luesch, Jiyong Hong
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmc.2017.03.071
Histone acetylation is an extensively investigated post-translational modification that plays an important role as an epigenetic regulator. It is controlled by histone acetyl transferases (HATs) and histone deacetylases (HDACs). The overexpression of HDACs and consequent hypoacetylation of histones have been observed in a variety of different diseases, leading to a recent focus of HDACs as attractive drug targets. The natural product largazole is one of the most potent natural HDAC inhibitors discovered so far and a number of largazole analogs have been prepared to define structural requirements for its HDAC inhibitory activity. However, previous structure–activity relationship studies have heavily investigated the macrocycle region of largazole, while there have been only limited efforts to probe the effect of various zinc-binding groups (ZBGs) on HDAC inhibition. Herein, we prepared a series of largazole analogs with various ZBGs and evaluated their HDAC inhibition and cytotoxicity. While none of the analogs tested were as potent or selective as largazole, the Zn2+-binding affinity of each ZBG correlated with HDAC inhibition and cytotoxicity. We expect that our findings will aid in building a deeper understanding of the role of ZBGs in HDAC inhibition as well as provide an important basis for the future development of new largazole analogs with non-thiol ZBGs as novel therapeutics for cancer.The overexpression of HDACs and consequent hypoacetylation of histones have been observed in a variety of different diseases, leading to a recent focus of HDACs as attractive drug targets. The natural product largazole is one of the most potent natural HDAC inhibitors discovered so far. To probe the effect of various zinc-binding groups (ZBGs) on HDAC inhibition. We prepared a series of largazole analogs with various ZBGs and evaluated their HDAC inhibition and cytotoxicity.Download high-res image (151KB)Download full-size image
Co-reporter:Do-Yeon Kwon; Hye Eun Lee; Douglas H. Weitzel; Kyunghye Park; Sun Hee Lee; Chen-Ting Lee; Tesia N. Stephenson; Hyeri Park; Michael C. Fitzgerald; Jen-Tsan Chi; Robert A. MookJr.; Mark W. Dewhirst; You Mie Lee
Journal of Medicinal Chemistry 2015 Volume 58(Issue 19) pp:7659-7671
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.jmedchem.5b01220
To cope with hypoxia, tumor cells have developed a number of adaptive mechanisms mediated by hypoxia-inducible factor 1 (HIF-1) to promote angiogenesis and cell survival. Due to significant roles of HIF-1 in the initiation, progression, metastasis, and resistance to treatment of most solid tumors, a considerable amount of effort has been made to identify HIF-1 inhibitors for treatment of cancer. Isolated from Saururus cernuus, manassantins A (1) and B (2) are potent inhibitors of HIF-1 activity. To define the structural requirements of manassantins for HIF-1 inhibition, we prepared and evaluated a series of manassantin analogues. Our SAR studies examined key regions of manassantin’s structure in order to understand the impact of these regions on biological activity and to define modifications that can lead to improved performance and drug-like properties. Our efforts identified several manassantin analogues with reduced structural complexity as potential lead compounds for further development. Analogues MA04, MA07, and MA11 down-regulated hypoxia-induced expression of the HIF-1α protein and reduced the levels of HIF-1 target genes, including cyclin-dependent kinase 6 (Cdk6) and vascular endothelial growth factor (VEGF). These findings provide an important framework to design potent and selective HIF-1α inhibitors, which is necessary to aid translation of manassantin-derived natural products to the clinic as novel therapeutics for cancers.
Co-reporter:Joseph B. Baker, Hyoungsu Kim, Jiyong Hong
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3120-3122
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.11.135
The tandem allylic oxidation/oxa-conjugate addition reaction promoted by the gem-disubstituent effect in conjunction with the NHC-mediated oxidative esterification was explored for the facile synthesis of clavosolide A.
Co-reporter:Megan L. Lanier, Amanda C. Kasper, Hyoungsu Kim, and Jiyong Hong
Organic Letters 2014 Volume 16(Issue 9) pp:2406-2409
Publication Date(Web):April 11, 2014
DOI:10.1021/ol500773w
Oxepanes are found in a wide range of natural products; however, they are challenging synthetic targets due to enthalpic and entropic barriers. Organocatalytic oxa-conjugate addition reactions promoted by the gem-disubstituent (Thorpe–Ingold) effect stereoselectively provided α,α′-trans-oxepanes. In addition, the potential of an organocatalytic tandem oxa-conjugate addition/α-oxidation was demonstrated in a rapid generation of molecular complexity. These organocatalytic oxa-conjugate addition reactions would provide powerful tools for the synthesis of natural products that contain highly functionalized oxepanes.
Co-reporter:Regina Lin, Hyoungsu Kim, Jiyong Hong, and Qi-Jing Li
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:485-490
Publication Date(Web):March 12, 2014
DOI:10.1021/ml4004809
Subglutinol A (1) is an immunosuppressive natural product isolated from Fusarium subglutinans, an endophytic fungus from the vine Tripterygium wilfordii. We show that 1 exerts multimodal immune-suppressive effects on activated T cells in vitro: subglutinol A (1) effectively blocks T cell proliferation and survival while profoundly inhibiting pro-inflammatory IFNγ and IL-17 production by fully differentiated effector Th1 and Th17 cells. Our data further reveal that 1 may exert its anti-inflammatory effects by exacerbating mitochondrial damage in T cells. Additionally, we demonstrate that 1 significantly reduces lymphocytic infiltration into the footpad and ameliorates footpad swelling in the mouse model of Th1-driven delayed-type hypersensitivity. These results suggest the potential of 1 as a novel therapeutic for inflammatory diseases.Keywords: immunosuppressive agent; inflammatory disease; mitochondrial dysfunction; Subglutinol; T cell activation;
Co-reporter:Bumki Kim, Heekwang Park, Lilibeth A. Salvador, Patrick E. Serrano, Jason C. Kwan, Sabrina L. Zeller, Qi-Yin Chen, Soyoung Ryu, Yanxia Liu, Seongrim Byeon, Hendrik Luesch, Jiyong Hong
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 16) pp:3728-3731
Publication Date(Web):15 August 2014
DOI:10.1016/j.bmcl.2014.07.006
Largazole is a potent class I selective histone deacetylase (HDAC) inhibitor. The majority of largazole analogues to date have modified the thiazole–thiazoline and the warhead moiety. In order to elucidate class I-specific structure–activity relationships, a series of analogues with modifications in the valine or the linker region were prepared and evaluated for their class I isoform selectivity. The inhibition profile showed that the C2 position of largazole has an optimal steric requirement for efficient HDAC inhibition and that substitution of the trans-alkene in the linker with an aromatic group results in complete loss of activity. This data will aid the design of class I isoform selective HDAC inhibitors.
Co-reporter:Jiyong Hong
Chemistry - A European Journal 2014 Volume 20( Issue 33) pp:10204-10212
Publication Date(Web):
DOI:10.1002/chem.201402804
Abstract
Nature has evolved to produce unique and diverse natural products that possess high target affinity and specificity. Natural products have been the richest sources for novel modulators of biomolecular function. Since the chemical synthesis of urea by Wöhler, organic chemists have been intrigued by natural products, leading to the evolution of the field of natural product synthesis over the past two centuries. Natural product synthesis has enabled natural products to play an essential role in drug discovery and chemical biology. With the introduction of novel, innovative concepts and strategies for synthetic efficiency, natural product synthesis in the 21st century is well poised to address the challenges and complexities faced by natural product chemistry and will remain essential to progress in biomedical sciences.
Co-reporter:Kiyoun Lee;Hyoungsu Kim
European Journal of Organic Chemistry 2012 Volume 2012( Issue 5) pp:1025-1032
Publication Date(Web):
DOI:10.1002/ejoc.201101549
Abstract
Tandem and organocatalytic oxa-Michael reactions of α,β-unsaturated aldehydes were explored for the stereoselective synthesis of structurally complex tetrahydropyrans. Thestereoselective synthesis of 2,6-trans-tetrahydropyrans, which are thermodynamically unfavorable, was accomplished through a reagent-controlled, organocatalytic oxa-Michael reaction. A temperature-dependent configurational switch allowed the preparation of both 2,3-trans-2,6-trans- and 2,3-cis-2,6-cis-tetrahydropyrans from a common substrate. This switch was then used to synthesize the precursors of the C21–C29 and C30–C37 fragments of ent-(+)-sorangicin A.
Co-reporter:Kiyoun Lee;Dr. Hyoungsu Kim;Dr. Jiyong Hong
Angewandte Chemie International Edition 2012 Volume 51( Issue 23) pp:5735-5738
Publication Date(Web):
DOI:10.1002/anie.201201653
Co-reporter:Yongcheng Ying, Hyoungsu Kim, and Jiyong Hong
Organic Letters 2011 Volume 13(Issue 4) pp:796-799
Publication Date(Web):January 20, 2011
DOI:10.1021/ol103064f
Both 2,6-cis- and 2,6-trans-piperidines were prepared from common substrates through organocatalytic aza-Michael reactions promoted by the gem-disubstituent effect in conjunction with dithiane coupling reactions. The organocatalytic aza-Michael reaction enabled a facile synthesis of (+)-myrtine and (−)-epimyrtine from a common substrate.
Co-reporter:Su-Ui Lee, Han Bok Kwak, Sung-Hee Pi, Hyung-Keun You, Seong Rim Byeon, Yongcheng Ying, Hendrik Luesch, Jiyong Hong, and Seong Hwan Kim
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 3) pp:248
Publication Date(Web):January 4, 2011
DOI:10.1021/ml1002794
Due to their capability of modifying chromatin structure and thereby regulating gene transcription, histone deacetylases (HDACs) have been reported to play important roles in osteogenesis and considered a promising potential therapeutic target for bone diseases, including osteoporosis. We showed that the novel marine-derived HDAC inhibitor largazole exhibits in vitro and in vivo osteogenic activity. Largazole significantly induced the expression of ALP and OPN. The osteogenic activity of largazole was mediated through the increased expression of Runx2 and BMPs. Importantly, largazole showed in vivo bone-forming efficacy in the mouse calvarial bone formation assay and the rabbit calvarial bone fracture healing model. The dual action of largazole to stimulate bone formation and inhibit bone resorption would be a useful feature in drug development for bone-related disorders.Keywords (keywords): bone morphogenetic protein; histone deacetylases; Largazole; osteogenic activity; Runx2
Co-reporter:Hyoungsu Kim, Seong Rim Byeon, Marina G.D. Leed, Jiyong Hong
Tetrahedron Letters 2011 Volume 52(Issue 19) pp:2468-2470
Publication Date(Web):11 May 2011
DOI:10.1016/j.tetlet.2011.03.008
Due to the high reactivity of the formyl group under either basic or acidic reaction conditions required for the direct generation of aldehyde enolates, intramolecular Michael additions of aldehyde enolates to α,β-unsaturated carbonyl compounds have been underexplored for the stereoselective synthesis of carbocyclic compounds. The intramolecular Michael reaction of aldehyde enolates generated by imidazolium carbenes was explored for the synthesis of cyclopentane aldehydes. The imidazolium carbenes were used as Brønsted bases to directly generate the aldehydes enolates.
Co-reporter:Hyoungsu Kim and Jiyong Hong
Organic Letters 2010 Volume 12(Issue 12) pp:2880-2883
Publication Date(Web):May 21, 2010
DOI:10.1021/ol101022z
The tandem allylic oxidation/oxa-Michael reaction promoted by the gem-disubstituent effect and the 2-methyl-6-nitrobenzoic anhydride (MNBA)-mediated dimerization were explored for the efficient and facile synthesis of cyanolide A.
Co-reporter:Hyoungsu Kim Dr.;JosephB. Baker;Yongho Park;Hyung-Bae Park;PatrickD. DeArmond;Seong Hwan Kim Dr.;MichaelC. Fitzgerald Dr.;Dong-Sup Lee Dr. Dr.
Chemistry – An Asian Journal 2010 Volume 5( Issue 8) pp:1902-1910
Publication Date(Web):
DOI:10.1002/asia.201000147
Abstract
Immunosuppressive drugs are used to prevent rejection of transplanted organs and treat autoimmune diseases. Clinically approved immunosuppressive drugs possess undesirable side effects, including acute neurological toxicity, chronic nephrotoxicity, and osteoporosis. As a result, considerable efforts have been devoted to the identification of immunosuppressive natural products that lack cytotoxicity and undesirable side effects on bone structure. Subglutinols A (1 a) and B (1 b) are diterpene pyrones isolated from Fusarium subglutinans. Compounds 1 a and 1 b are equipotent in the mixed lymphocyte reaction assay and thymocyte proliferation assay (IC50=0.1 μM). Owing to the lack of toxicity, 1 a and 1 b are expected to be promising new immunosuppressive drugs. Herein, we detail our efforts that have culminated in a stereoselective synthesis of 1 a and 1 b from the (S)-(+)-5-methyl-Wieland–Miescher ketone and determined their absolute stereochemistries. We also present initial biological data to show the great potential of 1 a as an immunosuppressive drug with dose-dependent osteogenic activity.
Co-reporter:Kiyoun Lee, Hyoungsu Kim and Jiyong Hong
Organic Letters 2009 Volume 11(Issue 22) pp:5202-5205
Publication Date(Web):October 13, 2009
DOI:10.1021/ol902125d
The tandem cross-metathesis/thermal SN2′ reaction was explored for the facile and efficient synthesis of 4-hydroxy-2,6-cis-tetrahydropyrans. The mildness of the thermal conditions allowed for the synthesis of 4-hydroxy-2,6-cis-tetrahydropyrans from base-sensitive substrates without the use of protecting groups. The tandem reaction enabled a protecting-group-free synthesis of (±)-diospongin A.
Co-reporter:Langtian Yuan, Jin-Soo Seo, Nam Sook Kang, Shahar Keinan, Sarah E. Steele, Gregory A. Michelotti, William C. Wetsel, David N. Beratan, Young-Dae Gong, Tong H. Lee and Jiyong Hong
Molecular BioSystems 2009 vol. 5(Issue 9) pp:927-930
Publication Date(Web):08 May 2009
DOI:10.1039/B903036K
From a screen of small molecule libraries to identify potential therapeutics for psychostimulant abuse, 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones were shown to be isoform-selective PKC-ζ inhibitors.
Co-reporter:Amanda C. Kasper, Eui Jung Moon, Xiangqian Hu, Yongho Park, Ceshea M. Wooten, Hyoungsu Kim, Weitao Yang, Mark W. Dewhirst, Jiyong Hong
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 14) pp:3783-3786
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmcl.2009.04.071
We have shown that manassantin A downregulated the HIF-1α expression and inhibited the secretion of VEGF. We have also demonstrated that the 2,3-cis-3,4-trans-4,5-cis-configuration of the tetrahydrofuran is critical to the HIF-1 inhibition of manassantin A.HIF-1 inhibition by manassantin A (1) and analogues with modified tetrahydrofuran configurations (11 and 13) is reported.
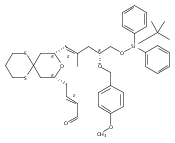
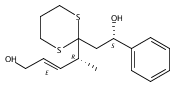
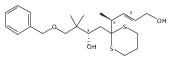
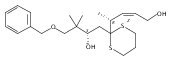
Co-reporter:Hyoungsu Kim Dr.;Yongho Park Dr.
Angewandte Chemie 2009 Volume 121( Issue 41) pp:7713-7717
Publication Date(Web):
DOI:10.1002/ange.200903690
Co-reporter:Hyoungsu Kim Dr.;Yongho Park Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 41) pp:7577-7581
Publication Date(Web):
DOI:10.1002/anie.200903690
.jpg)








![2-[2-[2-T-BOC-AMINOETHOXY]ETHOXY]ETHYL BROMIDE](http://img.cochemist.com/ccimg/166000/165963-71-3.png)
![2-[2-[2-T-BOC-AMINOETHOXY]ETHOXY]ETHYL BROMIDE](http://img.cochemist.com/ccimg/166000/165963-71-3_b.png)










![Benzenemethanol, a,a'-[[(2S,3R,4R,5S)-tetrahydro-3,4-dimethyl-2,5-furandiyl]bis[(2-methoxy-4,1-phenylene)oxy-(1R)-ethylidene]]bis[3,4-dimethoxy-,(aR,a'R)-](http://img.cochemist.com/ccimg/88500/88497-87-4.png)
![Benzenemethanol, a,a'-[[(2S,3R,4R,5S)-tetrahydro-3,4-dimethyl-2,5-furandiyl]bis[(2-methoxy-4,1-phenylene)oxy-(1R)-ethylidene]]bis[3,4-dimethoxy-,(aR,a'R)-](http://img.cochemist.com/ccimg/88500/88497-87-4_b.png)










































































































































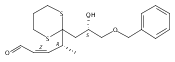













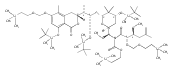





























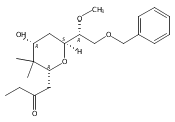









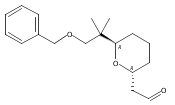




















































































































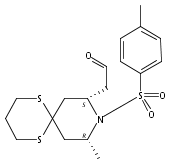


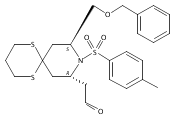


























































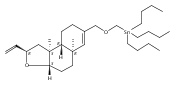






































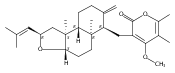













































![4,14,21,22-Tetraoxatricyclo[15.3.1.17,11]docosane-3,13-dione, 5,15-diethyl-9,19-dihydroxy-10,10,20,20-tetramethyl-, (1S,5R,7S,9S,11S,15R,17S,19S)-](/data/chemimg/174600/1229647-62-4.png)
![4,14,21,22-Tetraoxatricyclo[15.3.1.17,11]docosane-3,13-dione, 5,15-diethyl-9,19-dihydroxy-10,10,20,20-tetramethyl-, (1S,5R,7S,9S,11S,15R,17S,19S)-](/data/chemimg/174600/1229647-62-4_b.png)
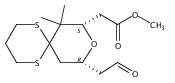


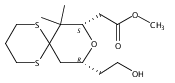





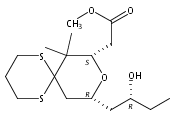












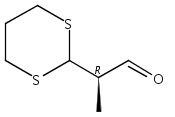
















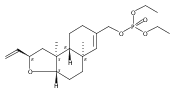






















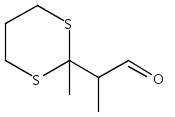









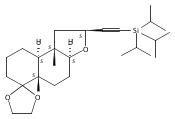











































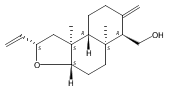
![6,21-Dioxabicyclo[15.3.1]heneicosa-2,8,10,14-tetraene-5-carboxaldehyde, 3,11-dimethyl-19-methylene-7,13-dioxo-, (1R,2E,5R,8E,10Z,14E,17R)-](/data/chemimg/120400/335059-30-8.png)
![6,21-Dioxabicyclo[15.3.1]heneicosa-2,8,10,14-tetraene-5-carboxaldehyde, 3,11-dimethyl-19-methylene-7,13-dioxo-, (1R,2E,5R,8E,10Z,14E,17R)-](/data/chemimg/120400/335059-30-8_b.png)
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0.png)
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0_b.png)








![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6.png)
![Propanamide, N-[(1R,2R)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-](http://img.cochemist.com/ccimg/192100/192060-67-6_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/178000/177971-32-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-, (2R)-](http://img.cochemist.com/ccimg/178000/177971-32-3_b.png)




![2H-Pyran-2-one,3-[[(2S,3aS,5aR,6R,9aR,9bS)-dodecahydro-5a,9b-dimethyl-7-methylene-2-(2-methyl-1-propen-1-yl)naphtho[2,1-b]furan-6-yl]methyl]-4-hydroxy-5,6-dimethyl-](http://img.cochemist.com/ccimg/169600/169532-90-5.png)
![2H-Pyran-2-one,3-[[(2S,3aS,5aR,6R,9aR,9bS)-dodecahydro-5a,9b-dimethyl-7-methylene-2-(2-methyl-1-propen-1-yl)naphtho[2,1-b]furan-6-yl]methyl]-4-hydroxy-5,6-dimethyl-](http://img.cochemist.com/ccimg/169600/169532-90-5_b.png)
![2H-Pyran-2-one,3-[[(2R,3aS,5aR,6R,9aR,9bS)-dodecahydro-5a,9b-dimethyl-7-methylene-2-(2-methyl-1-propen-1-yl)naphtho[2,1-b]furan-6-yl]methyl]-4-hydroxy-5,6-dimethyl-](http://img.cochemist.com/ccimg/169400/169397-88-0.png)
![2H-Pyran-2-one,3-[[(2R,3aS,5aR,6R,9aR,9bS)-dodecahydro-5a,9b-dimethyl-7-methylene-2-(2-methyl-1-propen-1-yl)naphtho[2,1-b]furan-6-yl]methyl]-4-hydroxy-5,6-dimethyl-](http://img.cochemist.com/ccimg/169400/169397-88-0_b.png)














![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3.png)
![2-Butenal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2E)-](http://img.cochemist.com/ccimg/119200/119125-30-3_b.png)
![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/113200/113123-37-8.png)
![2-Buten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (Z)-](http://img.cochemist.com/ccimg/113200/113123-37-8_b.png)




![a-L-Talofuranuronamide,1-deoxy-5-O-[4-deoxy-N-[(3S)-hexahydro-2-oxo-1H-azepin-3-yl]-b-L-erythro-hex-4-enopyranuronamidosyl]-1-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-O-methyl-](http://img.cochemist.com/ccimg/102800/102770-00-3.png)
![a-L-Talofuranuronamide,1-deoxy-5-O-[4-deoxy-N-[(3S)-hexahydro-2-oxo-1H-azepin-3-yl]-b-L-erythro-hex-4-enopyranuronamidosyl]-1-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-O-methyl-](http://img.cochemist.com/ccimg/102800/102770-00-3_b.png)






![3-[(1-hydroxy-2-oxoazepan-3-yl)amino]-1-methyl-3-oxopropyl N~6~-hydroxy-N~6~-[(2E)-octadec-2-enoyl]-N~2~-{[(2E)-2-(6-oxocyclohexa-2,4-dien-1-ylidene)-1,3-oxazolidin-4-yl]carbonyl}lysinate](http://img.cochemist.com/ccimg/26800/26769-11-9.png)
![3-[(1-hydroxy-2-oxoazepan-3-yl)amino]-1-methyl-3-oxopropyl N~6~-hydroxy-N~6~-[(2E)-octadec-2-enoyl]-N~2~-{[(2E)-2-(6-oxocyclohexa-2,4-dien-1-ylidene)-1,3-oxazolidin-4-yl]carbonyl}lysinate](http://img.cochemist.com/ccimg/26800/26769-11-9_b.png)


![Acetamide,N,N'-[[(2S,5S)-3,6-dioxo-2,5-piperazinediyl]di-3,1-propanediyl]bis[N-hydroxy-](http://img.cochemist.com/ccimg/19000/18928-00-2.png)
![Acetamide,N,N'-[[(2S,5S)-3,6-dioxo-2,5-piperazinediyl]di-3,1-propanediyl]bis[N-hydroxy-](http://img.cochemist.com/ccimg/19000/18928-00-2_b.png)