Co-reporter:Bo Qian, Shaowei Chen, Ting Wang, Xinhao Zhang, and Hongli Bao
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:13076-13076
Publication Date(Web):August 31, 2017
DOI:10.1021/jacs.7b06590
Intermolecular carboamination of olefins with general alkyl groups is an unsolved problem. Diastereoselective carboamination of acyclic olefins represents an additional challenge in intermolecular carboaminations. We have developed a general alkylamination of vinylarenes and the unprecedented diastereoselective anti-carboamination of unsaturated esters, generating amines and unnatural β-amino acids. This alkylamination is enabled by difunctional alkylating reagents and the iron catalyst. Alkyl diacyl peroxides, readily synthesized from aliphatic acids, serve as both alkylating reagents and internal oxidizing agents. A computational study suggests that addition of a nitrile to the carbocation is the diastereoselectivity-determining step, and hyperconjugation is proposed to account for the highly diastereoselective anti-carboamination.
Co-reporter:Nengbo Zhu, Ting Wang, Liang Ge, Yajun Li, Xinhao Zhang, and Hongli Bao
Organic Letters September 15, 2017 Volume 19(Issue 18) pp:
Publication Date(Web):September 1, 2017
DOI:10.1021/acs.orglett.7b01969
γ-Amino butyric acid (GABA) is the chief inhibitory neurotransmitter in the mammalian central nervous system. Many GABA derivatives are used clinically to prevent or treat neurodegenerative diseases. Copper-catalyzed carboamination of alkenes offers an efficient method to fashion the core structure of GABA derivatives from alkenes. In this reaction, acetonitrile serves as the source of the carbon and nitrogen functionalities used to difunctionalize alkenes. Experimental and density functional theory (DFT) studies were carried out to investigate the mechanism of the reaction, and a copper-catalyzed radical-polar crossover mechanism is proposed.
Co-reporter:Ping Chen, Linxing Zhang, Zi-Ling Xue, Yun-Dong Wu, and Xinhao Zhang
Inorganic Chemistry June 19, 2017 Volume 56(Issue 12) pp:7111-7111
Publication Date(Web):June 5, 2017
DOI:10.1021/acs.inorgchem.7b00713
The reactions of early-transition-metal complexes with H2O have been investigated. An understanding of these elementary steps promotes the design of precursors for the preparation of metal oxide materials or supported heterogeneous catalysts. Density functional theory (DFT) calculations have been conducted to investigate two elementary steps of the reactions between tungsten alkylidyne complexes and H2O, i.e., the addition of H2O to the W≡C bond and ligand hydrolysis. Four tungsten alkylidyne complexes, W(≡CSiMe3)(CH2SiMe3)3 (A-1), W(≡CSiMe3)(CH2tBu)3 (B-1), W(≡CtBu)(CH2tBu)3 (C-1), and W(≡CtBu)(OtBu)3 (D-1), have been compared. The DFT studies provide an energy profile of the two competing pathways. An additional H2O molecule can serve as a proton shuttle, accelerating the H2O addition reaction. The effect of atoms at the α and β positions has also been examined. Because the lone-pair electrons of an O atom at the α position can interact with the orbital of the proton, the barrier of the ligand-hydrolysis reaction for D-1 is dramatically reduced. Both the electronic and steric effects of the silyl group at the β position lower the barriers of both the H2O addition and ligand-hydrolysis reactions. These new mechanistic findings may lead to the further development of metal complex precursors.
Co-reporter:Heming Jiang, Tian-Yu Sun, Xiao Wang, Yaoming Xie, Xinhao Zhang, Yun-Dong Wu, and Henry F. Schaefer III
Organic Letters December 15, 2017 Volume 19(Issue 24) pp:6502-6502
Publication Date(Web):November 22, 2017
DOI:10.1021/acs.orglett.7b03167
2-Iodoxybenzoic acid (IBX) is an important species for the oxidation of alcohols to aldehydes or ketones. An often-cited mechanism involving a hypervalent twist as the rate-determining step (RDS) is inconsistent with kinetic isotope effect (KIE) experiments. The computations with larger basis sets reveal that the reductive elimination involving the C–H bond cleavage is the RDS (rate-determining step). Further computational/experimental studies suggest that the reactivity can be improved by adjusting the trans influence with Lewis acids.
Co-reporter:Xuefeng Liang, Liyan Zhou, Long Min, Weijian Ye, Wenli Bao, Wenjing Ma, Qianqian Yang, Fangfang Qiao, Xinhao Zhang, and Chi-Sing Lee
The Journal of Organic Chemistry April 7, 2017 Volume 82(Issue 7) pp:3463-3463
Publication Date(Web):March 2, 2017
DOI:10.1021/acs.joc.6b02921
The C8 and C9 stereogenic centers of the basiliolide/transtaganolide family have been established stereoselectively using a cyclopropane ring-opening strategy, which has been studied by DFT calculations of a variety of lithium-chelating models. The highly functionalized intermediates obtained in this strategy were successfully employed for the diastereoselective total synthesis of (±)-basiliolide B and (±)-epi-8-basiliolide B. The decalin core with a lactone bridge was constructed via a 2-pyrone Diels–Alder (DA) cycloaddition, and the unprecedented seven-membered acyl ketene acetal was established by a biomimetic intramolecular O-acylation cyclization.
Co-reporter:Xinhao Zhang, Lung Wa Chung, and Yun-Dong Wu
Accounts of Chemical Research 2016 Volume 49(Issue 6) pp:1302
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.accounts.6b00093
With new advances in theoretical methods and increased computational power, applications of computational chemistry are becoming practical and routine in many fields of chemistry. In organic chemistry, computational chemistry plays an indispensable role in elucidating reaction mechanisms and the origins of various selectivities, such as chemo-, regio-, and stereoselectivities. Consequently, mechanistic understanding improves synthesis and assists in the rational design of new catalysts. In this Account, we present some of our recent works to illustrate how computational chemistry provides new mechanistic insights for improvement of the selectivities of several organic reactions. These examples include not only explanations for the existing experimental observations, but also predictions which were subsequently verified experimentally.This Account consists of three sections discuss three different kinds of selectivities. The first section discusses the regio- and stereoselectivities of hydrosilylations of alkynes, mainly catalyzed by [Cp*Ru(MeCN)3]+ or [CpRu(MeCN)3]+. Calculations suggest a new mechanism that involves a key ruthenacyclopropene intermediate. This mechanism not only explains the unusual Markovnikov regio-selectivity and anti-addition stereoselectivity observed by Trost and co-workers, but also motivated further experimental investigations. New intriguing experimental observations and further theoretical studies led to an extension of the reaction mechanism. The second section includes three cases of meta-selective C–H activation of aryl compounds. In the case of Cu-catalyzed selective meta-C–H activation of aniline, a new mechanism that involves a Cu(III)-Ar-mediated Heck-like transition state, in which the Ar group acts as an electrophile, was proposed. This mechanism predicted a higher reactivity for more electron-deficient Ar groups, which was supported by experiments. For two template-mediated, meta-selective C–H bond activations catalyzed by Pd(II), different mechanisms were derived for the two templates. One involves a dimeric Pd–Pd or Pd–Ag active catalyst, and the other involves a monomeric Pd catalyst, in which a monoprotected amino acid coordinates in a bidentate fashion and serves as an internal base for C–H activation. The third section discusses a desymmetry strategy in asymmetric synthesis. The construction of rigid skeletons is critical for these catalysts to distinguish two prochiral groups. Overall, fruitful collaborations between computational and experimental chemists have provided new and comprehensive mechanistic understanding and insights into these useful reactions.
Co-reporter:Yong-Hui Sun, Tian-Yu Sun, Yun-Dong Wu, Xinhao Zhang and Yu Rao
Chemical Science 2016 vol. 7(Issue 3) pp:2229-2238
Publication Date(Web):03 Dec 2015
DOI:10.1039/C5SC03905C
A diversity-oriented synthesis of bioactive benzanilides via C(sp2)–H hydroxylation has been studied. Different regioselectivity was observed with Ru(II) and Pd(II) catalysts. The reaction demonstrates excellent regioselectivity, good tolerance of functional groups, and high yields. A wide range of ortho-hydroxylated-benzanilides can be readily synthesized with excellent regioselectivity via this new synthetic strategy. Computational investigations revealed that the regioselectivity was controlled mainly by both steric and electronic factors. Steric effects determine the regioselective outcomes in the Ru-catalyzed reaction, while electronic effects are dominant in the Pd-catalyzed reaction.
Co-reporter:Tian-Yu Sun, Xiao Wang, Hao Geng, Yaoming Xie, Yun-Dong Wu, Xinhao Zhang and Henry F. Schaefer III
Chemical Communications 2016 vol. 52(Issue 31) pp:5371-5374
Publication Date(Web):26 Feb 2016
DOI:10.1039/C6CC00384B
Togni's reagents have become very popular trifluoromethylating reagents in organic synthesis. The existing form of Togni's reagent I is a hypervalent iodine compound which lies much higher in energy than its ether isomer. The high-energy hypervalent iodine form makes Togni's reagent I very effective and versatile. The energy differences between the two forms correlate with the trans influence of the substituents. The five-membered ring in the benziodoxole-based scaffold is an important reason for its existence in the higher-energy form. The relation to Buchwald's 2014 research is discussed.
Co-reporter:Xiu-Mei Zhong, Gui-Juan Cheng, Ping Chen, Xinhao Zhang, and Yun-Dong Wu
Organic Letters 2016 Volume 18(Issue 20) pp:5240-5243
Publication Date(Web):October 4, 2016
DOI:10.1021/acs.orglett.6b02542
A combined mass spectrometric and computational study of the Pd/mono-N-protected amino acid (MPAA)-catalyzed vinyl–vinyl coupling reactions is reported. Computational study reveals that the reaction is initiated by C–H activation of the styrene followed by the insertion of acrylate. This is supported by mass spectrometry. The MPAA ligand facilitates the cross-coupling reaction between monosubstituted alkenes by stabilizing the active palladium catalyst and offering the N-protecting group as a stronger base than acetate. The E/Z selectivity is attributed to the stronger d−π interaction between the catalyst and the substrate in the transition state leading to E product.
Co-reporter:Li-Juan Song, Shengtao Ding, Yong Wang, Xinhao Zhang, Yun-Dong Wu, and Jianwei Sun
The Journal of Organic Chemistry 2016 Volume 81(Issue 15) pp:6157-6164
Publication Date(Web):May 27, 2016
DOI:10.1021/acs.joc.6b00854
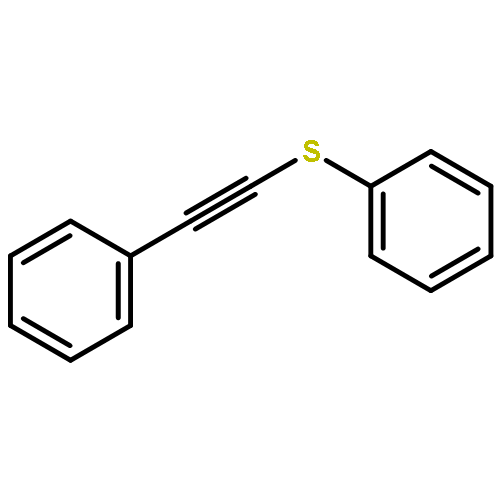
Iridium complexes are known catalysts for a range of silylation reactions. However, the exploitation for selective hydrosilylation of unsymmetrical internal alkynes has been limitedly known. Described here is a new example of this type. Specifically, [(cod)IrCl]2 catalyzes the efficient and mild hydrosilylation of thioalkynes by various silanes with excellent regio- and stereoselectivity. DFT studies suggested a new mechanism involving Ir(I) hydride as the key intermediate.
Co-reporter:Yong Wang;Li-Juan Song; Xinhao Zhang; Jianwei Sun
Angewandte Chemie International Edition 2016 Volume 55( Issue 33) pp:9704-9708
Publication Date(Web):
DOI:10.1002/anie.201603889
Abstract
A metal-free trimolecular [2+2+2] cycloaddition of internal ynamides and nitriles for de novo synthesis of fully substituted pyridines is disclosed. With the versatile Brønsted acid catalyst HNTf2, the mild intermolecular cyclotrimerization process proceeds with complementary chemoselectivity and excellent regioselectivity.
Co-reporter:Yong Wang;Li-Juan Song; Xinhao Zhang; Jianwei Sun
Angewandte Chemie 2016 Volume 128( Issue 33) pp:9856-9860
Publication Date(Web):
DOI:10.1002/ange.201603889
Abstract
A metal-free trimolecular [2+2+2] cycloaddition of internal ynamides and nitriles for de novo synthesis of fully substituted pyridines is disclosed. With the versatile Brønsted acid catalyst HNTf2, the mild intermolecular cyclotrimerization process proceeds with complementary chemoselectivity and excellent regioselectivity.
Co-reporter:Jialing Shi, Ting Wang, Yusha Huang, Xinhao Zhang, Yun-Dong Wu, and Qian Cai
Organic Letters 2015 Volume 17(Issue 4) pp:840-843
Publication Date(Web):February 6, 2015
DOI:10.1021/ol5036613
Employing a chiral spirodiphosphine monoxide ligand with 1,1′-spirobiindane backbone (SDP(O)), a desymmetrization strategy of Pd-catalyzed intramolecular asymmetric aryl C–O coupling of 2-(2-halophenoxy)propane-1,3-diols, was developed. The SDP(O) ligand shows much better results than its SDP counterpart. The protocol provides an efficient and highly enantioselective method for the synthesis of 2-hydroxymethyl-1,4-benzodioxanes. Density functional theory studies provide a model that accounts for the origin of the enantioselectivity.
Co-reporter:Gui-Juan Cheng;Ping Chen;Tian-Yu Sun;Dr. Xinhao Zhang;Dr. Jin-Quan Yu;Dr. Yun-Dong Wu
Chemistry - A European Journal 2015 Volume 21( Issue 31) pp:11180-11188
Publication Date(Web):
DOI:10.1002/chem.201501123
Abstract
A combined ion-mobility mass spectrometry (IM-MS) and DFT study has been employed to investigate the mechanism and the origin of selectivity of palladium/mono-N-protected amino acid (MPAA)-catalyzed enantioselective CH activation reactions of several prochiral substrates. We captured the [Pd(MPAA)(substrate)] complex at different stages, and demonstrated that the CH bond can be activated in the absence of an external base. DFT studies lead to the establishment of a significantly modified relay mechanism invoking a key conformational effect to account for the origin of enantioselectivity. This relay mechanism successfully accounts for the enantioselectivity for all the relevant reactions reported. The enantioselectivity originates from the rigid square-planar Pd coordination in the CH activation transition state: Bidentate MPAA and substrate coordination.
Co-reporter:Shengtao Ding;Li-Juan Song;Yong Wang; Xinhao Zhang; Lung Wa Chung; Yun-Dong Wu; Jianwei Sun
Angewandte Chemie International Edition 2015 Volume 54( Issue 19) pp:5632-5635
Publication Date(Web):
DOI:10.1002/anie.201500372
Abstract
A general and mild hydrosilylation of thioalkynes is described. With the cationic catalyst [Cp*Ru(MeCN)3]+ and the bulky silane (TMSO)3SiH, a range of thioalkynes underwent smooth hydrosilylation at room temperature with excellent α regioselectivity and syn stereoselectivity. DFT calculations provided important insight into the mechanism, particularly the unusual syn selectivity with the [Cp*Ru(MeCN)3]+ catalyst. The sulfenyl group in the substrates was found to provide important chelation stabilization to direct the reaction through a new mechanistic pathway.
Co-reporter:Shengtao Ding;Li-Juan Song;Yong Wang; Xinhao Zhang; Lung Wa Chung; Yun-Dong Wu; Jianwei Sun
Angewandte Chemie 2015 Volume 127( Issue 19) pp:5724-5727
Publication Date(Web):
DOI:10.1002/ange.201500372
Abstract
A general and mild hydrosilylation of thioalkynes is described. With the cationic catalyst [Cp*Ru(MeCN)3]+ and the bulky silane (TMSO)3SiH, a range of thioalkynes underwent smooth hydrosilylation at room temperature with excellent α regioselectivity and syn stereoselectivity. DFT calculations provided important insight into the mechanism, particularly the unusual syn selectivity with the [Cp*Ru(MeCN)3]+ catalyst. The sulfenyl group in the substrates was found to provide important chelation stabilization to direct the reaction through a new mechanistic pathway.
Co-reporter:Dr. Fengtao Zhou;Gui-Juan Cheng;Wenqiang Yang;Yan Long;Shasha Zhang;Dr. Yun-Dong Wu;Dr. Xinhao Zhang;Dr. Qian Cai
Angewandte Chemie 2014 Volume 126( Issue 36) pp:9709-9713
Publication Date(Web):
DOI:10.1002/ange.201405575
Abstract
The enantioselective construction of all-carbon quaternary stereocenters is one of the most challenging fields in asymmetric synthesis. An asymmetric desymmetrization strategy offers an indirect and efficient method for the formation of all-carbon stereocenters. An enantioselective formation of cyano-bearing all-carbon quaternary stereocenters in 1,2,3,4,-tetrahydroquinolines and 2,3,4,5-tetrahydro-1H-benzo[b]azepines by copper-catalyzed desymmetric N-arylation is demonstrated. The cyano group at the prochiral center plays a key role for the high enantioselectivity and works as an important functional group for further transformations. DFT studies provide a model which successfully accounts for the origin of enantioselectivity.
Co-reporter:Huan Sun, Chengming Wang, Yun-Fang Yang, Ping Chen, Yun-Dong Wu, Xinhao Zhang, and Yong Huang
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:11863-11872
Publication Date(Web):May 13, 2014
DOI:10.1021/jo500807d
Indole-containing polyaromatic scaffolds are widely found in natural products, pharmaceutical agents, and π-conjugated functional materials. Often, the synthesis of these highly valuable molecules requires a multistep sequence. Therefore, a simple, one-step protocol to access libraries of polyaromatic indole scaffolds is highly desirable. Herein we describe the direct synthesis of polysubstituted indolo[2,1-a]isoquinoline analogues via a double C–H annulation cascade using triazene as an internally cleavable directing group. Evidence from HRMS and theoretical calculations suggests that an unprecedented 1,2-alkyl migration might be responsible for the in situ cleavage of the directing group. Both kinetic isotope effects and DFT calculations suggested that the alkyne insertion step is rate-limiting for the second C,N annulation reaction.
Co-reporter:Dr. Fengtao Zhou;Gui-Juan Cheng;Wenqiang Yang;Yan Long;Shasha Zhang;Dr. Yun-Dong Wu;Dr. Xinhao Zhang;Dr. Qian Cai
Angewandte Chemie International Edition 2014 Volume 53( Issue 36) pp:9555-9559
Publication Date(Web):
DOI:10.1002/anie.201405575
Abstract
The enantioselective construction of all-carbon quaternary stereocenters is one of the most challenging fields in asymmetric synthesis. An asymmetric desymmetrization strategy offers an indirect and efficient method for the formation of all-carbon stereocenters. An enantioselective formation of cyano-bearing all-carbon quaternary stereocenters in 1,2,3,4,-tetrahydroquinolines and 2,3,4,5-tetrahydro-1H-benzo[b]azepines by copper-catalyzed desymmetric N-arylation is demonstrated. The cyano group at the prochiral center plays a key role for the high enantioselectivity and works as an important functional group for further transformations. DFT studies provide a model which successfully accounts for the origin of enantioselectivity.
Co-reporter:Gui-Juan Cheng;Li-Juan Song;Dr. Yun-Fang Yang;Dr. Xinhao Zhang;Dr. Olaf Wiest;Dr. Yun-Dong Wu
ChemPlusChem 2013 Volume 78( Issue 9) pp:943-951
Publication Date(Web):
DOI:10.1002/cplu.201300117
Abstract
A detailed computational study of a copper-catalyzed aerobic cross-dehydrogenative coupling reaction has been conducted. To select a reliable method to describe the thermochemistry of a single electron transfer (SET) process, benchmark calculations have been performed. M06/6-311+g(d,p) is appropriate to evaluate the thermochemistry of the SET process for the system involving iminium species. The computational results support an SET mechanism, but also uncover an alternative mechanism in which O2 is directly involved in a hydrogen-abstracting step. A comparative study with tert-butylhydroperoxide (TBHP) as the oxidant has also been performed. The computations reveal several competitive pathways, including a radical pathway, a CuIII pathway, and an SET mechanism for the Cu/TBHP system.
Co-reporter:Lizhi Zhu, Congshan Zhou, Wei Yang, Shuzhong He, Gui-Juan Cheng, Xinhao Zhang, and Chi-Sing Lee
The Journal of Organic Chemistry 2013 Volume 78(Issue 16) pp:7912-7929
Publication Date(Web):July 17, 2013
DOI:10.1021/jo401105q
A mild and efficient dual-mode Lewis acid induced Diels–Alder (DA)/carbocyclization cascade cyclization reaction has been developed for construction of the tricyclic core of ent-kaurenoids in one pot with the aid of a theoretical study on the π,σ-Lewis acidities of a variety of Lewis acids. With ZnBr2 as the dual-mode Lewis acid, a series of substituted enones and dienes underwent DA/carbocyclization cascade cyclization reaction smoothly at room temperature and provided the tricyclic cyclized products in one pot with good yields and high diastereoselectivity. The tricyclic cyclized product has been successfully utilized as a common intermediate for formal syntheses of (±)-platensimycin and (±)-platencin.
Co-reporter:Ping Chen, Brenda A. Dougan, Xinhao Zhang, Yun-Dong Wu, Zi-Ling Xue
Polyhedron 2013 58() pp: 30-38
Publication Date(Web):
DOI:10.1016/j.poly.2012.07.042
Co-reporter:Yun-Fang Yang;Gui-Juan Cheng;Dr. Jun Zhu;Dr. Xinhao Zhang;Dr. Shigeyoshi Inoue;Dr. Yun-Dong Wu
Chemistry - A European Journal 2012 Volume 18( Issue 24) pp:7516-7524
Publication Date(Web):
DOI:10.1002/chem.201103443
Abstract
Density functional theory calculations (B3LYP) have been carried out to investigate the 4π-electron systems of 2,4-disila-1,3-diphosphacyclobutadiene (compound 1) and the tetrasilacyclobutadiene dication (compound 2). The calculated nucleus-independent chemical shift (NICS) values for these two compounds are negative, which indicates that the core rings of compounds 1 and 2 have a certain amount of aromaticity. However, deep electronic analysis reveals that neither of these two formal 4π-electron four-membered ring systems is aromatic. Compound 1 has very weak, almost negligible antiaromaticity, and the amidinate ligands attached to the Si atoms play an important role in stabilizing this conjugated 4π-electron system. The monoanionic bidentate ligand interacts with the conjugated π system to cause π-orbital splitting. This ligand-induced π-orbital splitting effect provides an opportunity to manipulate the gap between occupied and unoccupied π orbitals in conjugated systems. Conversely, compound 2 is nonaromatic because its core ring does not have a conjugated π ring system and does not fulfill the requirements of a Hückel system.
Co-reporter:Tian-Yu Sun, Xiao Wang, Hao Geng, Yaoming Xie, Yun-Dong Wu, Xinhao Zhang and Henry F. Schaefer III
Chemical Communications 2016 - vol. 52(Issue 31) pp:NaN5374-5374
Publication Date(Web):2016/02/26
DOI:10.1039/C6CC00384B
Togni's reagents have become very popular trifluoromethylating reagents in organic synthesis. The existing form of Togni's reagent I is a hypervalent iodine compound which lies much higher in energy than its ether isomer. The high-energy hypervalent iodine form makes Togni's reagent I very effective and versatile. The energy differences between the two forms correlate with the trans influence of the substituents. The five-membered ring in the benziodoxole-based scaffold is an important reason for its existence in the higher-energy form. The relation to Buchwald's 2014 research is discussed.
Co-reporter:Yong-Hui Sun, Tian-Yu Sun, Yun-Dong Wu, Xinhao Zhang and Yu Rao
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN2238-2238
Publication Date(Web):2015/12/03
DOI:10.1039/C5SC03905C
A diversity-oriented synthesis of bioactive benzanilides via C(sp2)–H hydroxylation has been studied. Different regioselectivity was observed with Ru(II) and Pd(II) catalysts. The reaction demonstrates excellent regioselectivity, good tolerance of functional groups, and high yields. A wide range of ortho-hydroxylated-benzanilides can be readily synthesized with excellent regioselectivity via this new synthetic strategy. Computational investigations revealed that the regioselectivity was controlled mainly by both steric and electronic factors. Steric effects determine the regioselective outcomes in the Ru-catalyzed reaction, while electronic effects are dominant in the Pd-catalyzed reaction.

![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4.png)
![2-[(4s)-4-(2-methyl-2-propanyl)-4,5-dihydro-1,3-oxazol-2-yl]-5-(t Rifluoromethyl)pyridine](/data/chemimg/482100/1416819-91-4_b.png)







![3-(4-Chlorophenyl)benzo[d]isoxazole](http://img.cochemist.com/ccimg/1227200/1227180-50-8.png)
![3-(4-Chlorophenyl)benzo[d]isoxazole](http://img.cochemist.com/ccimg/1227200/1227180-50-8_b.png)









![Cyclopropanecarboxylicacid, 1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-phenyl-, (1S,2R)-](http://img.cochemist.com/ccimg/152000/151910-11-1.png)
![Cyclopropanecarboxylicacid, 1-[[(1,1-dimethylethoxy)carbonyl]amino]-2-phenyl-, (1S,2R)-](http://img.cochemist.com/ccimg/152000/151910-11-1_b.png)



![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0.png)
![Benzene, [(butylthio)ethynyl]-](http://img.cochemist.com/ccimg/127100/127060-60-0_b.png)








![L-Valine, N-[(2,2,2-trichloro-1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/66300/66270-38-0.png)
![L-Valine, N-[(2,2,2-trichloro-1,1-dimethylethoxy)carbonyl]-](http://img.cochemist.com/ccimg/66300/66270-38-0_b.png)















![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7.png)
![Palladium,hexakis[m-(acetato-kO:kO')]tri-, cyclo](http://img.cochemist.com/ccimg/53200/53189-26-7_b.png)





![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)










![Benzene, [(1-hexyn-1-ylthio)methyl]-](/data/chemimg/3765000/1355700-62-7.png)
![Benzene, [(1-hexyn-1-ylthio)methyl]-](/data/chemimg/3765000/1355700-62-7_b.png)













