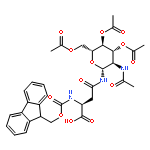
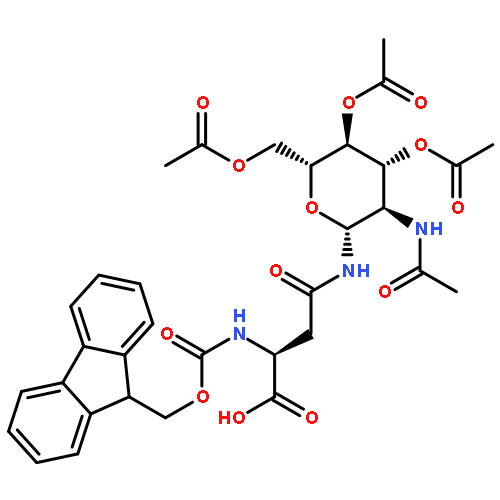
Co-reporter:Che-Hsiung Hsu; Sangho Park; David E. Mortenson; B. Lachele Foley; Xiaocong Wang; Robert J. Woods; David A. Case; Evan T. Powers; Chi-Huey Wong; H. Jane Dyson;Jeffery W. Kelly
Journal of the American Chemical Society 2016 Volume 138(Issue 24) pp:7636-7648
Publication Date(Web):June 1, 2016
DOI:10.1021/jacs.6b02879
Interactions between proteins and carbohydrates are ubiquitous in biology. Therefore, understanding the factors that determine their affinity and selectivity are correspondingly important. Herein, we have determined the relative strengths of intramolecular interactions between a series of monosaccharides and an aromatic ring close to the glycosylation site in an N-glycoprotein host. We employed the enhanced aromatic sequon, a structural motif found in the reverse turns of some N-glycoproteins, to facilitate face-to-face monosaccharide–aromatic interactions. A protein host was used because the dependence of the folding energetics on the identity of the monosaccharide can be accurately measured to assess the strength of the carbohydrate–aromatic interaction. Our data demonstrate that the carbohydrate–aromatic interaction strengths are moderately affected by changes in the stereochemistry and identity of the substituents on the pyranose rings of the sugars. Galactose seems to make the weakest and allose the strongest sugar–aromatic interactions, with glucose, N-acetylglucosamine (GlcNAc) and mannose in between. The NMR solution structures of several of the monosaccharide-containing N-glycoproteins were solved to further understand the origins of the similarities and differences between the monosaccharide–aromatic interaction energies. Peracetylation of the monosaccharides substantially increases the strength of the sugar–aromatic interaction in the context of our N-glycoprotein host. Finally, we discuss our results in light of recent literature regarding the contribution of electrostatics to CH−π interactions and speculate on what our observations imply about the absolute conservation of GlcNAc as the monosaccharide through which N-linked glycans are attached to glycoproteins in eukaryotes.
Co-reporter:Wentao Chen, Leopold Kong, Stephen Connelly, Julia M. Dendle, Yu Liu, Ian A. Wilson, Evan T. Powers, and Jeffery W. Kelly
ACS Chemical Biology 2016 Volume 11(Issue 7) pp:1852
Publication Date(Web):April 29, 2016
DOI:10.1021/acschembio.5b01035
Monoclonal antibodies (mAbs) exhibiting highly selective binding to a protein target constitute a large and growing proportion of the therapeutics market. Aggregation of mAbs results in the loss of their therapeutic efficacy and can result in deleterious immune responses. The CH2 domain comprising part of the Fc portion of Immunoglobulin G (IgG) is typically the least stable domain in IgG-type antibodies and therefore influences their aggregation propensity. We stabilized the CH2 domain by engineering an enhanced aromatic sequon (EAS) into the N-glycosylated C′E loop and observed a 4.8 °C increase in the melting temperature of the purified IgG1 Fc fragment. This EAS-stabilized CH2 domain also conferred enhanced stability against thermal and low pH induced aggregation in the context of a full-length monoclonal IgG1 antibody. The crystal structure of the EAS-stabilized (Q295F/Y296A) IgG1 Fc fragment confirms the design principle, i.e., the importance of the GlcNAc1•F295 interaction, and surprisingly reveals that the core fucose attached to GlcNAc1 also engages in an interaction with F295. Inhibition of core fucosylation confirms the contribution of the fucose–Phe interaction to the stabilization. The Q295F/Y296A mutations also modulate the binding affinity of the full-length antibody to Fc receptors by decreasing the binding to low affinity Fc gamma receptors (FcγRIIa, FcγRIIIa, and FcγRIIIb), while maintaining wild-type binding affinity to FcRn and FcγRI. Our results demonstrate that engineering an EAS into the N-glycosylated reverse turn on the C′E loop leads to stabilizing N-glycan–protein interactions in antibodies and that this modification modulates antibody–Fc receptor binding.
Co-reporter:Amber N. Murray, Wentao Chen, Aristotelis Antonopoulos, Sarah R. Hanson, R. Luke Wiseman, Anne Dell, Stuart M. Haslam, David L. Powers, Evan T. Powers, Jeffery W. Kelly
Chemistry & Biology 2015 Volume 22(Issue 8) pp:1052-1062
Publication Date(Web):20 August 2015
DOI:10.1016/j.chembiol.2015.06.017
•Aromatic amino acids two residues before sequons increase glycosylation efficiency•Aromatic amino acids two residues before sequons decrease N-glycan heterogeneity•Increased glycan occupancy results from oligosaccharyltransferase preferences•Decreased N-glycan heterogeneity results from suppressed Golgi glycan remodelingN-Glycosylation plays an important role in protein folding and function. Previous studies demonstrate that a phenylalanine residue introduced at the n-2 position relative to an Asn-Xxx-Thr/Ser N-glycosylation sequon increases the glycan occupancy of the sequon in insect cells. Here, we show that any aromatic residue at n-2 increases glycan occupancy in human cells and that this effect is dependent upon oligosaccharyltransferase substrate preferences rather than differences in other cellular processing events such as degradation or trafficking. Moreover, aromatic residues at n-2 alter glycan processing in the Golgi, producing proteins with less complex N-glycan structures. These results demonstrate that manipulating the sequence space surrounding N-glycosylation sequons is useful both for controlling glycosylation efficiency, thus enhancing glycan occupancy, and for influencing the N-glycan structures produced.Figure optionsDownload full-size imageDownload high-quality image (170 K)Download as PowerPoint slide
Co-reporter:Evan T. Powers
&
William E. Balch
Nature Reviews Molecular Cell Biology 2013 14(4) pp:237
Publication Date(Web):2013-03-06
DOI:10.1038/nrm3542
Although the sequence of a protein largely determines its function, proteins can adopt different folding states in response to changes in the environment, some of which may be deleterious to the organism. All organisms —Bacteria, Archaea and Eukarya — have evolved a protein homeostasis, or proteostasis, network comprising chaperones and folding factors, degradation components, signalling pathways and specialized compartmentalized modules that manage protein folding in response to environmental stimuli and variation. Surveying the origins of proteostasis networks reveals that they have co-evolved with the proteome to regulate the physiological state of the cell, reflecting the unique stresses that different cells or organisms experience, and that they have a key role in driving evolution by closely managing the link between the phenotype and the genotype.
Co-reporter:Wentao Chen ; Sebastian Enck ; Joshua L. Price ; David L. Powers ; Evan T. Powers ; Chi-Huey Wong ; H. Jane Dyson ;Jeffery W. Kelly
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9877-9884
Publication Date(Web):June 7, 2013
DOI:10.1021/ja4040472
Carbohydrate–aromatic interactions mediate many biological processes. However, the structure–energy relationships underpinning direct carbohydrate–aromatic packing interactions in aqueous solution have been difficult to assess experimentally and remain elusive. Here, we determine the structures and folding energetics of chemically synthesized glycoproteins to quantify the contributions of the hydrophobic effect and CH−π interactions to carbohydrate–aromatic packing interactions in proteins. We find that the hydrophobic effect contributes significantly to protein–carbohydrate interactions. Interactions between carbohydrates and aromatic amino acid side chains, however, are supplemented by CH−π interactions. The strengths of experimentally determined carbohydrate CH−π interactions do not correlate with the electrostatic properties of the involved aromatic residues, suggesting that the electrostatic component of CH−π interactions in aqueous solution is small. Thus, tight binding of carbohydrates and aromatic residues is driven by the hydrophobic effect and CH−π interactions featuring a dominating dispersive component.
Co-reporter:Joshua L. Price, Evan T. Powers, and Jeffery W. Kelly
ACS Chemical Biology 2011 Volume 6(Issue 11) pp:1188
Publication Date(Web):September 22, 2011
DOI:10.1021/cb200277u
The intrinsic stabilization of therapeutic proteins by N-glycosylation can endow them with increased shelf and serum half-lives owing to lower populations of misfolded and unfolded states, which are susceptible to aggregation and proteolysis. Conjugation of poly(ethylene glycol) (PEG) oligomers to nucleophilic groups on the surfaces of folded proteins (i.e., PEGylation) is a chemical alternative to N-glycosylation, in that it can also enhance the pharmacologic attributes of therapeutic proteins. However, the energetic consequences of PEGylation are currently not predictable. We find that PEGylation of an Asn residue in reverse turn 1 of the Pin WW domain is intrinsically stabilizing in several sequence contexts, unlike N-glycosylation, which is only stabilizing in a particular sequence context. Our thermodynamic data are consistent with the hypothesis that PEGylation destabilizes the protein denatured state ensemble via an excluded volume effect, whereas N-glycosylation-associated stabilization results primarily from native state interactions between the N-glycan and the protein.
Co-reporter:Elizabeth K. Culyba;Joshua L. Price;Sarah R. Hanson;Apratim Dhar;Chi-Huey Wong;Martin Gruebele;Jeffery W. Kelly
Science 2011 Volume 331(Issue 6017) pp:571-575
Publication Date(Web):04 Feb 2011
DOI:10.1126/science.1198461
Protein reverse turns that interact with a phenlyalanine group allow stable introduction of glycan groups at asparagine residues.
Co-reporter:Sarah R. Hanson;Elizabeth K. Culyba;Tsui-Ling Hsu;Chi-Huey Wong;Jeffery W. Kelly
PNAS 2009 Volume 106 (Issue 9 ) pp:3131-3136
Publication Date(Web):2009-03-03
DOI:10.1073/pnas.0810318105
The folding energetics of the mono-N-glycosylated adhesion domain of the human immune cell receptor cluster of differentiation
2 (hCD2ad) were studied systematically to understand the influence of the N-glycan on the folding energy landscape. Fully
elaborated N-glycan structures accelerate folding by 4-fold and stabilize the β-sandwich structure by 3.1 kcal/mol, relative
to the nonglycosylated protein. The N-glycan's first saccharide unit accounts for the entire acceleration of folding and for
2/3 of the native state stabilization. The remaining third of the stabilization is derived from the next 2 saccharide units.
Thus, the conserved N-linked triose core, ManGlcNAc2, improves both the kinetics and the thermodynamics of protein folding. The native state stabilization and decreased activation
barrier for folding conferred by N-glycosylation provide a powerful and potentially general mechanism for enhancing folding
in the secretory pathway.
Co-reporter:Amy R. Hurshman Babbes, Evan T. Powers and Jeffery W. Kelly
Biochemistry 2008 Volume 47(Issue 26) pp:
Publication Date(Web):June 7, 2008
DOI:10.1021/bi800636q
Urea denaturation studies were carried out as a function of transthyretin (TTR) concentration to quantify the thermodynamically linked quaternary and tertiary structural stability and to improve our understanding of the relationship between mutant folding energetics and amyloid disease phenotype. Urea denaturation of TTR involves at least two equilibria: dissociation of tetramers into folded monomers and monomer unfolding. To deal with the thermodynamic linkage of these equilibria, we analyzed concentration-dependent denaturation data by globally fitting them to an equation that simultaneously accounts for the two-step denaturation process. Using this method, the quaternary and tertiary structural stabilities of well-behaved TTR sequences, wild-type (WT) TTR and the disease-associated variant V122I, were scrutinized. The V122I variant is linked to late onset familial amyloid cardiomyopathy, the most common familial TTR amyloid disease. V122I TTR exhibits a destabilized quaternary structure and a stable tertiary structure relative to those of WT TTR. Three other variants of TTR were also examined, L55P, V30M, and A25T TTR. The L55P mutation is associated with the most aggressive familial TTR amyloid disease. L55P TTR has a complicated denaturation pathway that includes dimers and trimers, so globally fitting its concentration-dependent urea denaturation data yielded error-laden estimates of stability parameters. Nevertheless, it is clear that L55P TTR is substantially less stable than WT TTR, primarily because its tertiary structure is unstable, although its quaternary structure is destabilized as well. V30M is the most common mutation associated with neuropathic forms of TTR amyloid disease. V30M TTR is certainly destabilized relative to WT TTR, but like L55P TTR, it has a complex denaturation pathway that cannot be fit to the aforementioned two-step denaturation model. Literature data suggest that V30M TTR has stable quaternary structure but unstable tertiary structure. The A25T mutant, associated with central nervous system amyloidosis, is highly aggregation-prone and exhibits drastically reduced quaternary and tertiary structural stabilities. The observed differences in stability among the disease-associated TTR variants highlight the complexity and heterogeneity of TTR amyloid disease, an observation that has important implications for the treatment of these maladies.
Co-reporter:Darren M. Hutt, Evan T. Powers, William E. Balch
FEBS Letters (20 August 2009) Volume 583(Issue 16) pp:2639-2646
Publication Date(Web):20 August 2009
DOI:10.1016/j.febslet.2009.07.014
Protein function is regulated by the proteostasis network (PN) [Balch, W.E., Morimoto, R.I., Dillin, A. and Kelly, J.W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919], an integrated biological system that generates and protects the protein fold. The composition of the PN is regulated by signaling pathways including the unfolded protein response (UPR), the heat-shock response (HSR), the ubiquitin proteasome system (UPS) and epigenetic programs. Mismanagement of protein folding and function during membrane trafficking through the exocytic and endocytic pathways of eukaryotic cells by the PN is responsible for a wide range of diseases that include, among others, lysosomal storage diseases, myelination diseases, cystic fibrosis, systemic amyloidoses such as light chain myeloma, and neurodegenerative diseases including Alzheimer’s. Toxicity from misfolding can be cell autonomous (affect the producing cell) or cell non-autonomous (affect a non-producing cell) or both, and have either a loss-of-function or gain-of-toxic function phenotype. Herein, we review the role of the PN and its regulatory transcriptional circuitry likely to be operational in managing the protein fold and function during membrane trafficking. We emphasize the enabling principle of a ‘proteostasis boundary (PB)’ [Powers, E.T., Morimoto, R.T., Dillin, A., Kelly, J.W., and Balch, W.E. (2009) Biochemical and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991]. The PB is defined by the combined effects of the kinetics and thermodynamics of folding and the kinetics of misfolding, which are linked to the variable and adjustable PN capacity found different cell types. Differences in the PN account for the versatility of protein folding and function in health, and the cellular and tissue response to mutation and environmental challenges in disease. We discuss how manipulation of the folding energetics or the PB through metabolites and pharmacological intervention provides multiple routes for restoration of biological function in trafficking disease.
Co-reporter:Evan T. Powers, David L. Powers
Biophysical Journal (15 January 2008) Volume 94(Issue 2) pp:
Publication Date(Web):15 January 2008
DOI:10.1529/biophysj.107.117168
The formation of protein fibrils, and in particular amyloid fibrils, underlies many human diseases. Understanding fibril formation mechanisms is important for understanding disease pathology, but fibril formation kinetics can be complicated, making the relationship between experimental observables and specific mechanisms unclear. Here we examine one often-proposed fibril formation mechanism, nucleated polymerization with off-pathway aggregation. We use the characteristics of this mechanism to derive three tests that can be performed on experimental data to identify it. We also find that this mechanism has an especially striking feature: although increasing protein concentrations generally cause simple nucleated polymerizations to reach completion faster, they cause nucleated polymerizations with off-pathway aggregation to reach completion more slowly when the protein concentration becomes too high.
Co-reporter:Steven M. Johnson, Stephen Connelly, Colleen Fearns, Evan T. Powers, Jeffery W. Kelly
Journal of Molecular Biology (10 August 2012) Volume 421(Issues 2–3) pp:185-203
Publication Date(Web):10 August 2012
DOI:10.1016/j.jmb.2011.12.060
Transthyretin (TTR) is one of the many proteins that are known to misfold and aggregate (i.e., undergo amyloidogenesis) in vivo. The process of TTR amyloidogenesis causes nervous system and/or heart pathology. While several of these maladies are associated with mutations that destabilize the native TTR quaternary and/or tertiary structure, wild-type TTR amyloidogenesis also leads to the degeneration of postmitotic tissue. Over the past 20 years, much has been learned about the factors that influence the propensity of TTR to aggregate. This biophysical information led to the development of a therapeutic strategy, termed “kinetic stabilization,” to prevent TTR amyloidogenesis. This strategy afforded the drug tafamidis which was recently approved by the European Medicines Agency for the treatment of TTR familial amyloid polyneuropathy, the most common familial TTR amyloid disease. Tafamidis is the first and currently the only medication approved to treat TTR familial amyloid polyneuropathy. Here we review the biophysical basis for the kinetic stabilization strategy and the structure-based drug design effort that led to this first-in-class pharmacologic agent.Download high-res image (335KB)Download full-size imageHighlights► Wild-type and mutant TTRs cause amyloid diseases in humans. ► Amyloid formation requires tetramer dissociation (rate limiting) and monomer misfolding. ► TTR tetramers can be kinetically stabilized by binding small molecules. ► Tafamidis, a kinetic stabilizer of TTR, is the first drug approved to treat TTR amyloidosis.
.jpg)




![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0.png)
![[(2R,3R,4S,5S,6R)-3,4,5-triacetyloxy-6-azidooxan-2-yl]methyl acetate](http://img.cochemist.com/ccimg/65900/65864-60-0_b.png)













![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,4',5'-bis(1,3,2-dithiarsolan-2-yl)-3',6'-dihydroxy-](/data/chemimg/119300/212118-77-9.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,4',5'-bis(1,3,2-dithiarsolan-2-yl)-3',6'-dihydroxy-](/data/chemimg/119300/212118-77-9_b.png)








![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)