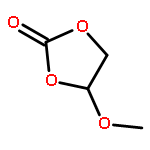
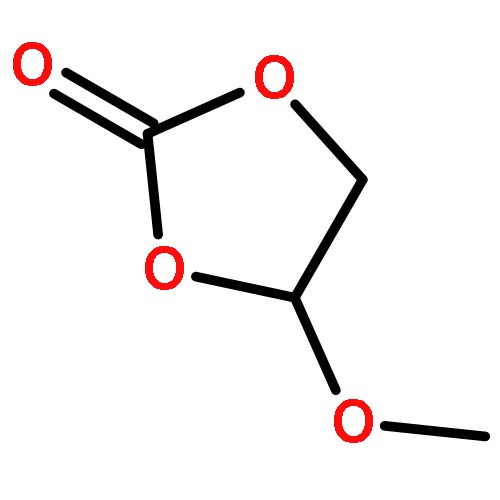
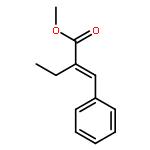
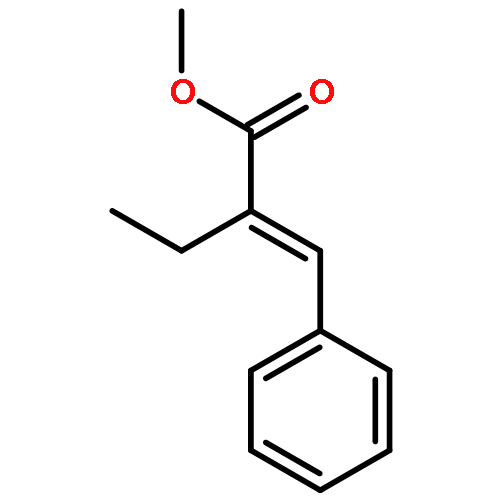
Co-reporter:Liang Zhao, Bi-Zhi Xu, Jianfeng Jia, Hai-Shun Wu
Computational Materials Science 2017 Volume 137(Volume 137) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.commatsci.2017.05.017
•The hydrogen storage capacities of newly designed Sc and Ti doped COFs were investigated with DFT calculations.•The C4B2H6Sc2 blocks are proper for hydrogen storage under ambient condition, while the C4B2H6Ti2 blocks are not.•The designed (C6H3)2(B2C4H4)3Sc6 frameworks can adsorb 24 hydrogen molecules with the binding energies of 0.21–0.34 eV.The hydrogen storage properties of newly designed Sc- and Ti-decorated covalent organic frameworks, (C6H3)2(B2C4H4)3M6 (MSc, Ti), were investigated using density functional theory method. We found that each doped Sc atom can effectively adsorb 4 H2 with the average adsorption energy being in a range from 0.21 to 0.34 eV. On the doped Ti atoms, the first H2 is dissociated and other H2 molecules have small, or even negative consecutive adsorption energy. Our calculation results showed that the Sc-decorated (C6H3)2(B2C4H4)3Sc6 covalent organic frameworks can adsorb 24 hydrogen molecules, which give a hydrogen storage capacity of 7.02 wt% under ambient condition.Download high-res image (226KB)Download full-size image
Co-reporter:Li-Juan Ma, Min Han, Jianfeng Wang, Jianfeng Jia, Hai-Shun Wu
International Journal of Hydrogen Energy 2017 Volume 42, Issue 49(Volume 42, Issue 49) pp:
Publication Date(Web):7 December 2017
DOI:10.1016/j.ijhydene.2017.10.093
•Hydrogen gravimetric density of TMC2H2 (TM = Sc-Ni) is 4.54–12.43%.•H2 storage capacity of TiC2H2 is 12.00%, regardless of CH2 group formation.•Formation of CH2 make H2 storage capacity of ScC2H2 reduce from 12.43% to 10.13%.Different aspects of the hydrogen storage properties of TMC2H2 (TM = Sc-Ni) were evaluated using density functional theory and ab initio molecular dynamics calculations. Hydrogen saturation conformations indicate that the hydrogen gravimetric density of TMC2H2 (TM = Sc-Ni) is 4.54–12.43%. The free energy profiles and first-principles molecular-dynamics (MD) simulations showed that when the first H2 molecule attaches to TMC2H2 (TM = Sc, Ti), the H2 molecule is first dissociated over Ti/Sc, and then, one of the H atoms goes to a carbon atom, forming a CH2 group. That is, there are two types of hydrogen bonding in TiC2H2 and ScTiC2H2: the first set of hydrogen binds to C, and the subsequent set of hydrogen binds to Ti/Sc. Importantly, TiC2H2 is able to bind five H2 molecules with the hydrogen gravimetric density of 12.00%, regardless of CH2 group formation. However, ScC2H3H can bind only three H2 molecules, reducing the gravimetric uptake capacity of ScC2H2 from 12.43% to 10.13%. For TMC2H2 (TM = V, Cr, Mn), the first hydrogen molecule is dissociated, but there is no “spillover effect”. For TMC2H2 (TM = Fe, Co, Ni), the first hydrogen molecule is bound quasi-molecularly. Most of our findings, such as those regarding H2 dissociation and TMC2H3H formation, should be valid for other TM-decorated nanostructures.
Co-reporter:Li-Juan Ma, Min Han, Jianfeng Wang, Jianfeng Jia, Hai-Shun Wu
International Journal of Hydrogen Energy 2017 Volume 42, Issue 20(Volume 42, Issue 20) pp:
Publication Date(Web):18 May 2017
DOI:10.1016/j.ijhydene.2017.04.114
•C2H2V monomer can adsorb five H2 with a binding energy of 0.13–0.46 eV/H2.•Oligomers are possible for Vanadium-acetylene complexes.•Steric condition besides the 18-electron rule affects the number of adsorbed H2.•Back donation is the only determinant of adsorption state of H2.•Changing the charge of V can regulate the back-donation and the state of adsorbed H2.A systematic computational study of oligomers and hydrogen storage properties of Vanadium-acetylene complexes has been performed by using the density-functional B3LYP method with 6-311++G (3df, 3pd) basis sets. C2H2V monomers can trap up to five hydrogen molecules with a binding energy of 0.13–0.46 eV/H2, reaching gravimetric uptake capacity as high as 11.57%. In condensed phase, oligomers are possible. Because the maximum number of H2 molecules that can be stored by (C2H2)nVm (n = 1–4; m = 1, 2) complexes not only depends on the coordination number of a single metal atom but also on the steric condition, the corresponding gravimetric uptake capacity reduce from 11.57% (C2H2V) to 2.85% ((C2H2)4V2). Both monomers and oligomers of Vanadium-acetylene complexes can coordinate multiple hydrogen molecules by the Kubas-typed orbital interaction, and back donation is the only determinant of the hydrogen adsorption state.Download high-res image (207KB)Download full-size image
Co-reporter:Xiaonan Wang;Haiyan Zhou;Zhi Yan;Xiaoyang Zhang
Theoretical Chemistry Accounts 2017 Volume 136( Issue 8) pp:90
Publication Date(Web):25 July 2017
DOI:10.1007/s00214-017-2121-z
The well-dispersed single-metal atom with high activity on the support is the important prerequisite to the application of the single-atom catalysts (SACs). In this study, we find that the inexpensive metal, iron (Fe), onto boron (B)-doped graphene based on n–p codoping approach can effectively form stale and high-activity SACs. As a prototype example, we check CO oxidation reaction on B-/Fe-codoped graphene. It is firstly shown that well-dispersed Fe atom can be realized with the help of stronger electrostatic attraction from the n-type Fe- and p-type B-doped graphene. Secondly, the maximum energy barrier of CO oxidation on B-/Fe-codoped graphene is 0.692 eV by the Eley–Rideal mechanism. Further analysis indicates that spin state of the O2 plays an important role in the CO oxidation on B-/Fe-codoped graphene.
Co-reporter:Tianjun Hu, Ying Wang, Qing Liu, Lina Zhang, ... Jianfeng Jia
International Journal of Hydrogen Energy 2017 Volume 42, Issue 41(Volume 42, Issue 41) pp:
Publication Date(Web):12 October 2017
DOI:10.1016/j.ijhydene.2017.08.160
•The PdPdO-CoOx was prepared via a simple, mild, green and efficient method.•CoOx promoted the generation of oxygenated species located at Pd particle surface.•The generation of oxygenated species enhance the activity and stability.•The performance relies on the electronic interaction between Pd and PdO-CoOx.•The PdPdO-CoOx (1:4) have a higher performance than the commercial Pt/C.To achieve the better electrocatalytic activity and stability of Pd-base catalysts for ethylene glycol and glycerol oxidation reactions, a novel Pd-base binary PdCo oxides nanoparticles (PdPdO-CoOx) was synthesized by in-situ oxidation of PdCo precursor. The strategy was simple, mild, green and efficient. The prepared nanoparticles exhibited a mutually connected, fused irregular nanoparticles in TEM. The as-synthesized PdPdO-CoOx (1:4) nanoparticles displayed prominent catalytic activity (5.82 A mgPd−1 for ethylene glycol and 5.16 A mgPd−1 for glycerol) for ethylene glycol and glycerol oxidation reactions in alkaline solution compared to the commercial Pt/C (1.64 A mgPt−1 for ethylene glycol and 1.48 A mgPt−1 for glycerol) catalyst. The improved electrocatalytic activity of PdPdO-CoOx catalyst mainly ascribes to the producing Strong Metal-Support Interactions (SMSI) between PdO-CoOx and Pd nanoparticles, the synergistic effect between PdO and CoOx and the presence of CoOx promoved hydroxyl adsorption at lower potentials. Combined with the simple synthetic method, lower cost and good performance, PdPdO-CoOx is a promising catalyst for direct fuel cells.
Co-reporter:Wenjuan Liang, Jianfeng Jia, Jin Lv, Haishun Wu
Journal of Molecular Structure 2016 Volume 1108() pp:92-95
Publication Date(Web):15 March 2016
DOI:10.1016/j.molstruc.2015.11.057
•The magnetic nature of the clusters M@(BN)48 significantly changed when doping with Mo atom, except for Co@(BN)48.•Only the magnetic moment for the CrMo@(BN)48 cluster was decreased to zero.•M@(BN)48 clusters can be selected as the model system to detect Mo atom by the change of the magnetic moment.The structure and magnetic properties of Mo-doped M@(BN)48 (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu) clusters were calculated at BPW91/LanL2DZ level. The magnetic nature of the clusters M@(BN)48 significantly changed when doping with Mo atom, except for Co@(BN)48. Only the magnetic moment for the CrMo@(BN)48 cluster was decreased to zero. Thus, M@(BN)48 clusters can be selected as the model system to detect Mo atom by the change of the magnetic moment.Comparative magnetic moments for M@(BN)48 and MMo@(BN)48 clusters.
Co-reporter:Fei Tang, Jianfeng Jia, Hai-Shun Wu
Computational Materials Science 2016 Volume 112(Part A) pp:327-332
Publication Date(Web):1 February 2016
DOI:10.1016/j.commatsci.2015.11.005
•The structures and hydrogen storage properties of C6−nBnH6Sc (n = 0–6) units were theoretically investigated.•The double hybrid method of MPW2PLYPD was found more suitable for Kubas interaction.•Introduction of the B atom enhances the interaction between Sc atom and the C6−nBnH6.•C5B6H6Sc, C4B2H6Sc, C2B4H6Sc, and B6H6Sc can effectively adsorb four, four, four and three H2 molecules, respectively.The structures and hydrogen adsorption properties of C6−nBnH6Sc (n = 0–6) were investigated with density functional methods and MP2 method. The double hybrid method of MPW2PLYPD was found more suitable for this system. Our calculations showed that the local geometries of C6−nBn change from planar ring to three-dimensional structure with the increasing n. Introduction of the B atoms enhances the interaction between Sc and the C6−nBnH6 substrate. The number of B atoms also affects the H2 adsorption properties of C6−nBnH6Sc. Our calculations show that C5B6H6Sc, C4B2H6Sc, C2B4H6Sc, and B6H6Sc can adsorb four, four, four and three H2 molecules, respectively. The H2 adsorption energies of C3B3H6Sc and CB5H6Sc are remarkably smaller than these of others. The natural bond orbital analysis indicated that there is no positive correlation between the adsorption energy and the charge on Sc atom. Electronic structure analysis was performed to explain the poor H2 adsorption energies of C3B3H6Sc and CB5H6Sc.
Co-reporter:Yan-Bin He, Jian-Feng Jia, Hai-Shun Wu
Applied Surface Science 2015 Volume 339() pp:36-45
Publication Date(Web):1 June 2015
DOI:10.1016/j.apsusc.2015.02.136
Highlights
- •
A series of Ni-based surface alloy films for hydrazine adsorption are proposed.
- •
The doped atoms of Ir, Rh and Fe provide stronger adsorption sites.
- •
The doped atoms of Pt and Pd provide weaker adsorption sites.
- •
Ni8Fe8, Ni8Rh8 and Ni15Ir1 seem to be better candidates for hydrazine catalysis.
Co-reporter:Yan Bin He, Jian Feng Jia, Hai Shun Wu
Applied Surface Science 2015 Volume 327() pp:462-469
Publication Date(Web):1 February 2015
DOI:10.1016/j.apsusc.2014.12.007
Highlights
- •
We propose a model suitable for simulating the adsorption of hydrazine on rhodium nanoparticles.
- •
We found that inclusion of dispersion correction results in significant enhancement for the adsorption to the Rh(1 1 1) surface.
- •
Nanoparticles surface with lower-coordinated sites are more reactive than those with almost saturated surface sites.
Co-reporter:Li-Juan Ma, Jianfeng Jia, Hai-Shun Wu
International Journal of Hydrogen Energy 2015 Volume 40(Issue 1) pp:420-428
Publication Date(Web):5 January 2015
DOI:10.1016/j.ijhydene.2014.10.136
•Synthesized C2H2Sc can adsorb six H2 with a binding energy of 0.14–1.35 eV/H2.•Maximal retrievable hydrogen storage density at 77–298.15 K is 10.20 wt%.•Operation mechanism of hydrogen storage on C2H2Sc was profoundly anatomized.•Estimating the adsorption/desorption temperatures at 1 atm.•Changing the charge of Sc can regulate the adsorption energy.The hydrogen storage capacities of synthesized Scandium–Acetylene systems (Sc–η2–(C2H2) and HCC–ScH) are tested by using density functional theory (DFT) and the coupled-cluster theory (CCSD (T)) with 6–311++G (3df, 3pd) basis sets. Both the energy profile and natural bond orbital analysis predict that Sc–η2–C2H2 and HCC–ScH complexes are promising hydrogen storage materials. The Sc–η2–(C2H2) and HCC–ScH complexes can trap up to six hydrogen molecules, reaching gravimetric uptake capacities as high as 14.56 wt%. Thermo-chemistry calculations indicate two H2 in Sc–η2–C2H2(H2)6 and four H2 in HCC–ScH(H2)6 can be readily adsorbed at 77 K and desorbed at 298.15 K under atmospheric pressure, corresponding to the maximal reversible hydrogen storage abilities of 5.37 and 10.20 wt%, respectively. The further comparison between HCC–ScH(H2) and HCC–ScH− (2H) reveals that the charged state of Sc atom has a great influence on the hydrogen adsorption state and adsorption energy. Moreover, dimers may form in case of scandium-acetylene systems. The most stable (C2H2Sc)2 can adsorb ten H2 molecules, reaching the hydrogen storage capacity of 12.43 wt%. Thermo-chemistry calculations indicate the maximal reversible hydrogen storage capacities of Sc(C2H2)2 and (C2H2Sc)2 are 7.67 and 7.85 wt%, respectively.
Co-reporter:Rui-Li Ding, Jianfeng Jia, Hai-Shun Wu
Applied Surface Science 2015 Volume 359() pp:729-735
Publication Date(Web):30 December 2015
DOI:10.1016/j.apsusc.2015.10.164
Highlights
- •
The structure of supported Aun clusters (n = 1–20) on TiO2 (1 1 0) surface was investigated based on density functional theory calculations.
- •
The supported Aun clusters were found to prefer the planar or quasi-planar structures.
- •
The dispersion interaction dominates the interactions between the Aun clusters and the TiO2 surface.
- •
The obvious charge nonuniformities of the Aun clusters with even n were observed.
Co-reporter:Wenjuan Liang, Jianfeng Jia, Jin Lv, Haishun Wu
Chemical Physics Letters 2015 Volume 622() pp:57-62
Publication Date(Web):16 February 2015
DOI:10.1016/j.cplett.2014.12.020
•Using the Gaussian 09 program package, M1–4@(BN)48 clusters are calculated at BPW91/LanL2DZ level.•The M1–4 clusters generally prefer an off-center position and near the hexagonal rings in the (BN)48 cages.•The (BN)48 cages can increase the stability of these small magnetic clusters.•The (BN)48 cages can protect the magnetic nature of M and M2 clusters.The geometrical structure and magnetic properties of M1–4(M = Fe, Co and Ni) clusters within a (BN)48 cage were calculated at the BPW91/LanL2DZ level. The small M1–4 clusters generally prefer an off-centered position near the hexagonal rings in the (BN)48 cages. The (BN)48 cages can increase the stability of these small magnetic clusters while protecting the magnetic nature of M and M2 clusters.
Co-reporter:Yan-Bin He
The Journal of Physical Chemistry C 2015 Volume 119(Issue 16) pp:8763-8774
Publication Date(Web):March 31, 2015
DOI:10.1021/acs.jpcc.5b01605
We have used density functional theory (DFT) with dispersion correction to investigate the adsorption and first dissociation step of hydrazine on Fe3Ni(111), FeNi(111), and FeNi3(111) surfaces. The calculations have shown that the FeNi3(111) surface offers the strongest binding of molecular hydrazine adsorbed on the top of an iron atom, whereas the strongest adsorption of NH2 fragment bridging between iron atoms is obtained on the Fe3Ni(111) surface; both molecular hydrazine and the NH2 fragment can be adsorbed strongly on the FeNi(111) surface.The first dissociation step of hydrazine on the FeNi(111) surface is found to be exothermic by −1.19 eV and presents an activation energy barrier of only 0.15 eV. A similar energy barrier is found on the Fe3Ni(111) surface, but a higher reaction energy of −1.47 eV is released on this surface. Furthermore, the electronic structures of the molecular and dissociative adsorptions are discussed, which is intended to shed some light on the binding nature of the adsorption and stability among conformations of the adsorbed molecule on these surfaces. It is expected that our results will provide useful information for the development of a catalyst for hydrazine dissociation.
Co-reporter:Li-Juan Ma, Jianfeng Jia, Hai-Shun Wu, Ying Ren
International Journal of Hydrogen Energy 2013 Volume 38(Issue 36) pp:16185-16192
Publication Date(Web):13 December 2013
DOI:10.1016/j.ijhydene.2013.09.151
•Ti–η2-(C2H2) and HCC–TiH complexes show an uptake capacity of 14.06 wt%.•Ti–η2-(C2H2)(6H2) and HCC–TiH(6H2) are energetically favorable below 315 K, 275 K.•The importance of Gibbs free energy correction on the adsorption energy was shown.•The adsorption energy is affected by the theory used and dependent on the complex.The hydrogen storage capacities of π complex (Ti–η2-(C2H2)) and the corresponding ethynyl metal hydrides (HCC–TiH) complex were tested using second order Møller Plesset method with 6-311++G(3df,3pd) basis sets. Both on Ti–η2-(C2H2) and HCC–TiH, up to six hydrogen molecules can be absorbed with a binding energy of 0.20–0.42 eV/H2, corresponding the maximal retrievable hydrogen storage density of 14.06 wt%. A comparative study of various density functional theory calculations showed that the averaged H2 adsorption energy is not only considerably affected by the level of theory used but also dependent on the complex of the material dealing with. Dimerizations of the Ti–acetylene complexes were also discussed.
Co-reporter:Ying Ren, Jianfeng Jia, Wenxian Liu, and Hai-Shun Wu
Organometallics 2013 Volume 32(Issue 1) pp:52-62
Publication Date(Web):December 31, 2012
DOI:10.1021/om300797n
The Pd-catalyzed dearomatization of chloromethylnaphthalene with allenyltributylstannane has been investigated at the B3LYP density functional level of theory. The calculations indicate that the monophosphine complex is catalytically more active than the bisphosphine complex for oxidative addition. The transmetalation step is a crucial step for determining the dearomatized products due to the formation of two stable bis-π-complexes. It is found that reductive elimination occurs by coupling the terminal carbons of the η1-propargyl ligand and η1-allenyl ligand with the para-carbons of the η3-methylnaphthalene ligands in η3-methylnaphthalene-η1-propargyl-Pd(PH3) and η3-methylnaphthalene-η1-allenyl-Pd(PH3) to form the corresponding allenylated and propargylated dearomatization products. For comparison, various C–C coupling pathways in reductive elimination have also been studied.
Co-reporter:Ying Ren, Jianfeng Jia, Ting-Ting Zhang, Hai-Shun Wu, and Wenxian Liu
Organometallics 2012 Volume 31(Issue 3) pp:1168-1179
Publication Date(Web):February 1, 2012
DOI:10.1021/om201248t
The detailed mechanism of the Pd-catalyzed coupling of naphthalene allyl chloride with allenyltributylstannane, resulting in the dearomatization of the naphthalene group, has been studied using density functional theory (DFT) calculations at the B3LYP level. The catalyst cycle can be divided into three main stages involving oxidative addition, transmetalation, and reductive elimination, none of which contains significantly large barriers. It is found that the oxidative addition takes place through a monophosphine pathway. The transmetalation step is responsible for the formation of the propargylic dearomatized product, due to the orientation of the metal-coordinated allenyl ligand. Reductive elimination of the dearomatized product from the intermediate (η3-allylnaphthalene)(η1-allenyl)PdPH3 occurs by coupling of the terminal carbon of the η1-allenyl ligand with the ortho carbon of the η3-naphthalene ligand. Furthermore, it is shown that dichloromethane as solvent does not change the mechanistic picture significantly.