Co-reporter:Jenna Scotcher, David J. Clarke, C. Logan Mackay, Ted Hupp, Peter J. Sadler and Pat R. R. Langridge-Smith
Chemical Science 2013 vol. 4(Issue 3) pp:1257-1269
Publication Date(Web):03 Jan 2013
DOI:10.1039/C2SC21702C
The p53 tumour suppressor protein promotes cell cycle arrest or apoptosis in response to several forms of cellular stress and DNA damage. p53 acts as a transcription factor by binding to specific DNA sequences and regulating the expression of a myriad of target genes. p53 activity is known to be influenced by several post-translational modifications and is dependent on coordination of a zinc ion within the protein core domain. Several lines of evidence have demonstrated that oxidative modification of cysteine residues within p53 can also influence the protein's activity. However, little is known regarding the molecular details of p53 oxidation. Here we analyze oxidation pathways in the p53 core domain by Fourier transform ion cyclotron resonance mass spectrometry and top-down fragmentation. Firstly, we show that p53 core domain is sensitive to oxidation by the reactive oxygen species (ROS) hydrogen peroxide and that the zinc-coordination site is the initial target for ROS-induced oxidation. Two disulfide bonds are formed involving Cys182 and the three cysteines which coordinate to zinc (Cys176, 238 and 242). This disulfide bond formation is accompanied by loss of zinc from the binding site. Secondly, an additional cysteine, Cys277, is prone to oxidation via a ROS-independent mechanism. This residue undergoes S-glutathionylation at biologically relevant reduction potentials. We discuss our findings in the context of redox regulation of p53 activity and in comparison to other redox regulated proteins.
Co-reporter:Jenna Scotcher, David J. Clarke, Pat R.R. Langridge-Smith
Analytical Biochemistry 2012 Volume 420(Issue 1) pp:96-98
Publication Date(Web):1 January 2012
DOI:10.1016/j.ab.2011.08.049
Oxidation of cysteine is now known to serve as a fundamental mechanism to control protein function or activity. Many redox-regulated proteins do not oxidize to homogeneity, resulting in a mixture of reduced and oxidized species which cannot be separated chromatographically. Here we describe a protocol for the separation of reduced and oxidized forms of the tumor suppressor protein p53. This purification method relies on the reversible labeling of thiol groups with biotin and exploitation of the ultrastrong biotin–avidin interaction. This purification procedure can be applied to other cysteine-containing proteins where enrichment of the oxidized form is required.
Co-reporter:Jenna Scotcher;David J. Clarke
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 5) pp:888-897
Publication Date(Web):2011 May
DOI:10.1007/s13361-011-0088-x
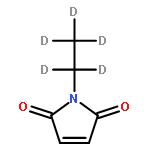
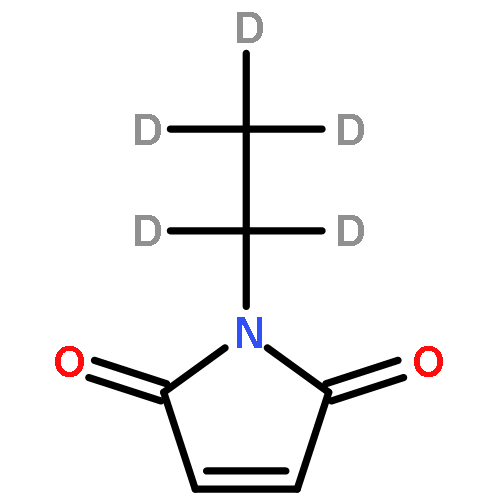
The tumor suppressor p53 is a redox-regulated transcription factor involved in cell cycle arrest, apoptosis and senescence in response to multiple forms of stress, as well as many other cellular processes such as DNA repair, glycolysis, autophagy, oxidative stress and differentiation. The discovery of cysteine-targeting compounds that cause re-activation of mutant p53 and the death of tumor cells in vivo has emphasized the functional importance of p53 thiols. Using a combination of top-down and middle-down FTICR mass spectrometry, we show that of the 10 Cys residues in the core domain of wild-type p53, Cys182 and Cys277 exhibit a remarkable preference for modification by the alkylating reagent N-ethylmaleimide. The assignment of Cys182 and Cys277 as the two reactive Cys residues was confirmed by site-directed mutagenesis. Further alkylation of p53 beyond Cys182 and Cys277 was found to trigger co-operative modification of the remaining seven Cys residues and protein unfolding. This study highlights the power of top-down FTICR mass spectrometry for analysis of the cysteine reactivity and redox chemistry in multiple cysteine-containing proteins.
Co-reporter:David J. Clarke;Euan Murray;Ted Hupp
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 8) pp:1432-1440
Publication Date(Web):2011 August
DOI:10.1007/s13361-011-0155-3
Noncovalent protein–ligand and protein–protein complexes are readily detected using electrospray ionization mass spectrometry (ESI MS). Furthermore, recent reports have demonstrated that careful use of electron capture dissociation (ECD) fragmentation allows covalent backbone bonds of protein complexes to be dissociated without disruption of noncovalent protein–ligand interactions. In this way the site of protein–ligand interfaces can be identified. To date, protein–ligand complexes, which have proven tractable to this technique, have been mediated by ionic electrostatic interactions, i.e., ion pair interactions or salt bridging. Here we extend this methodology by applying ECD to study a protein–peptide complex that contains no electrostatics interactions. We analyzed the complex between the 21 kDa p53-inhibitor protein anterior gradient-2 and its hexapeptide binding ligand (PTTIYY). ECD fragmentation of the 1:1 complex occurs with retention of protein–peptide binding and analysis of the resulting fragments allows the binding interface to be localized to a C-terminal region between residues 109 and 175. These finding are supported by a solution-phase competition assay, which implicates the region between residues 108 and 122 within AGR2 as the PTTIYY binding interface. Our study expands previous findings by demonstrating that top-down ECD mass spectrometry can be used to determine directly the sites of peptide–protein interfaces. This highlights the growing potential of using ECD and related top-down fragmentation techniques for interrogation of protein–protein interfaces.
Co-reporter:Jenna Scotcher, David J. Clarke, C. Logan Mackay, Ted Hupp, Peter J. Sadler and Pat R. R. Langridge-Smith
Chemical Science (2010-Present) 2013 - vol. 4(Issue 3) pp:NaN1269-1269
Publication Date(Web):2013/01/03
DOI:10.1039/C2SC21702C
The p53 tumour suppressor protein promotes cell cycle arrest or apoptosis in response to several forms of cellular stress and DNA damage. p53 acts as a transcription factor by binding to specific DNA sequences and regulating the expression of a myriad of target genes. p53 activity is known to be influenced by several post-translational modifications and is dependent on coordination of a zinc ion within the protein core domain. Several lines of evidence have demonstrated that oxidative modification of cysteine residues within p53 can also influence the protein's activity. However, little is known regarding the molecular details of p53 oxidation. Here we analyze oxidation pathways in the p53 core domain by Fourier transform ion cyclotron resonance mass spectrometry and top-down fragmentation. Firstly, we show that p53 core domain is sensitive to oxidation by the reactive oxygen species (ROS) hydrogen peroxide and that the zinc-coordination site is the initial target for ROS-induced oxidation. Two disulfide bonds are formed involving Cys182 and the three cysteines which coordinate to zinc (Cys176, 238 and 242). This disulfide bond formation is accompanied by loss of zinc from the binding site. Secondly, an additional cysteine, Cys277, is prone to oxidation via a ROS-independent mechanism. This residue undergoes S-glutathionylation at biologically relevant reduction potentials. We discuss our findings in the context of redox regulation of p53 activity and in comparison to other redox regulated proteins.




![N-[6-(Biotinamido)hexyl]-3'-(2'-pyridyldithio)propionamide](http://img.cochemist.com/ccimg/129200/129179-83-5.png)
![N-[6-(Biotinamido)hexyl]-3'-(2'-pyridyldithio)propionamide](http://img.cochemist.com/ccimg/129200/129179-83-5_b.png)