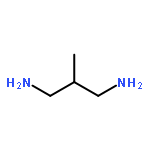
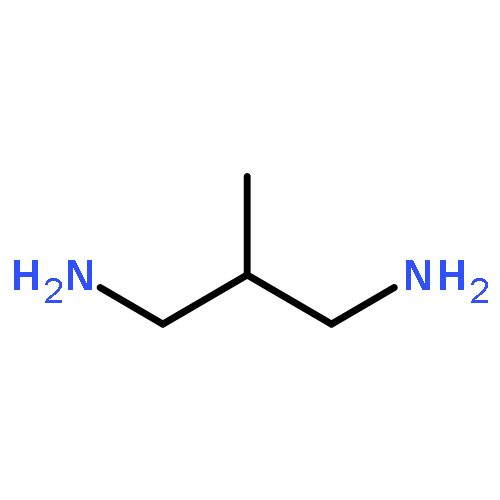
Co-reporter:Li Wang;Ignacio Moraleda;Isabel Iriepa;Alejandro Romero;Francisco López-Muñoz;Mourad Chioua;Manuela Bartolini;José Marco-Contelles
MedChemComm (2010-Present) 2017 vol. 8(Issue 6) pp:1307-1317
Publication Date(Web):2017/06/21
DOI:10.1039/C7MD00143F
The synthesis, cholinesterase inhibition, molecular modelling and ADME properties of novel tacrine–neocryptolepine heterodimers are described. Compound 3 [5-methyl-N-(8-(5,6,7,8-tetrahydroacridin-9-ylamino)octyl)-5H-indolo[2,3-b]quinolin-11-amine], showing a moderate inhibition of the Aβ1–42 self-aggregation (26.5% at a 1 : 5 ratio with Aβ1–42), and a calculated log BB value (0.27) indicating excellent potential BBB penetration, is a highly potent human cholinesterase inhibitor [IC50 (hAChE) = 0.95 ± 0.04 nM; IC50 (hBuChE) = 2.29 ± 0.14 nM] which can be listed among the most potent hAChE inhibitors so far identified, and is not hepatotoxic in vitro at the concentrations at which the ChEs are inhibited. A molecular modeling study was also undertaken in order to elucidate the AChE and the BuChE bind modes of all the new compounds. The docking results show that all of them bind to AChE in extended conformations and to BuChE in folded conformations. Moreover, these studies revealed that the length of the linker is crucial to binding both the catalytic anionic site and the peripheral anionic site.
Co-reporter:Ming-Yu Wu, Gerard Esteban, Simone Brogi, Masahi Shionoya, Li Wang, Giuseppe Campiani, Mercedes Unzeta, Tsutomu Inokuchi, Stefania Butini, Jose Marco-Contelles
European Journal of Medicinal Chemistry 2016 Volume 121() pp:864-879
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2015.10.001
•Development of multifunctional compounds.•Potential therapeutic agents for the treatment of AD.•Scaffold hopping approach.Currently available drugs against Alzheimer's disease (AD) are only able to ameliorate the disease symptoms resulting in a moderate improvement in memory and cognitive function without any efficacy in preventing and inhibiting the progression of the pathology. In an effort to obtain disease-modifying anti-Alzheimer's drugs (DMAADs) following the multifactorial nature of AD, we have recently developed multifunctional compounds. We herein describe the design, synthesis, molecular modeling and biological evaluation of a new series of donepezil-related compounds possessing metal chelating properties, and being capable of targeting different enzymatic systems related to AD (cholinesterases, ChEs, and monoamine oxidase A, MAO-A). Among this set of analogues compound 5f showed excellent ChEs inhibition potency and a selective MAO-A inhibition (vs MAO-B) coupled to strong complexing properties for zinc and copper ions, both known to be involved in the progression of AD. Moreover, 5f exhibited moderate antioxidant properties as found by in vitro assessment. This compound represents a novel donepezil–hydroxyquinoline hybrid with DMAAD profile paving the way to the development of a novel class of drugs potentially able to treat AD.
Co-reporter:Masashi Okada;Zhen-Wu Mei;Md. Imran Hossain;Li Wang
Medicinal Chemistry Research 2016 Volume 25( Issue 5) pp:879-892
Publication Date(Web):2016 May
DOI:10.1007/s00044-016-1508-z
A plant-derived neocryptolepine core, the 5-Me-indolo[2,3-b]quinoline skeleton, was emblazoned with substituents at C11 and C2 and then tested against various cancer cell lines to find potent anticancer agents. In the in vitro antiproliferative activity assay against the breast cancer MDA-MB-453 cell line, the attachment of alkylamino substituents at C11 of the 5-Me-indolo[2,3-b]quinoline induced improved activities. Specifically, 11-(3-aminopropylamino) and 11-(4-aminobutylamino) derivatives indicated the highest activity and selectivity against MDA-MB-453 (IC50 = 0.3–0.5 μM) and also exhibited a higher cytotoxicity against the colon adenocarcinoma (WiDr) and ovarian cancer (SKOv3) cell lines. A synergistic effect by attachment of substituents at C2 was favorably observed with an electron-donating group, such as CH3O, and unfavorably observed with an electron-withdrawing one, such as F and CF3. Further modification of the terminal free amino group of the lariat attachment at C11 into the corresponding acylamides and 2,3-dihydrobenzo[e][1,3]thiazin-4-ones was not effective for the antiproliferative activity. The computer-assisted database analysis, COMPARE, suggested that 14e and 13b have a mode of action similar to actinomycin D and 13c has a mode of actions similar to vindesine sulfate or aclarubicin hydrochloride. However, the new compounds may have other unique mode of actions since the correlation coefficients (r) were in relatively low levels.
Co-reporter:Ning Wang, Marta Świtalska, Ming-Yu Wu, Kento Imai, Tran Anh Ngoc, Cui-Qing Pang, Li Wang, Joanna Wietrzyk, Tsutomu Inokuchi
European Journal of Medicinal Chemistry 2014 Volume 78() pp:314-323
Publication Date(Web):6 May 2014
DOI:10.1016/j.ejmech.2014.03.038
•11N–H and 11N–Me indolo[3,2-c]quinolines were tested.•The 3-aminopropylamino groups at C6 enhanced cytotoxicity.•High cytotoxicity was achieved with 11N–Me derivatives.•The nitro group at C2 best contributed to cytotoxicity.•11N–Me group slightly enhances the DNA intercalating constant.A series of 6-amino-11H- indolo[3,2-c]quinoline derivatives with various substituents on the quinoline ring were synthesized. A methyl group introduced to N-11 of the intermediate 4 to elaborate novel analog 7. The cytotoxic effect of these 6-amino-substituted 11H- and 11-methyl-indolo[3,2-c]quinoline derivatives in vitro were tested against MV4-11 (human leukemia), A549 (non-small cell lung cancer) and HCT116 (colon cancer) and BALB/3T3 (normal murine fibroblasts). All the N-11 methylated compounds significantly increased the cytotoxicity. Compound 7p was most active with the IC50 value of 0.052 μM against the MV4-11 cell line, and also exhibited a selective activity against A549, HCT116 and BALB/3T3 cell line, with the respective IC50 values of 0.112, 0.007 and 0.083 μM, which were higher or comparable to those of the anticancer drug doxorubicin HCl. The binding constants of 5g and 7h to salmon fish sperm DNA were also evaluated using UV–vis absorption spectroscopy, indicating intercalation binding with constants of 1.05 × 106 L/mol and 4.84 × 106 L/mol.
Co-reporter:Ning Wang, Kathryn J. Wicht, Kento Imai, Ming-qi Wang, Tran Anh Ngoc, Ryo Kiguchi, Marcel Kaiser, Timothy J. Egan, Tsutomu Inokuchi
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 9) pp:2629-2642
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmc.2014.03.030
A series of indolo[3,2-c]quinolines were synthesized by modifying the side chains of the ω-aminoalkylamines at the C6 position and introducing substituents at the C2 position, such as F, Cl, Br, Me, MeO and NO2, and a methyl group at the N11 position for an SAR study. The in vitro antiplasmodial activities of the derivative agents against two different strains (CQS: NF54 and CQR: K1) and the cytotoxic activity against normal L6 cells were evaluated. The test results showed that compounds 6k and 6l containing the branched methyl groups of 3-aminopropylamino at C6 with a Cl atom at C2 exhibited a very low cytotoxicity with IC50 values above 4000 nM, high antimalarial activities with IC50 values of about 11 nM for CQS (NF54), IC50 values of about 17 nM for CQR (K1), and RI resistance indices of 1.6. Furthermore, the compounds were tested for β-haematic inhibition, and QSAR revealed an interesting linear correlation between the biological activity of CQS (NF54) and three contributing factors, namely solubility, hydrophilic surface area, and β-haematin inhibition for this series. In vivo testing of 6l showed a reduction in parasitaemia on day 4 with an activity of 38%.
Co-reporter:Ning Wang, Kathryn J. Wicht, Elkhabiry Shaban, Tran Anh Ngoc, Ming-Qi Wang, Ikuya Hayashi, Md. Imran Hossain, Yoshihiko Takemasa, Marcel Kaiser, Ibrahim El Tantawy El Sayed, Timothy J. Egan and Tsutomu Inokuchi
MedChemComm 2014 vol. 5(Issue 7) pp:927-931
Publication Date(Web):12 May 2014
DOI:10.1039/C4MD00091A
Hybrids of artesunate–indolo[2,3-b]quinoline, –indolo[3,2-c]quinoline, and –indolo[3,2-b]quinoline were synthesized and screened for their antiplasmodial activity against two different malaria strains (CQS and CQR) and their cytotoxic activities against normal cells were evaluated. All the synthesized hybrids showed a decreased cytotoxicity and increased antimalarial activity relative to the individual, non-hybridized compounds. Furthermore, these hybrids were stronger β-haematin inhibitors than the corresponding molecules from which they were derived. The most effective antimalarial hybrid showed an IC50 value of 0.45 nM against the CQS strain. At the same time this hybrid also showed effective activity against the CQR strain, with an IC50 value of 0.42 nM and an RI value of 0.93. With the dosing of the artesunate–indolo[2,3-b]quinoline set at 10 mg kg−1 once a day for four consecutive days, parasitemia was significantly reduced on day 4, with an antiparasitic activity of 89.6%, and a mean mouse survival time of 7.7 days.
Co-reporter:Li Wang;Wen-Jie Lu;Tomohito Odawara;Ryuhei Misumi;Zhen-Wu Mei;Wei Peng;Ibrahim El-Tantawy El-Sayed
Journal of Heterocyclic Chemistry 2014 Volume 51( Issue 4) pp:1106-1114
Publication Date(Web):
DOI:10.1002/jhet.1617
Various 11-chloro-5-methyl-5H-indolo[2,3-b]quinolines (neocryptolepines) with different substituents on the quinoline ring, key intermediates for antimalaria agents, are prepared from the substituted N-methylanilines, easily accessible by the N-methylation of anilines, and indole-3-carboxylate as a counterpart. This protocol is benign in terms of the reduced number of steps to reach the target, compared with the known method using anilines, and easy product purification. Alternatively, their 6-methyl congener is prepared by N-methylation of the indole moiety of 2-arylaminoindole-3-carboxylate followed by successive cyclization and chlorination. 11-Chloroneocryptolepines are found more reactive than their 6-methyl congener in the nucleophilic substitution at the C11 position.
Co-reporter:Zhen-Wu Mei ; Li Wang ; Wen-Jie Lu ; Cui-Qing Pang ; Tsukasa Maeda ; Wei Peng ; Marcel Kaiser ; Ibrahim El Sayed
Journal of Medicinal Chemistry 2013 Volume 56(Issue 4) pp:1431-1442
Publication Date(Web):January 29, 2013
DOI:10.1021/jm300887b
To obtain a high antimalarial activity with neocryptolepine derivatives, modifying and changing the side chains at the C11 position with varying the substituents of an electron-withdrawing or electron-donating nature at the C2 position for a SAR study were executed. Installation of alkylamino and ω-aminoalkylamino groups at the C11 position of the neocryptolepine core was successful. For further variation, the aminoalkylamino substituents were transformed into the corresponding acyclic or cyclic carbamides or thiocarbamides. These side chain modified neocryptolepine derivatives were tested for antimalarial activity against CQR (K1) and CQS (NF54) of Plasmodium falciparum in vitro and for cytotoxicity toward mammalian L6 cells. Among the tested compounds, the compound 17f showed an IC50 of 2.2 nM for CQS (NF54) and a selectivity index of 1400, and 17i showed an IC50 of 2.2 nM for CQR (K1), a selectivity index of 1243, and a resistance index of 0.5.
Co-reporter:Md. Imran Hossain, Marta Świtalska, Wei Peng, Mariko Takashima, Ning Wang, Marcel Kaiser, Joanna Wietrzyk, Shingo Dan, Takao Yamori, Tsutomu Inokuchi
European Journal of Medicinal Chemistry 2013 Volume 69() pp:294-309
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.08.008
•High yielded 1,2,4-trioxanes were synthesized via photooxidation.•The aromatic groups at C-2 and C-7 increased the antiproliferative activity.•The 1,2,4-trioxane ring is the key for anticancer activities.•The IC50 0.5 μM against human leukemia (MV4-11) cell line was achieved.•A sharp hypochromic shifts were observed for the oxidative damage study.A novel series of 1,2,4-trioxanes were synthesized from 2H-pyrans via photooxidation, and their antiproliferative and growth factor inhibitory activity has been investigated across a variety of human cancer cell lines. Compounds 5k, 5l, 5s, 7a and 7c exhibited the highest activity and selectivity against a human leukemia (MV4-11) cell line (IC50 = 0.5 μM). Compound 5o showed the highest growth factor inhibitory activity against a melanoma (LOX-IMVI) cancer cell line (GI50 = 1.0 μM). A SAR study has confirmed the importance of the 1,2,4-trioxane unit as a pharmacophore for anticancer activity. The computer-assisted database analysis, COMPARE, has suggested that the compounds have unique mechanisms of actions that were different from those of known anticancer drugs. Some of the selected trioxanes were tested against the NF54 strain, albeit showing weak antiplasmodial activity. The molecular docking of trioxanes and hemin reveals that a short distance (1.30 Å) leads to their physical contact. The UV–vis spectroscopic analysis ensured the definite complexation between 1,2,4-trioxanes and hemin. The role of hemin–trioxane interaction in the hemin-induced oxidative damage has been studied using methylene blue as a substrate by UV–vis spectroscopy.
Co-reporter:Wei Peng, Marta Świtalska, Li Wang, Zhen-Wu Mei, Yoshiki Edazawa, Cui-Qing Pang, Ibrahim El-Tantawy El-Sayed, Joanna Wietrzyk, Tsutomu Inokuchi
European Journal of Medicinal Chemistry 2012 Volume 58() pp:441-451
Publication Date(Web):December 2012
DOI:10.1016/j.ejmech.2012.10.023
To search for new biological activities of the chromeno[2,3-b]indoles, the 5-oxa analog of the indolo[2,3-b]quinolines that are known to have a potent antitumor activity, a series of 11-amino derivatives with various substituents at the C-2 position were prepared. The synthesis of the chromeno[2,3-b]indole structure involves the cyclization of 2-phenoxy-3-indolecarboxylates 3, accessible from the indole-3-carboxylate 1 and phenols 2, producing the chromeno[2,3-b]indol-11(6H)-ones 4, which is followed by dehydroxychlorination with phosphorus oxychloride to afford the 11-chlorochromeno[2,3-b]indoles 5. The treatment of 5 with various amines produced the corresponding 11-aminated chromeno[2,3-b]indoles 6, while some of the 11-ω-aminoalkylamino derivatives 6 were transformed into the 11-ω-sulfonylaminoalkylamino derivatives 8. The antiproliferative activity of these 11-aminochromeno[2,3-b]indoles 6 and 8in vitro were tested using MV4-11 (human leukemia), A549 (lung cancer), HCT116 (colon cancer), and the normal mice fibroblast (BALB/3T3) and their potencies were described.Graphical abstractHighlights► In vitro antiproliferative activity of thirty chromeno[2,3-b]indoles were tested. ► 11-(ω-Aminoalkylamino) substituents contributed highly to the activity. ► Most 11-aminoalkylamino derivatives were more active and less toxic than cisplatin. ► Alkylmonoamino substituents at the C-11 were less contributing to activity. ► The C-2 substituents brought synergy effect in combination with the C-11 groups.
Co-reporter:Li Wang, Marta Świtalska, Zhen-Wu Mei, Wen-Jie Lu, Yoshito Takahara, Xing-Wen Feng, Ibrahim El-Tantawy El-Sayed, Joanna Wietrzyk, Tsutomu Inokuchi
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4820-4829
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.05.054
The present report describes the synthesis and antiproliferative evaluation of certain 11-aminoalkylamino-substituted 5H- and 6H-indolo[2,3-b]quinolines and their methylated derivatives. These 5-Me- and 6-Me-indolo[2,3-b]quinoline derivatives 10–14, 20 were prepared by amination at the C-11 position of the 11-chloro-5-methyl-5H- and 11-chloro-6-methyl-6H-indolo[2,3-b]quinolines with different substituents on the quinoline ring. The 11-aminoalkylaminomethylated 23, the homologue of 11, was prepared from the same intermediate for a further SAR study. These intermediates are accessible from 4-substituted anilines or their N-methylated analogues and methyl indole-3-carboxylate as a counterpart. The in vitro antiproliferative assay indicated that the 5-methylated derivatives 10–14 are more cytotoxic than their respective 6-methylated 6H-indolo[2,3-b]quinoline derivatives 20. Among them, N-(3-aminopropyl)-2-bromo-5-methyl-5H-indolo[2,3-b]quinolin-11-amine 12f was the most cytotoxic with a mean IC50 value of 0.12 μM against human leukemia MV4-11 cell line, and also exhibited selective cytotoxicities against A549 (lung cancer), HCT116 (colon cancer) cell lines and normal fibroblast BALB/3T3 with IC50 values of 0.543, 0.274 and 0.869 μM, respectively. The binding constant of products 12f and 20f to salmon fish sperm DNA were also evaluated using UV–vis absorption spectroscopy, indicating intercalation binding with a constant of 2.93 × 105 and 3.28 × 105 L mol−1, respectively.
Co-reporter:Wei Peng;Toshiaki Hirabaru;Hiroyuki Kawafuchi
European Journal of Organic Chemistry 2011 Volume 2011( Issue 28) pp:5469-5474
Publication Date(Web):
DOI:10.1002/ejoc.201100780
Abstract
A facile access to 2,3,6-trisubstituted 2H-pyran-5-carboxylates is developed by employing 2-alkyl-2-enals as reactants with acetoacetates. The reaction involves Knoevenagel condensation followed by a 6π-electrocyclization, in which the presence of the C2 alkyl substituent in the enals favors the formation of (E)-Knoevenagel adducts for the ensuing electrocyclization. The resulting 2H-pyrans are hydrogenated to form 3,4-dihydro-2H-pyrans and converted into the endoperoxides by singlet-oxygen cycloaddition.
Co-reporter:Li-Jian Ma and Tsutomu Inokuchi
Chemical Communications 2010 vol. 46(Issue 37) pp:7037-7039
Publication Date(Web):25 Aug 2010
DOI:10.1039/C0CC00859A
Microwave irradiation of chloroacetylarenes and enamines induced an aza-Darzens reaction followed by rearrangement of the aziridinium intermediate to give (2-amino-3-alkenoyl)arenes that were reduced selectively to syn-β-aminoalcohols as pseudo-ephedrine analogues.
Co-reporter:Wei Peng, Kazumi Ashida, Toshiaki Hirabaru, Li-Jian Ma, Tsutomu Inokuchi
Tetrahedron 2010 66(51) pp: 9714-9720
Publication Date(Web):
DOI:10.1016/j.tet.2010.10.034
Co-reporter:Tsutomu Inokuchi, Hiroyuki Kawafuchi and Junzo Nokami
Chemical Communications 2005 (Issue 4) pp:537-539
Publication Date(Web):06 Dec 2004
DOI:10.1039/B414129F
Owing to the unprecedented stability of O-acylTEMPOs towards hydride-transferring and metallic alkylating reagents such as LiAlH4 and RMgX, chemoselective transformation of diacid mixed alkyl/TEMP-1-yl esters, where O-acylTEMPOs remained intact, is achieved with these reagents, giving the corresponding carbinols, respectively.
Co-reporter:Li-Jian Ma and Tsutomu Inokuchi
Chemical Communications 2010 - vol. 46(Issue 37) pp:NaN7039-7039
Publication Date(Web):2010/08/25
DOI:10.1039/C0CC00859A
Microwave irradiation of chloroacetylarenes and enamines induced an aza-Darzens reaction followed by rearrangement of the aziridinium intermediate to give (2-amino-3-alkenoyl)arenes that were reduced selectively to syn-β-aminoalcohols as pseudo-ephedrine analogues.

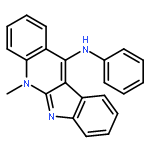
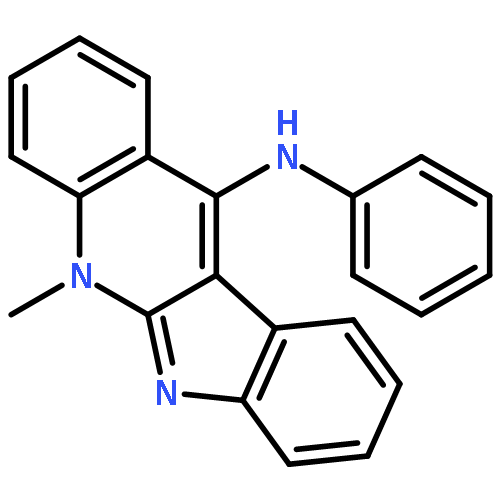
![5-[[METHYL(PROP-2-YNYL)AMINO]METHYL]QUINOLIN-8-OL](http://img.cochemist.com/ccimg/766500/766454-72-2.png)
![5-[[METHYL(PROP-2-YNYL)AMINO]METHYL]QUINOLIN-8-OL](http://img.cochemist.com/ccimg/766500/766454-72-2_b.png)

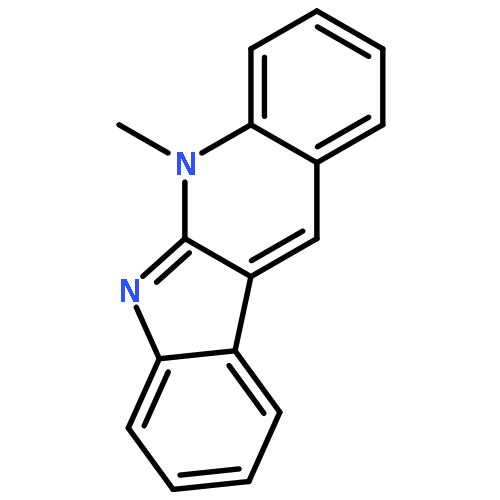
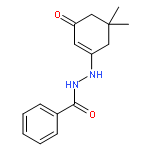
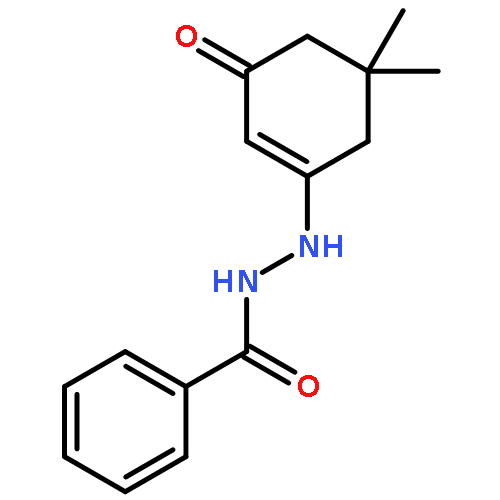
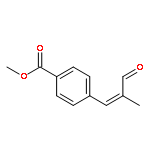
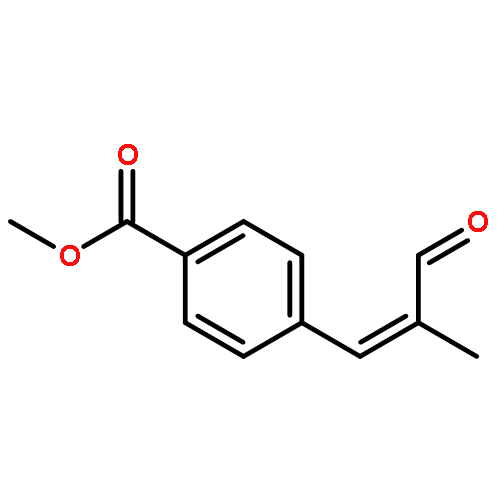
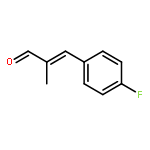
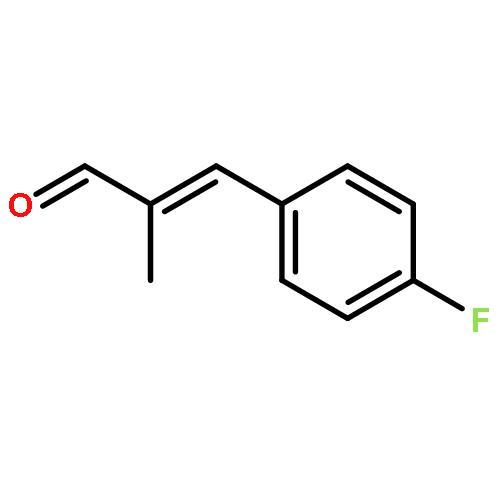

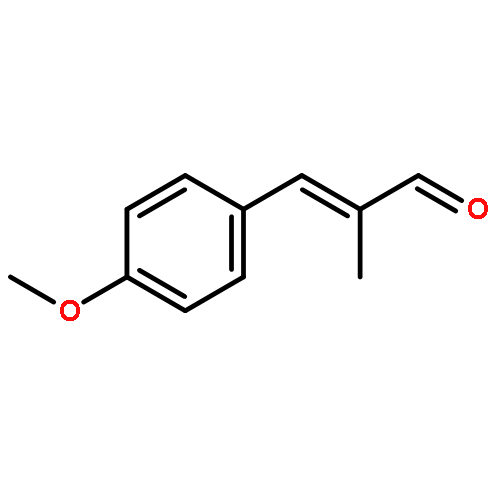
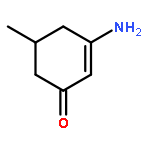
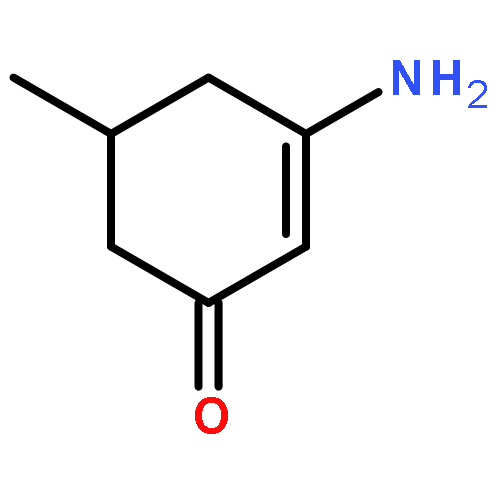
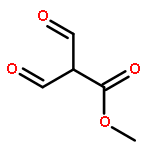
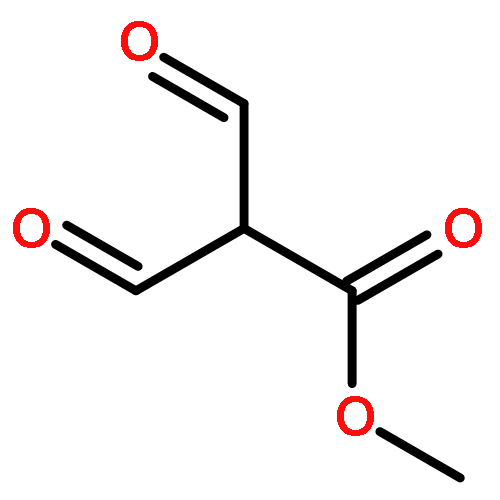


![N-[4-(METHYLAMINO)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/40000/39970-48-4.png)
![N-[4-(METHYLAMINO)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/40000/39970-48-4_b.png)
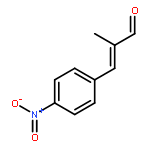

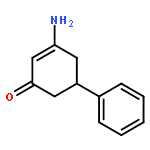
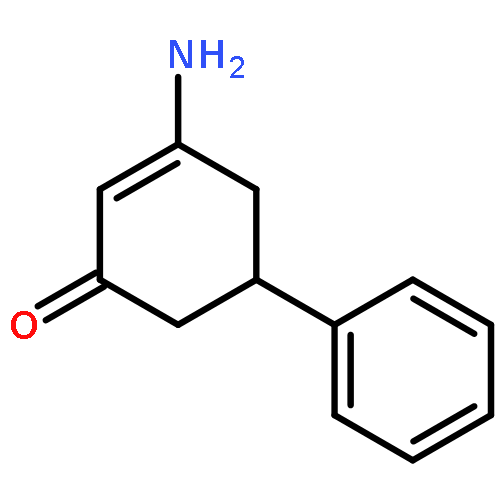
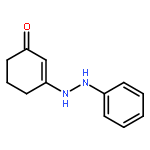
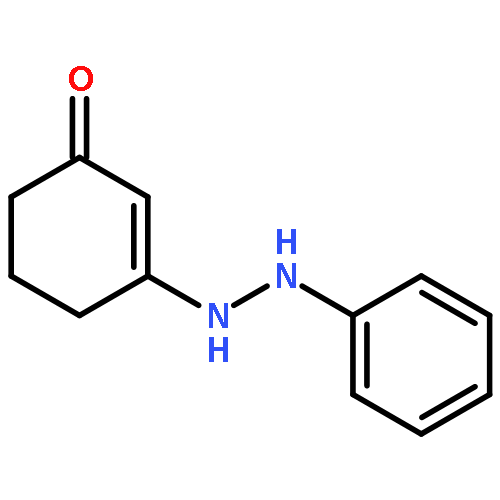
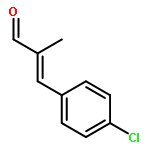
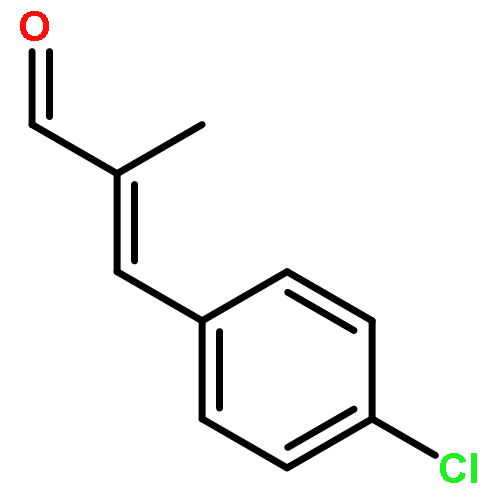
![2-Propyn-1-amine,N-[3-(2,4-dichlorophenoxy)propyl]-N-methyl-](http://img.cochemist.com/ccimg/17800/17780-72-2.png)
![2-Propyn-1-amine,N-[3-(2,4-dichlorophenoxy)propyl]-N-methyl-](http://img.cochemist.com/ccimg/17800/17780-72-2_b.png)


![11H-Indolo[3,2-c]quinoline, 6-chloro-2-methoxy-](http://img.cochemist.com/ccimg/649800/649749-00-8.png)
![11H-Indolo[3,2-c]quinoline, 6-chloro-2-methoxy-](http://img.cochemist.com/ccimg/649800/649749-00-8_b.png)
![11H-Indolo[3,2-c]quinoline, 6-chloro-2-methyl-](http://img.cochemist.com/ccimg/649800/649748-98-1.png)
![11H-Indolo[3,2-c]quinoline, 6-chloro-2-methyl-](http://img.cochemist.com/ccimg/649800/649748-98-1_b.png)
![6H-Indolo[3,2-c]quinolin-6-one, 5,11-dihydro-2-methoxy-](http://img.cochemist.com/ccimg/649800/649748-96-9.png)
![6H-Indolo[3,2-c]quinolin-6-one, 5,11-dihydro-2-methoxy-](http://img.cochemist.com/ccimg/649800/649748-96-9_b.png)
![2-METHYL-5,11-DIHYDROINDOLO[3,2-C]QUINOLIN-6-ONE](http://img.cochemist.com/ccimg/543800/543745-39-7.png)
![2-METHYL-5,11-DIHYDROINDOLO[3,2-C]QUINOLIN-6-ONE](http://img.cochemist.com/ccimg/543800/543745-39-7_b.png)
![11H-Indolo[3,2-c]quinoline, 2,6-dichloro-](http://img.cochemist.com/ccimg/484000/483995-53-5.png)
![11H-Indolo[3,2-c]quinoline, 2,6-dichloro-](http://img.cochemist.com/ccimg/484000/483995-53-5_b.png)
![6H-Indolo[3,2-c]quinolin-6-one, 2-chloro-5,11-dihydro-](http://img.cochemist.com/ccimg/484000/483995-48-8.png)
![6H-Indolo[3,2-c]quinolin-6-one, 2-chloro-5,11-dihydro-](http://img.cochemist.com/ccimg/484000/483995-48-8_b.png)
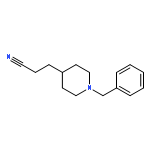

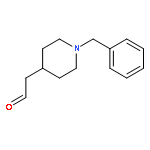
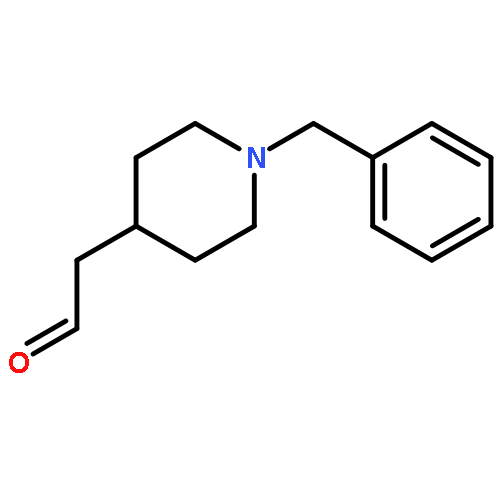
![11H-Indolo[3,2-c]quinoline, 6-chloro-](http://img.cochemist.com/ccimg/108900/108832-13-9.png)
![11H-Indolo[3,2-c]quinoline, 6-chloro-](http://img.cochemist.com/ccimg/108900/108832-13-9_b.png)
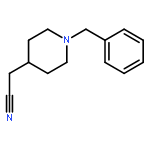
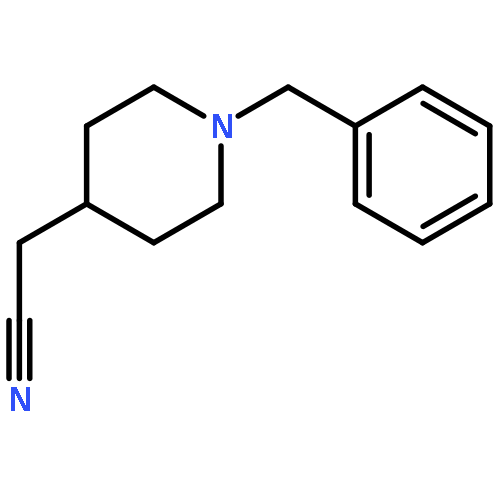

![6H-Indolo[3,2-c]quinolin-6-one,5,11-dihydro-](http://img.cochemist.com/ccimg/18800/18735-98-3.png)
![6H-Indolo[3,2-c]quinolin-6-one,5,11-dihydro-](http://img.cochemist.com/ccimg/18800/18735-98-3_b.png)