Co-reporter:Svante Hedström, Adam J. Matula, and Victor S. Batista
The Journal of Physical Chemistry C September 7, 2017 Volume 121(Issue 35) pp:19053-19053
Publication Date(Web):August 15, 2017
DOI:10.1021/acs.jpcc.7b05749
Organic, conjugated donor–acceptor (D–A) systems are essential components of photovoltaic devices. Design and optimization of D–A systems is typically based on trial-and-error experimentation methods that would benefit from fundamental physical insights on structure–function relationships at the molecular level. Here, we implement a nonequilibrium Green’s function methodology at the density functional theory (DFT) level and examine charge-transport and rectification properties of a series of conjugated D–A systems. We investigate 42 molecular junctions formed by D–A dyads bridging model gold electrodes, showing clearly how transport properties are determined by chemical composition, symmetry of frontier orbitals, and molecular conformation. Key properties are compared to experimental data. Notably, an inverse correlation between conductance and rectification is found, with relatively large rectification ratios caused by the asymmetry of frontier orbitals near the Fermi level. We discuss design principles that should be valuable for the rational design of molecular D–A systems with appropriate transport properties.
Co-reporter:Pablo Garrido-Barros, Carolina Gimbert-Suriñach, Dooshaye Moonshiram, Antonio Picón, Pere Monge, Victor S. Batista, and Antoni Llobet
Journal of the American Chemical Society September 20, 2017 Volume 139(Issue 37) pp:12907-12907
Publication Date(Web):August 30, 2017
DOI:10.1021/jacs.7b06828
A molecular water oxidation catalyst based on the copper complex of general formula [(Lpy)CuII]2–, 22–, (Lpy is 4-pyrenyl-1,2-phenylenebis(oxamidate) ligand) has been rationally designed and prepared to support a more extended π-conjugation through its structure in contrast with its homologue, the [(L)CuII]2– water oxidation catalyst, 12– (L is o-phenylenebis(oxamidate)). The catalytic performance of both catalysts has been comparatively studied in homogeneous phase and in heterogeneous phase by π-stacking anchorage to graphene-based electrodes. In the homogeneous system, the electronic perturbation provided by the pyrene functionality translates into a 150 mV lower overpotential for 22– with respect to 12– and an impressive increase in the kcat from 6 to 128 s–1. Upon anchorage, π-stacking interactions with the graphene sheets provide further π-delocalization that improves the catalytic performance of both catalysts. In this sense, 22– turned out to be the most active catalyst due to the double influence of both the pyrene and the graphene, displaying an overpotential of 538 mV, a kcat of 540 s–1 and producing more than 5300 TONs.
Co-reporter:Samuel M. Greene and Victor S. Batista
Journal of Chemical Theory and Computation September 12, 2017 Volume 13(Issue 9) pp:4034-4034
Publication Date(Web):August 1, 2017
DOI:10.1021/acs.jctc.7b00608
We introduce the “tensor-train split-operator Fourier transform” (TT-SOFT) method for simulations of multidimensional nonadiabatic quantum dynamics. TT-SOFT is essentially the grid-based SOFT method implemented in dynamically adaptive tensor-train representations. In the same spirit of all matrix product states, the tensor-train format enables the representation, propagation, and computation of observables of multidimensional wave functions in terms of the grid-based wavepacket tensor components, bypassing the need of actually computing the wave function in its full-rank tensor product grid space. We demonstrate the accuracy and efficiency of the TT-SOFT method as applied to propagation of 24-dimensional wave packets, describing the S1/S2 interconversion dynamics of pyrazine after UV photoexcitation to the S2 state. Our results show that the TT-SOFT method is a powerful computational approach for simulations of quantum dynamics of polyatomic systems since it avoids the exponential scaling problem of full-rank grid-based representations.
Co-reporter:Benjamin Rudshteyn, Hunter B. Vibbert, Richard May, Eric Wasserman, Ingolf Warnke, Michael D. Hopkins, and Victor S. Batista
ACS Catalysis September 1, 2017 Volume 7(Issue 9) pp:6134-6134
Publication Date(Web):July 14, 2017
DOI:10.1021/acscatal.7b01636
The thermodynamic and structural factors that influence the redox properties of an extensive set of tungsten–alkylidyne complexes (W(CR)L4X) are analyzed by combining synthesis, electrochemistry, and computational modeling based on free energy calculations of oxidation potentials at the density functional theory level. The observed linear correlations among oxidation potentials, HOMO energies, and gas-phase ionization energies are found to be consistent with the approximately constant solvation free energy differences between reduced and oxidized species over the complete set. The W–X bond length, trans to the alkylidyne ligand, is found to be a good descriptor of the positioning of the key frontier orbitals that regulate the redox properties of the complexes.Keywords: density functional theory; descriptor; photoredox chromophores; redox potentials; tungsten−alkylidyne complex;
Co-reporter:Benjamin Rudshteyn, Christian F. A. Negre, Robson S. Oliboni, Adriano Monti, Jeffrey Chen, Robert H. Crabtree, Luis G. C. Rego, and Victor S. Batista
The Journal of Physical Chemistry C June 8, 2017 Volume 121(Issue 22) pp:11985-11985
Publication Date(Web):May 10, 2017
DOI:10.1021/acs.jpcc.7b01272
We explore the protonation states of benzohydroxamic acid adsorbates bound to the {101} facet of TiO2 anatase by using a combination of density functional theory, simulations of UV–vis spectra based on a tight-binding Hamiltonian, and direct comparisons to experimental measurements. We find that the characteristic red-shifted spectrum of nonmethylated, relative to the methylated, hydroxamic acids can only be explained by proposing a monodeprotonated monodentate mode as the main adsorption mode. The reported analysis suggests a simple, yet general, spectroscopic method based on UV–vis absorption measurements and tight-binding calculations for inferring changes of pKa of molecular adsorbates interacting with semiconductor electrode surfaces.
Co-reporter:Yiren Zhong, Ke R. Yang, Wen Liu, Peng He, Victor Batista, and Hailiang Wang
The Journal of Physical Chemistry C July 6, 2017 Volume 121(Issue 26) pp:14222-14222
Publication Date(Web):June 15, 2017
DOI:10.1021/acs.jpcc.7b04170
The design and development of materials for electrochemical energy storage and conversion devices requires fundamental understanding of chemical interactions at electrode/electrolyte interfaces. For Li–S batteries that hold the promise for outperforming the current generation of Li ion batteries, the interactions of lithium polysulfide (LPS) intermediates with the electrode surface strongly influence the efficiency and cycle life of the sulfur cathode. While metal oxides have been demonstrated to be useful in trapping LPS, the actual binding modes of LPS on 3d transition metal oxides and their dependence on the metal element identity across the periodic table remain poorly understood. Here, we investigate the chemical interactions between LPS and oxides of Mn, Fe, Co, and Cu by combining X-ray photoelectron spectroscopy and density functional theory calculations. We find that Li–O interactions dominate LPS binding to the oxides (Mn3O4, Fe2O3, and Co3O4), with increasing strength from Mn to Fe to Co. For Co3O4, LPS binding also involves metal–sulfur interactions. We also find that the metal oxides exhibit different binding preferences for different LPS, with Co3O4 binding shorter-chain LPS more strongly than Mn3O4. In contrast to the other oxides, CuO undergoes intense reduction and dissolution reactions upon interaction with LPS. The reported findings are thus particularly relevant to the design of LPS/oxide interfaces for high-performance Li–S batteries.
Co-reporter:Gary N. Lim, Svante Hedström, Kenneth A. Jung, Pierce A. D. Smith, Victor S. Batista, Francis D’Souza, Art van der Est, and Prashanth K. Poddutoori
The Journal of Physical Chemistry C July 13, 2017 Volume 121(Issue 27) pp:14484-14484
Publication Date(Web):June 17, 2017
DOI:10.1021/acs.jpcc.7b04197
Two self-assembled photoanodes have been constructed by exploiting the unique optical and structural properties of aluminum(III) porphyrin (AlPor) in conjunction with TiO2 nanoparticles as an electron acceptor and bis(p-anisole)aminopyridine (BAA–Py) as an electron donor. AlPor is bound to the TiO2 surface by either: (i) a benzohydroxamic acid bridge, in which the hydroxamic acid acts as an anchor or (ii) direct covalent binding of Al via an ether bond. The open sixth coordination site of the Al center is then used to coordinate BAA–Py through Lewis acid–base interactions, which results in donor–photosensitizer–semiconductor constructs that can be used as photoanodes. The two photoanodes were characterized by steady-state and transient spectroscopic techniques as well as computational methods. Transient-absorption studies show that in the absence of BAA–Py both the photoanodes exhibit electron injection from AlPor to the conduction band of TiO2. However, the injection efficiencies and kinetics are strongly dependent on the linker with faster and more efficient injection occurring when the porphyrin is directly bound. Kinetic results also suggest that the recombination is faster in directly bound AlPor than benzohydroxamic acid bridged AlPor. When BAA–Py is coordinated to AlPor, electron injection from AlPor to TiO2 is followed by electron transfer from BAA–Py to the oxidized AlPor. The injection efficiencies modeled using density functional theory and semiempirical tight-binding calculations are consistent with experimentally observed trends.
Co-reporter:Subhajyoti Chaudhuri, Svante Hedström, Dalvin D. Méndez-Hernández, Heidi P. Hendrickson, Kenneth A. Jung, Junming Ho, and Victor S. Batista
Journal of Chemical Theory and Computation December 12, 2017 Volume 13(Issue 12) pp:6000-6000
Publication Date(Web):November 2, 2017
DOI:10.1021/acs.jctc.7b00513
Understanding the effect of vibronic coupling on electron transfer (ET) rates is a challenge common to a wide range of applications, from electrochemical synthesis and catalysis to biochemical reactions and solar energy conversion. The Marcus–Jortner–Levich (MJL) theory offers a model of ET rates based on a simple analytic expression with a few adjustable parameters. However, the MJL equation in conjunction with density functional theory (DFT) has yet to be established as a predictive first-principles methodology. A framework is presented for calculating transfer rates modulated by molecular vibrations, that circumvents the steep computational cost which has previously necessitated approximations such as condensing the vibrational manifold into a single empirical frequency. Our DFT–MJL approach provides robust and accurate predictions of ET rates spanning over 4 orders of magnitude in the 106–1010 s–1 range. We evaluate the full MJL equation with a Monte Carlo sampling of the entire active space of thermally accessible vibrational modes, while using no empirical parameters. The contribution to the rate of individual modes is illustrated, providing insight into the interplay between vibrational degrees of freedom and changes in electronic state. The reported findings are valuable for understanding ET rates modulated by multiple vibrational modes, relevant to a broad range of systems within the chemical sciences.
Co-reporter:Yueshen Wu, Benjamin Rudshteyn, Almagul Zhanaidarova, Jesse D. Froehlich, Wendu Ding, Clifford P. Kubiak, and Victor S. Batista
ACS Catalysis August 4, 2017 Volume 7(Issue 8) pp:5282-5282
Publication Date(Web):June 29, 2017
DOI:10.1021/acscatal.7b01109
Dramatic enhancement of electrochemical CO2 conversion to CO, catalyzed by [Ni(cyclam)](PF6)2 is observed on mercury/gold electrodes. We find that Hg provides favorable noncovalent dispersive interactions with the cyclam ligand. As a result, the Hg surface destabilizes the poisoned CO-bound form of the catalyst, leading to enhanced reaction kinetics. These findings are particularly relevant to the design of ligands that improve the electrocatalytic performance of transition-metal complexes on interaction with metallic surfaces under cell operating conditions.Keywords: adsorption; CO poisoning; CO2 electroreduction; dispersive interaction; nickel cyclam;
Co-reporter:Aimin Ge, Pablo E. Videla, Gwendolynne L. Lee, Benjamin Rudshteyn, Jia Song, Clifford P. Kubiak, Victor S. Batista, and Tianquan Lian
The Journal of Physical Chemistry C August 31, 2017 Volume 121(Issue 34) pp:18674-18674
Publication Date(Web):August 7, 2017
DOI:10.1021/acs.jpcc.7b05563
Interfacial electric fields play crucial roles in electrochemistry, catalysis, and solar energy conversion. Understanding of the interfacial electric field effects has been hindered by the lack of a direct spectroscopic method to probe of the interfacial field at the molecular level. Here, we report the characterization of the field and interfacial structure at Au/diisocyanide/aqueous electrolyte interfaces, using a combination of in situ electrochemical vibrational sum frequency generation (SFG) spectroscopy, density functional theory (DFT) calculations, and molecular dynamics (MD) simulations. For 1,4-phenylene diisocyanide (PDI), 4,4′-biphenyl diisocyanide (BPDI), and 4,4″-p-terphenyl diisocyanide (TPDI), our results reveal that the frequency of the gold-bound NC stretch mode of the diisocyanide self-assembled monolayer (SAM) increases linearly with the applied potential, suggesting that SFG can be an in situ probe of the strength of the electric field at electrode/electrolyte interfaces. Using DFT-computed Stark tuning rates of model complexes, the electric field strength at the metal/SAM/electrolyte interfaces is estimated to be 108–109 V/m. The linear dependence of the vibrational frequency (and field) with applied potential is consistent with an electrochemical double-layer structure that consists of a Helmholtz layer in contact with a diffused layer. The Helmholtz layer thickness is approximately the same as the molecular length for PDI, suggesting a well-ordered SAM with negligible electrolyte penetration. For BPDI and TPDI, we found that the Helmholtz layer is thinner than the monolayer of molecular adsorbates, indicating that the electrolyte percolates into the SAM, as shown by molecular dynamics simulations of the Au/PDI/electrolyte interface. The reported analysis demonstrates that a combination of in situ SFG probes and computational modeling provides a powerful approach to elucidate the structure of electrochemical interfaces at the detailed molecular level.
Co-reporter:Jimin Wang, Mikhail Askerka, Gary W. BrudvigVictor S. Batista
ACS Energy Letters - New in 2016 2017 Volume 2(Issue 2) pp:
Publication Date(Web):January 12, 2017
DOI:10.1021/acsenergylett.6b00626
Understanding structure–function relations in photosystem II (PSII) is important for the development of biomimetic photocatalytic systems. X-ray crystallography, computational modeling, and spectroscopy have played central roles in elucidating the structure and function of PSII. Recent breakthroughs in femtosecond X-ray crystallography offer the possibility of collecting diffraction data from the X-ray free electron laser (XFEL) before radiation damage of the sample, thereby overcoming the main challenge of conventional X-ray diffraction methods. However, the interpretation of XFEL data from PSII intermediates is challenging because of the issues regarding data-processing, uncertainty on the precise positions of light oxygen atoms next to heavy metal centers, and different kinetics of the S-state transition in microcrystals compared to solution. Here, we summarize recent advances and outstanding challenges in PSII structure–function determination with emphasis on the implementation of quantum mechanics/molecular mechanics techniques combined with isomorphous difference Fourier maps, direct methods, and high-resolution spectroscopy.
Co-reporter:Mikhail Askerka, Gary W. BrudvigVictor S. Batista
Accounts of Chemical Research 2017 Volume 50(Issue 1) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acs.accounts.6b00405
ConspectusEfficient photoelectrochemical water oxidation may open a way to produce energy from renewable solar power. In biology, generation of fuel due to water oxidation happens efficiently on an immense scale during the light reactions of photosynthesis. To oxidize water, photosynthetic organisms have evolved a highly conserved protein complex, Photosystem II. Within that complex, water oxidation happens at the CaMn4O5 inorganic catalytic cluster, the so-called oxygen-evolving complex (OEC), which cycles through storage “S” states as it accumulates oxidizing equivalents and produces molecular oxygen. In recent years, there has been significant progress in understanding the OEC as it evolves through the catalytic cycle. Studies have combined conventional and femtosecond X-ray crystallography with extended X-ray absorption fine structure (EXAFS) and quantum mechanics/molecular mechanics (QM/MM) methods and have addressed changes in protonation states of μ-oxo bridges and the coordination of substrate water through the analysis of ammonia binding as a chemical analog of water. These advances are thought to be critical to understanding the catalytic cycle since protonation states regulate the relative stability of different redox states and the geometry of the OEC. Therefore, establishing the mechanism for substrate water binding and the nature of protonation/redox state transitions in the OEC is essential for understanding the catalytic cycle of O2 evolution.The structure of the dark-stable S1 state has been a target for X-ray crystallography for the past 15 years. However, traditional X-ray crystallography has been hampered by radiation-induced reduction of the OEC. Very recently, a revolutionary X-ray free electron laser (XFEL) technique was applied to PSII to reveal atomic positions at 1.95 Å without radiation damage, which brought us closer than ever to establishing the ultimate structure of the OEC in the S1 state. However, the atom positions in this crystal structure are still not consistent with high-resolution EXAFS spectroscopy, partially due to the poorly resolved oxygen positions next to Mn centers and partial reduction due to extended dark adaptation of the sample. These inconsistencies led to the new models of the OEC with an alternative low oxidation state and raised questions on the protonation state of the cluster, especially the O5 μ-oxo bridge. This Account summarizes the most recent models of the OEC that emerged from QM/MM, EXAFS and femtosecond X-ray crystallography methods.When PSII in the S1 state is exposed to light, the S1 state is advanced to the higher oxidation states and eventually binds substrate water molecules. Identifying the substrate waters is of paramount importance for establishing the water-oxidation mechanism but is complicated by a large number of spectroscopically similar waters. Water analogues can, therefore, be helpful because they serve as spectroscopic markers that help to track the motion of the substrate waters. Due to a close structural and electronic similarity to water, ammonia has been of particular interest. We review three competing hypotheses on substrate water/ammonia binding and compile theoretical and experimental evidence to support them. Binding of ammonia as a sixth ligand to Mn4 during the S1 → S2 transition seems to satisfy most of the criteria, especially the most compelling recent EPR data on D1-D61A mutated PSII. Such a binding mode suggests delivery of water from the “narrow” channel through a “carousel” rearrangement of waters around Mn4 upon the S2 → S3 transition. An alternative hypothesis suggests water delivery through the “large” channel on the Ca side. However, both water delivery paths lead to a similar S3 structure, seemingly reaching consensus on the nature of the last detectable S-state intermediate in the Kok cycle before O2 evolution.
Co-reporter:Dr. Jianbing Jiang;Kelly L. Materna;Dr. Svante Hedström;Dr. Ke R. Yang; Robert H. Crabtree; Victor S. Batista; Gary W. Brudvig
Angewandte Chemie International Edition 2017 Volume 56(Issue 31) pp:9111-9115
Publication Date(Web):2017/07/24
DOI:10.1002/anie.201704700
AbstractMain-group complexes are shown to be viable electrocatalysts for the H2-evolution reaction (HER) from acid. A series of antimony porphyrins with varying axial ligands were synthesized for electrocatalysis applications. The proton-reduction catalytic properties of TPSb(OH)2 (TP=5,10,15,20-tetra(p-tolyl)porphyrin) with two axial hydroxy ligands were studied in detail, demonstrating catalytic H2 production. Experiments, in conjunction with quantum chemistry calculations, show that the catalytic cycle is driven via the redox activity of both the porphyrin ligand and the Sb center. This study brings insight into main group catalysis and the role of redox-active ligands during catalysis.
Co-reporter:Zachary S. Fishman;Yulian He;Ke R. Yang;Amanda W. Lounsbury;Junqing Zhu;Thanh Minh Tran;Julie B. Zimmerman;Lisa D. Pfefferle
Nanoscale (2009-Present) 2017 vol. 9(Issue 35) pp:12984-12995
Publication Date(Web):2017/09/14
DOI:10.1039/C7NR03522E
Understanding how nano-dimensionality impacts iron oxide based catalysis is central to a wide range of applications. Here, we focus on hematite nanosheets, nanowires and nanoparticles as applied to catalyze the reverse water gas shift (RWGS) probe reaction. We introduce a novel approach to synthesize ultrathin (4–7 nm) hematite nanosheets using copper oxide nanosheets as a hard template and propose a reaction mechanism based on density functional theory (DFT) calculations. Hematite nanowires and nanoparticles were also synthesized and characterized. H2 temperature programmed reduction (H2-TPR) and RWGS reactions were performed to glean insights into the mechanism of CO2 conversion to CO over the iron oxide nanomaterials and were compared to H2 binding energy calculations based on density functional theory. While the nanosheets did exhibit high CO2 conversion, 28% at 510 °C, we found that the iron oxide nanowires had the highest CO2 conversion, reaching 50% at 750 °C under atmospheric pressure. No products besides CO and H2O were detected.
Co-reporter:James M. Lipchock, Heidi P. HendricksonBonnie B. Douglas, Kelly E. Bird, Patrick S. Ginther, Ivan Rivalta, Nicholas S. TenVictor S. Batista, J. Patrick Loria
Biochemistry 2017 Volume 56(Issue 1) pp:
Publication Date(Web):December 13, 2016
DOI:10.1021/acs.biochem.6b01025
Protein tyrosine phosphatase 1B (PTP1B) is a known regulator of the insulin and leptin signaling pathways and is an active target for the design of inhibitors for the treatment of type II diabetes and obesity. Recently, cichoric acid (CHA) and chlorogenic acid (CGA) were predicted by docking methods to be allosteric inhibitors that bind distal to the active site. However, using a combination of steady-state inhibition kinetics, solution nuclear magnetic resonance experiments, and molecular dynamics simulations, we show that CHA is a competitive inhibitor that binds in the active site of PTP1B. CGA, while a noncompetitive inhibitor, binds in the second aryl phosphate binding site, rather than the predicted benzfuran binding pocket. The molecular dynamics simulations of the apo enzyme and cysteine–phosphoryl intermediate states with and without bound CGA suggest CGA binding inhibits PTP1B by altering hydrogen bonding patterns at the active site. This study provides a mechanistic understanding of the allosteric inhibition of PTP1B.
Co-reporter:Dr. Jianbing Jiang;Kelly L. Materna;Dr. Svante Hedström;Dr. Ke R. Yang; Robert H. Crabtree; Victor S. Batista; Gary W. Brudvig
Angewandte Chemie 2017 Volume 129(Issue 31) pp:9239-9243
Publication Date(Web):2017/07/24
DOI:10.1002/ange.201704700
AbstractMain-group complexes are shown to be viable electrocatalysts for the H2-evolution reaction (HER) from acid. A series of antimony porphyrins with varying axial ligands were synthesized for electrocatalysis applications. The proton-reduction catalytic properties of TPSb(OH)2 (TP=5,10,15,20-tetra(p-tolyl)porphyrin) with two axial hydroxy ligands were studied in detail, demonstrating catalytic H2 production. Experiments, in conjunction with quantum chemistry calculations, show that the catalytic cycle is driven via the redox activity of both the porphyrin ligand and the Sb center. This study brings insight into main group catalysis and the role of redox-active ligands during catalysis.
Co-reporter:Subhajyoti Chaudhuri, Benjamin Rudshteyn, Mirabelle Prémont-Schwarz, Dina Pines, Ehud Pines, Dan Huppert, Erik T.J. Nibbering, Victor S. Batista
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2017.03.080
•Ultrafast fluorescence quenching of naphthols induced by halocarbon solvents.•Quenching mechanism based on photo-induced charge transfer to halocarbon solvents.•Multiple timescales support electron-transfer coupled to solute reorganization.•Solvent interacting with the naphthol π-electronic structure governs charge transfer.We explore the fluorescence quenching of 1-naphthol and 2-naphthol in halocarbon solvents by using time-correlated single-photon-counting, femtosecond IR-spectroscopy and quantum chemistry computations. We find that halocarbon solvents facilitate a de-excitation mechanism via solute-solvent electron transfer. Decay rates are modulated by close contact interactions between the π-electronic structure of naphthols and halocarbon molecules in their first solvation shell. 1-naphthol exhibits faster decay rates than 2-naphthol due to closer interactions with the solvent.Download high-res image (35KB)Download full-size image
Co-reporter:Wen Liu;Jianbing Jiang;Ke R. Yang;Yingying Mi;Piranavan Kumaravadivel;Yiren Zhong;Qi Fan;Zhe Weng;Zishan Wu;Judy J. Cha;Henghui Zhou;Gary W. Brudvig;Hailiang Wang
PNAS 2017 114 (14 ) pp:3578-3583
Publication Date(Web):2017-04-04
DOI:10.1073/pnas.1620809114
Lithium–sulfur batteries (Li–S batteries) have attracted intense interest because of their high specific capacity and low
cost, although they are still hindered by severe capacity loss upon cycling caused by the soluble lithium polysulfide intermediates.
Although many structure innovations at the material and device levels have been explored for the ultimate goal of realizing
long cycle life of Li–S batteries, it remains a major challenge to achieve stable cycling while avoiding energy and power
density compromises caused by the introduction of significant dead weight/volume and increased electrochemical resistance.
Here we introduce an ultrathin composite film consisting of naphthalimide-functionalized poly(amidoamine) dendrimers and graphene
oxide nanosheets as a cycling stabilizer. Combining the dendrimer structure that can confine polysulfide intermediates chemically
and physically together with the graphene oxide that renders the film robust and thin (<1% of the thickness of the active
sulfur layer), the composite film is designed to enable stable cycling of sulfur cathodes without compromising the energy
and power densities. Our sulfur electrodes coated with the composite film exhibit very good cycling stability, together with
high sulfur content, large areal capacity, and improved power rate.
Co-reporter:Shengju Li, Lucky Ahmed, Ruina Zhang, Yi Pan, Hiroaki Matsunami, Jessica L. Burger, Eric Block, Victor S. Batista, and Hanyi Zhuang
Journal of the American Chemical Society 2016 Volume 138(Issue 40) pp:13281-13288
Publication Date(Web):September 23, 2016
DOI:10.1021/jacs.6b06983
Mammalian survival depends on ultrasensitive olfactory detection of volatile sulfur compounds, since these compounds can signal the presence of rancid food, O2 depleted atmospheres, and predators (through carnivore excretions). Skunks exploit this sensitivity with their noxious spray. In commerce, natural and liquefied gases are odorized with t-BuSH and EtSH, respectively, as warnings. The 100-million-fold difference in olfactory perception between structurally similar EtSH and EtOH has long puzzled those studying olfaction. Mammals detect thiols and other odorants using odorant receptors (ORs), members of the family of seven transmembrane G-protein-coupled receptors (GPCRs). Understanding the regulator cofactors and response of ORs is particularly challenging due to the lack of X-ray structural models. Here, we combine computational modeling and site-directed mutagenesis with saturation transfer difference (STD) NMR spectroscopy and measurements of the receptor response profiles. We find that human thiol receptor OR2T11 responds specifically to gas odorants t-BuSH and EtSH requiring ionic copper for its robust activation and that this role of copper is mimicked by ionic and nanoparticulate silver. While copper is both an essential nutrient for life and, in excess, a hallmark of various pathologies and neurodegenerative diseases, its involvement in human olfaction has not been previously demonstrated. When screened against a series of alcohols, thiols, sulfides, and metal-coordinating ligands, OR2T11 responds with enhancement by copper to the mouse semiochemical CH3SCH2SH and derivatives, to four-membered cyclic sulfide thietane and to one- to four-carbon straight- and branched-chain and five-carbon branched-chain thiols but not to longer chain thiols, suggesting compact receptor dimensions. Alcohols are unreactive.
Co-reporter:Zachary S. Fishman, Benjamin Rudshteyn, Yulian He, Bolun Liu, Subhajyoti Chaudhuri, Mikhail Askerka, Gary L. Haller, Victor S. Batista, and Lisa D. Pfefferle
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10978-10985
Publication Date(Web):July 25, 2016
DOI:10.1021/jacs.6b05332
CuO is a nonhazardous, earth-abundant material that has exciting potential for use in solar cells, photocatalysis, and other optoelectronic applications. While progress has been made on the characterization of properties and reactivity of CuO, there remains significant controversy on how to control the precise band gap by tuning conditions of synthetic methods. Here, we combine experimental and theoretical methods to address the origin of the wide distribution of reported band gaps for CuO nanosheets. We establish reaction conditions to control the band gap and reactivity via a high-temperature treatment in an oxygen-rich environment. SEM, TEM, XRD, and BET physisorption reveals little to no change in nanostructure, crystal structure, or surface area. In contrast, UV–vis spectroscopy shows a modulation in the material band gap over a range of 330 meV. A similar trend is found in H2 temperature-programmed reduction where peak H2 consumption temperature decreases with treatment. Calculations of the density of states show that increasing the oxygen to copper coverage ratio of the surface accounts for most of the observed changes in the band gap. An oxygen exchange mechanism, supported by 18O2 temperature-programmed oxidation, is proposed to be responsible for changes in the CuO nanosheet oxygen to copper stoichiometry. The changes induced by oxygen depletion/deposition serve to explain discrepancies in the band gap of CuO, as reported in the literature, as well as dramatic differences in catalytic performance.
Co-reporter:Ke R. Yang; Adam J. Matula; Gihan Kwon; Jiyun Hong; Stafford W. Sheehan; Julianne M. Thomsen; Gary W. Brudvig; Robert H. Crabtree; David M. Tiede; Lin X. Chen
Journal of the American Chemical Society 2016 Volume 138(Issue 17) pp:5511-5514
Publication Date(Web):April 18, 2016
DOI:10.1021/jacs.6b01750
The solution structures of highly active Ir water-oxidation catalysts are elucidated by combining density functional theory, high-energy X-ray scattering (HEXS), and extended X-ray absorption fine structure (EXAFS) spectroscopy. We find that the catalysts are Ir dimers with mono-μ-O cores and terminal anionic ligands, generated in situ through partial oxidation of a common catalyst precursor. The proposed structures are supported by 1H and 17O NMR, EPR, resonance Raman and UV–vis spectra, electrophoresis, etc. Our findings are particularly valuable to understand the mechanism of water oxidation by highly reactive Ir catalysts. Importantly, our DFT-EXAFS-HEXS methodology provides a new in situ technique for characterization of active species in catalytic systems.
Co-reporter:Kelly L. Materna, Benjamin Rudshteyn, Bradley J. Brennan, Morgan H. Kane, Aaron J. Bloomfield, Daria L. Huang, Dimitar Y. Shopov, Victor S. Batista, Robert H. Crabtree, and Gary W. Brudvig
ACS Catalysis 2016 Volume 6(Issue 8) pp:5371
Publication Date(Web):July 8, 2016
DOI:10.1021/acscatal.6b01101
A pentamethylcyclopentadienyl (Cp*) iridium water-oxidation precatalyst was modified to include a silatrane functional group for covalent attachment to metal oxide semiconductor surfaces. The heterogenized catalyst was found to perform electrochemically driven water oxidation at an overpotential of 462 mV with a turnover number of 304 and turnover frequency of 0.035 s–1 in a 0.1 M KNO3 electrolyte at pH 5.8. Computational modeling of the experimental IR spectra suggests that the catalyst retains its Cp* group during the first hour of catalysis and likely remains monomeric.Keywords: alternative energy; electrocatalysis; iridium; metal oxide; silatrane; surface binding; water oxidation
Co-reporter:Bradley J. Brennan, Jeffrey Chen, Benjamin Rudshteyn, Subhajyoti Chaudhuri, Brandon Q. Mercado, Victor S. Batista, Robert H. Crabtree and Gary W. Brudvig
Chemical Communications 2016 vol. 52(Issue 14) pp:2972-2975
Publication Date(Web):12 Jan 2016
DOI:10.1039/C5CC09857B
Hydroxamate binding modes and protonation states have yet to be conclusively determined. Molecular titanium(IV) phenylhydroxamate complexes were synthesized as structural and spectroscopic models, and compared to functionalized TiO2 nanoparticles. In a combined experimental–theoretical study, we find that the predominant binding form is monodeprotonated, with evidence for the chelate mode.
Co-reporter:Srinivasan Ramakrishnan, Kate M. Waldie, Ingolf Warnke, Antonio G. De Crisci, Victor S. Batista, Robert M. Waymouth, and Christopher E. D. Chidsey
Inorganic Chemistry 2016 Volume 55(Issue 4) pp:1623-1632
Publication Date(Web):February 2, 2016
DOI:10.1021/acs.inorgchem.5b02556
The ruthenium hydride [RuH(CNN)(dppb)] (1; CNN = 2-aminomethyl-6-tolylpyridine, dppb = 1,4-bis(diphenylphosphino)butane) reacts rapidly and irreversibly with CO2 under ambient conditions to yield the corresponding Ru formate complex 2. In contrast, the Ru hydride 1 reacts with acetone reversibly to generate the Ru isopropoxide, with the reaction free energy ΔG°298 K = −3.1 kcal/mol measured by 1H NMR in tetrahydrofuran-d8. Density functional theory (DFT), calibrated to the experimentally measured free energies of ketone insertion, was used to evaluate and compare the mechanism and energetics of insertion of acetone and CO2 into the Ru–hydride bond of 1. The calculated reaction coordinate for acetone insertion involves a stepwise outer-sphere dihydrogen transfer to acetone via hydride transfer from the metal and proton transfer from the N–H group on the CNN ligand. In contrast, the lowest energy pathway calculated for CO2 insertion proceeds by an initial Ru–H hydride transfer to CO2 followed by rotation of the resulting N–H-stabilized formate to a Ru–O-bound formate. DFT calculations were used to evaluate the influence of the ancillary ligands on the thermodynamics of CO2 insertion, revealing that increasing the π acidity of the ligand cis to the hydride ligand and increasing the σ basicity of the ligand trans to it decreases the free energy of CO2 insertion, providing a strategy for the design of metal hydride systems capable of reversible, ergoneutral interconversion of CO2 and formate.
Co-reporter:Jianbing Jiang, John R. Swierk, Svante Hedström, Adam J. Matula, Robert H. Crabtree, Victor S. Batista, Charles A. Schmuttenmaer and Gary W. Brudvig
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 28) pp:18678-18682
Publication Date(Web):30 Jun 2016
DOI:10.1039/C6CP04377A
Interfacial electron transfer dynamics of a series of photosensitizers bound to TiO2via linkers of varying conjugation strength are explored by spectroscopic and computational techniques. Injection and recombination depend on the extent of conjugation in the linker, where the LUMO delocalization determines the injection dynamics but both the HOMO and HOMO−1 are involved in recombination.
Co-reporter:Mikhail Askerka, Jimin Wang, David J. Vinyard, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2016 Volume 55(Issue 7) pp:981-984
Publication Date(Web):February 5, 2016
DOI:10.1021/acs.biochem.6b00041
The oxygen-evolving complex (OEC) of photosystem II has been studied in the S3 state by electron paramagnetic resonance, extended X-ray absorption fine structure (EXAFS), and femtosecond X-ray diffraction (XRD). However, the actual structure of the OEC in the S3 state has yet to be established. Here, we apply hybrid quantum mechanics/molecular mechanics methods and propose a structural model that is consistent with EXAFS and XRD. The model supports binding of water ligands to the cluster in the S2 → S3 transition through a carousel rearrangement around Mn4, inspired by studies of ammonia binding.
Co-reporter:Xiangmeng Kong, Andreas Markmann, and Victor S. Batista
The Journal of Physical Chemistry A 2016 Volume 120(Issue 19) pp:3260-3269
Publication Date(Web):February 4, 2016
DOI:10.1021/acs.jpca.5b12192
A rigorous method for simulations of quantum dynamics is introduced on the basis of concatenation of semiclassical thawed Gaussian propagation steps. The time-evolving state is represented as a linear superposition of closely overlapping Gaussians that evolve in time according to their characteristic equations of motion, integrated by fourth-order Runge–Kutta or velocity Verlet. The expansion coefficients of the initial superposition are updated after each semiclassical propagation period by implementing the Husimi Transform analytically in the basis of closely overlapping Gaussians. An advantage of the resulting time-sliced thawed Gaussian (TSTG) method is that it allows for full-quantum dynamics propagation without any kind of multidimensional integral calculation, or inversion of overlap matrices. The accuracy of the TSTG method is demonstrated as applied to simulations of quantum tunneling, showing quantitative agreement with benchmark calculations based on the split-operator Fourier transform method.
Co-reporter:Hilary M. Chase, Benjamin Rudshteyn, Brian T. Psciuk, Mary Alice Upshur, Benjamin F. Strick, Regan J. Thomson, Victor S. Batista, and Franz M. Geiger
The Journal of Physical Chemistry B 2016 Volume 120(Issue 8) pp:1919-1927
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jpcb.5b09769
We assess the capabilities of eight popular density functional theory (DFT) functionals, in combination with several basis sets, as applied to calculations of vibrational sum frequency generation (SFG) spectra of the atmospherically relevant isoprene oxidation product trans-β-isoprene epoxydiol (IEPOX) and one of its deuterated isotopologues at the fused silica/vapor interface. We use sum of squared differences (SSD) and total absolute error (TAE) calculations to estimate the performance of each functional/basis set combination in producing SFG spectra that match experimentally obtained spectra from trans-β-IEPOX and one of its isotopologues. Our joined SSD/TAE analysis shows that while the twist angle of the methyl C3v symmetry axis of trans-β-IEPOX relative to the surface is sensitive to the choice of DFT functional, the calculated tilt angle relative to the surface normal is largely independent of the functional and basis set. Moreover, we report that hybrid functionals such as B3LYP, ωB97X-D, PBE0, and B97-1 in combination with a modest basis set, such as 6-311G(d,p), provides good agreement with experimental data and much better performance than pure functionals such as PBE and BP86. However, improving the quality of the basis set only improves agreement with experimental data for calculations based on pure functionals. A conformational analysis, based on comparisons of calculated and experimental SFG spectra, suggests that trans-β-IEPOX points all of its oxygen atoms toward the silica/vapor interface.
Co-reporter:Junming Ho
The Journal of Physical Chemistry C 2016 Volume 120(Issue 23) pp:12578-12589
Publication Date(Web):May 17, 2016
DOI:10.1021/acs.jpcc.6b03158
A rotationally fluid state of α-pinene at fused silica/vapor interfaces is revealed by computational and experimental vibrational sum frequency generation (SFG) studies. We report the first assignment of the vibrational modes in the notoriously congested C–H stretching region of α-pinene and identify its bridge methylene group on the four-membered ring (“βCH2”) as the origin of its dominant spectral feature. We find that the spectra are perfused with Fermi resonances that need to be accounted for explicitly in the computation of vibrational spectra of strained hydrocarbons such as α-pinene. The preferred orientations of α-pinene are consistent with optimization of van der Waals contacts with the silica surface that results in a bimodal distribution of highly fluxional orientations in which the βCH2 group points “towards” or “away from” the surface. Classical molecular dynamics simulations further provide rotational diffusion constants of 49 ± 1 ps and 2580 ± 60 ps, which are attributed to two broad types of adsorption modes on silica. The reported findings are particularly relevant to the exposure of α-pinene to primary oxidants in heterogeneous catalytic pathways that exploit α-pinene as a sustainable feedstock for fine chemicals and polymers.
Co-reporter:Aimin Ge, Benjamin Rudshteyn, Brian T. Psciuk, Dequan Xiao, Jia Song, Chantelle L. Anfuso, Allen M. Ricks, Victor S. Batista, and Tianquan Lian
The Journal of Physical Chemistry C 2016 Volume 120(Issue 37) pp:20970-20977
Publication Date(Web):May 31, 2016
DOI:10.1021/acs.jpcc.6b03165
Vibrational sum frequency generation (SFG) spectroscopy has been utilized to study the spatial orientation and alignment of Re(CO)3Cl(dcbpy) (dcbpy = 4,4′-dicarboxy-2,2′-bipyridine) (or ReC0A) on the (001) and (110) surfaces of rutile single-crystalline TiO2. The SFG intensity of the CO stretching modes shows an isotropic distribution on the (001) surface and an anisotropic distribution on the (110) surfaces with respect to the in-plane rotation of the crystal relative to the surface normal (or the incident laser beam plane). By combining these results with ab initio SFG simulations and with modeling of ReC0A–TiO2 cluster binding structures at the density functional theory level, we reveal that the origin of the optical anisotropy for ReC0A on the TiO2(110) surface is associated with the binding preference of ReC0A along the [−110] axis. Along this direction, the binding structure is energetically favorable, because of the formation of proper hydrogen bonding between the carboxylate group and passivating water molecules adsorbed on the TiO2(110) surface. Simulations of dimers of ReC0A molecules binding close together with full nearest-neighbor effects give a structure that reproduces the experimental SFG polar plot. The tilt angle, defined by the bpy ring angle relative to the surface normal, of the catalyst is found to be 26° for one monomer and 18° for the other, which corresponds to an aggregate at high surface coverage.
Co-reporter:Jianbing Jiang, John R. Swierk, Kelly L. Materna, Svante HedströmShin Hee Lee, Robert H. Crabtree, Charles A. Schmuttenmaer, Victor S. Batista, Gary W. Brudvig
The Journal of Physical Chemistry C 2016 Volume 120(Issue 51) pp:28971-28982
Publication Date(Web):December 3, 2016
DOI:10.1021/acs.jpcc.6b10350
We report CF3-substituted porphyrins and evaluate their use as photosensitizers in water-splitting dye-sensitized photoelectrochemical cells (WS-DSPECs) by characterizing interfacial electron transfer on metal oxide surfaces. By using (CF3)2C6H3 instead of C6F5 substituents at the meso positions, we obtain the desired high potentials while avoiding the sensitivity of C6F5 substituents to nucleophilic substitution, a process that limits the types of synthetic reactions that can be used. Both the number of CF3 groups and the central metal tune the ground and excited-state potentials. A pair of porphyrins bearing carboxylic acids as anchoring groups were deposited on SnO2 and TiO2 surfaces, and the interfacial charge-injection and charge-recombination kinetics were characterized by using a combination of computational modeling, terahertz measurements, and transient absorption spectroscopy. We find that both free-base and metalated porphyrins inject into SnO2 and that recombination is slower for the latter case. These findings demonstrate that (CF3)2C6H3-substituted porphyrins are promising photosensitizers for use in WS-DSPECs.
Co-reporter:Ivan Rivalta, George P. Lisi, Ning-Shiuan Snoeberger, Gregory Manley, J. Patrick Loria, and Victor S. Batista
Biochemistry 2016 Volume 55(Issue 47) pp:
Publication Date(Web):October 31, 2016
DOI:10.1021/acs.biochem.6b00859
Allosteric enzymes regulate a wide range of catalytic transformations, including biosynthetic mechanisms of important human pathogens, upon binding of substrate molecules to an orthosteric (or active) site and effector ligands at distant (allosteric) sites. We find that enzymatic activity can be impaired by small molecules that bind along the allosteric pathway connecting the orthosteric and allosteric sites, without competing with endogenous ligands. Noncompetitive allosteric inhibitors disrupted allostery in the imidazole glycerol phosphate synthase (IGPS) enzyme from Thermotoga maritima as evidenced by nuclear magnetic resonance, microsecond time-scale molecular dynamics simulations, isothermal titration calorimetry, and kinetic assays. The findings are particularly relevant for the development of allosteric antibiotics, herbicides, and antifungal compounds because IGPS is absent in mammals but provides an entry point to fundamental biosynthetic pathways in plants, fungi, and bacteria.
Co-reporter:Roc Matheu; Mehmed Z. Ertem; Jordi Benet-Buchholz; Eugenio Coronado; Victor S. Batista; Xavier Sala;Antoni Llobet
Journal of the American Chemical Society 2015 Volume 137(Issue 33) pp:10786-10795
Publication Date(Web):July 30, 2015
DOI:10.1021/jacs.5b06541
We introduce a new family of complexes with the general formula [Run(tda)(py)2]m+ (n = 2, m = 0, 1; n = 3, m = 1, 2+; n = 4, m = 2, 32+), with tda2– being [2,2′:6′,2″-terpyridine]-6,6″-dicarboxylate, including complex [RuIV(OH)(tda-κ-N3O)(py)2]+, 4H+, which we find to be an impressive water oxidation catalyst, formed by hydroxo coordination to 32+ under basic conditions. The complexes are synthesized, isolated, and thoroughly characterized by analytical, spectroscopic (UV–vis, nuclear magnetic resonance, electron paramagnetic resonance), computational, and electrochemical techniques (cyclic voltammetry, differential pulse voltammetry, coulometry), including solid-state monocrystal X-ray diffraction analysis. In oxidation state IV, the Ru center is seven-coordinated and diamagnetic, whereas in oxidation state II, the complex has an unbonded dangling carboxylate and is six-coordinated while still diamagnetic. With oxidation state III, the coordination number is halfway between the coordination of oxidation states II and IV. Species generated in situ have also been characterized by spectroscopic, computational, and electrochemical techniques, together with the related species derived from a different degree of protonation and oxidation states. 4H+ can be generated potentiometrically, or voltammetrically, from 32+, and both coexist in solution. While complex 32+ is not catalytically active, the catalytic performance of complex 4H+ is characterized by the foot of the wave analysis, giving an impressive turnover frequency record of 8000 s–1 at pH 7.0 and 50 000 s–1 at pH 10.0. Density functional theory calculations provide a complete description of the water oxidation catalytic cycle of 4H+, manifesting the key functional role of the dangling carboxylate in lowering the activation free energies that lead to O–O bond formation.
Co-reporter:Dimitar Y. Shopov; Benjamin Rudshteyn; Jesús Campos; Victor S. Batista; Robert H. Crabtree;Gary W. Brudvig
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7243-7250
Publication Date(Web):May 19, 2015
DOI:10.1021/jacs.5b04185
The preparation of the facial and meridional isomers of [Ir(pyalk)3] (pyalk = 2-(2-pyridyl)isopropanoate), as model complexes for a powerful water oxidation catalyst, is reported. The strongly donating N3O3 ligand set is very oxidation-resistant, yet promotes facile metal-centered oxidation to form stable Ir(IV) compounds. The IrIII/IV reduction potentials of the two isomers differ by 340 mV despite the identical ligand set. A ligand field rationalization is advanced and supported by DFT calculations.
Co-reporter:Jinquan Chen; Kaifeng Wu; Benjamin Rudshteyn; Yanyan Jia; Wendu Ding; Zhao-Xiong Xie; Victor S. Batista;Tianquan Lian
Journal of the American Chemical Society 2015 Volume 138(Issue 3) pp:884-892
Publication Date(Web):December 29, 2015
DOI:10.1021/jacs.5b10354
Pyridine and derivatives have been reported as efficient and selective catalysts for the electrochemical and photoelectrochemical reduction of CO2 to methanol. Although the catalytic mechanism remains a subject of considerable recent debate, most proposed models involve interfacial proton coupled electron transfer (PCET) to electrode-bound catalysts. We report a combined experimental and theoretical study of the photoreduction of 4,4′-bipyridium (bPYD) using CdSe quantum dots (QDs) as a model system for interfacial PCET. We observed ultrafast photoinduced PCET from CdSe QDs to form doubly protonated [bPYDH2]+• radical cations at low pH (4–6). Through studies of the dependence of PCET rate on isotopic substitution, pH and bPYD concentration, the radical formation mechanism was identified to be a sequential interfacial electron and proton transfer (ET/PT) process with a rate-limiting pH independent electron transfer rate constant, kint, of 1.05 ± 0.13 × 1010 s–1 between a QD and an adsorbed singly protonated [bPYDH]+. Theoretical studies of the adsorption of [bPYDH]+ and methylviologen on QD surfaces revealed important effects of hydrogen bonding with the capping ligand (3-mercaptopropionic acid) on binding geometry and interfacial PCET. In the presence of sacrificial electron donors, this system was shown to be capable of generating [bPYDH2]+• radical cations under continuous illumination at 405 nm with a steady-state photoreduction quantum yield of 1.1 ± 0.1% at pH 4. The mechanism of bPYD photoreduction reported in this work may provide useful insights into the catalytic roles of pyridine and pyridine derivatives in the electrochemical and photoelectrochemical reduction of CO2.
Co-reporter:Sahr Khan, Ke R. Yang, Mehmed Z. Ertem, Victor S. Batista, and Gary W. Brudvig
ACS Catalysis 2015 Volume 5(Issue 12) pp:7104
Publication Date(Web):October 22, 2015
DOI:10.1021/acscatal.5b01976
The biomimetic oxomanganese complex [MnIII/IV2(μ-O)2(terpy)2(OH2)2](NO3)3 (1; terpy = 2,2′:6′,2″-terpyridine) catalyzes O2 evolution from water when activated by oxidants, such as oxone (2KHSO5·KHSO4·K2SO4). The mechanism of this reaction has never been characterized, due to the fleeting nature of the intermediates. In the present study, we elucidate the underlying reaction mechanism through experimental and theoretical analyses of competitive kinetic oxygen isotope effects (KIEs) during catalytic turnover conditions. The experimental 18O KIE is a sensitive probe of the highest transition state in the O2-evolution mechanism and provides a strict constraint for calculated mechanisms. The 18O kinetic isotope effect of 1.013 ± 0.003 measured using natural abundance reactants is consistent with the calculated isotope effect of peroxymonosulfate binding to the complex, as described by density functional theory (DFT). This provides strong evidence for peroxymonosulfate binding being both the first irreversible and rate-determining step during turnover, in contrast to the previously held assumption that formation of a high-valent Mn-oxo/oxyl species is the highest barrier step that controls the rate of O2 evolution by this complex. The comparison of the measured and calculated KIEs supplements previous kinetic studies, enabling us to describe the complete mechanism of O2 evolution, starting from when the oxidant first binds to the manganese complex to when O2 is released. The reported findings lay the groundwork for understanding O2 evolution catalyzed by other biomimetic oxomanganese complexes, with features common to those of the O2-evolving complex of photosystem II, providing experimental and theoretical diagnostics of oxygen isotope effects that could reveal the nature of elusive reaction intermediates.Keywords: density functional theory; manganese complex; oxygen evolution mechanism; oxygen isotope effects; peroxymonosulfate
Co-reporter:Ivan Rivalta, Ke R. Yang, Gary W. Brudvig, and Victor S. Batista
ACS Catalysis 2015 Volume 5(Issue 4) pp:2384
Publication Date(Web):March 5, 2015
DOI:10.1021/acscatal.5b00048
Photosynthetic oxygen evolution involves water splitting into triplet oxygen, protons, and electrons, as follows: 2H2O → 3O2 + 4e–+ 4H+. The reaction is catalyzed by the oxomanganese complex of photosystem II, embedded in the thylakoid membrane of green plant chloroplasts and internal membranes of cyanobacteria. Biomimetic synthetic complexes have been developed over the years, although the reactivity of most of these complexes remains to be established. Here, we report a computational study of water splitting catalyzed by the mixed-valent oxomanganese dimer [H2O(terpy)MnIII(μ-oxo)2MnIV(terpy)OH2]3+ (terpy = 2,2′:6′,2″-terpyridine) in acetate buffer, with emphasis on the origin of triplet oxygen and the noninnocent role of carboxylate ligands in the underlying reaction mechanism. Our calculations suggest triplet oxygen evolution from an end-on (η1) Mn(III)-superoxo species, which forms from a hydroperoxo intermediate generated by nucleophilic attack of substrate water onto an oxyl radical Mn(IV)–O•. Carboxylate groups of acetate facilitate formation of the oxyl radical by shifting the redox potential of the Mn complex upon exchange with water ligands, and catalyze the O–O bond formation by deprotonating the nucleophilic water molecule. These findings provide valuable insights on the origin of triplet oxygen and on the regulatory role of the environment surrounding the inorganic core of oxomanganese complexes during catalytic oxygen evolution.Keywords: acid/base cofactors; artificial photosynthesis; biomimetic oxomanganese complexes; O−O bond formation; water oxidation
Co-reporter:Roc Matheu, Laia Francàs, Petko Chernev, Mehmed Z. Ertem, Victor Batista, Michael Haumann, Xavier Sala, and Antoni Llobet
ACS Catalysis 2015 Volume 5(Issue 6) pp:3422
Publication Date(Web):April 23, 2015
DOI:10.1021/acscatal.5b00132
Electrochemical reduction of the dizaonium complex, [RuII(bda)(NO)(N–N2)2]3+, 23+ (N–N22+ is 4-(pyridin-4-yl) benzenediazonium and bda2– is [2,2′-bipyridine]-6,6′-dicarboxylate), in acetone produces the covalent grafting of this molecular complex onto glassy carbon (GC) electrodes. Multiple cycling voltammetric experiments on the GC electrode generates hybrid materials labeled as GC-4, with the corresponding Ru-aqua complex anchored on the graphite surface. GC-4 has been characterized at pH = 7.0 by electrochemical techniques and X-ray absorption spectroscopy (XAS) and has been shown to act as an active catalyst for the oxidation of water to dioxygen. This new hybrid material has a lower catalytic performance than its counterpart in homogeneous phase and progressively decomposes to form RuO2 at the electrode surface. Nevertheless the resulting metal oxide attached at the GC electrode surface, GC-RuO2, is a very fast and rugged heterogeneous water oxidation catalyst with TOFis of 300 s–1 and TONs > 45 000. The observed performance is comparable to the best electrocatalysts reported so far, at neutral pH.Keywords: electrocatalysis; heterogeneous water oxidation catalysis; modified graphite electrodes; Ru complexes; RuO2; water oxidation catalysis; water splitting
Co-reporter:Wendu Ding, Matthieu Koepf, Christopher Koenigsmann, Arunabh Batra, Latha Venkataraman, Christian F. A. Negre, Gary W. Brudvig, Robert H. Crabtree, Charles A. Schmuttenmaer, and Victor S. Batista
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 12) pp:5888-5896
Publication Date(Web):November 3, 2015
DOI:10.1021/acs.jctc.5b00823
We report a systematic computational search of molecular frameworks for intrinsic rectification of electron transport. The screening of molecular rectifiers includes 52 molecules and conformers spanning over 9 series of structural motifs. N-Phenylbenzamide is found to be a promising framework with both suitable conductance and rectification properties. A targeted screening performed on 30 additional derivatives and conformers of N-phenylbenzamide yielded enhanced rectification based on asymmetric functionalization. We demonstrate that electron-donating substituent groups that maintain an asymmetric distribution of charge in the dominant transport channel (e.g., HOMO) enhance rectification by raising the channel closer to the Fermi level. These findings are particularly valuable for the design of molecular assemblies that could ensure directionality of electron transport in a wide range of applications, from molecular electronics to catalytic reactions.
Co-reporter:Mikhail Askerka, David J. Vinyard, Jimin Wang, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2015 Volume 54(Issue 9) pp:1713-1716
Publication Date(Web):February 24, 2015
DOI:10.1021/acs.biochem.5b00089
A recent femtosecond X-ray diffraction study produced the first high-resolution structural model of the oxygen-evolving complex of photosystem II that is free of radiation-induced manganese reduction (Protein Data Bank entries 4UB6 and 4UB8). We find, however, that the model does not match extended X-ray absorption fine structure and QM/MM data for the S1 state. This is attributed to uncertainty about the positions of oxygen atoms that remain partially unresolved, even at 1.95 Å resolution, next to the heavy manganese centers. In addition, the photosystem II crystals may contain significant amounts of the S0 state, because of extensive dark adaptation prior to data collection.
Co-reporter:Mikhail Askerka, David J. Vinyard, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2015 Volume 54(Issue 38) pp:
Publication Date(Web):September 17, 2015
DOI:10.1021/acs.biochem.5b00974
Ammonia binds directly to the oxygen-evolving complex of photosystem II (PSII) upon formation of the S2 intermediate, as evidenced by electron paramagnetic resonance spectroscopy. We explore the binding mode by using quantum mechanics/molecular mechanics methods and simulations of extended X-ray absorption fine structure spectra. We find that NH3 binds as an additional terminal ligand to the dangling Mn4, instead of exchanging with terminal water. Because water and ammonia are electronic and structural analogues, these findings suggest that water binds analogously during the S2 → S3 transition, leading to rearrangement of ligands in a carrousel around Mn4.
Co-reporter:Leslie Vogt, Mehmed Z. Ertem, Rhitankar Pal, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2015 Volume 54(Issue 3) pp:820-825
Publication Date(Web):January 2, 2015
DOI:10.1021/bi5011706
The oxygen-evolving complex of photosystem II can function with either Ca2+ or Sr2+ as the heterocation, but the reason for different turnover rates remains unresolved despite reported X-ray crystal structures for both forms. Using quantum mechanics/molecular mechanics (QM/MM) calculations, we optimize structures with each cation in both the resting state (S1) and in a series of reduced states (S0, S–1, and S–2). Through comparison with experimental data, we determine that the X-ray crystal structures with either Ca2+ or Sr2+ are most consistent with the S–2 state (i.e., Mn4[III,III,III,II] with O4 and O5 protonated). As expected, the QM/MM models show that Ca2+/Sr2+ substitution results in the elongation of the heterocation bonds and the displacement of terminal waters W3 and W4. The optimized structures also show that hydrogen-bonded W5 is displaced in all S states with Sr2+ as the heterocation, suggesting that this water may play a critical role during water oxidation.
Co-reporter:Kuo Liao;Mikhail Askerka;Elizabeth L. Zeitler;Andrew B. Bocarsly
Topics in Catalysis 2015 Volume 58( Issue 1) pp:23-29
Publication Date(Web):2015 February
DOI:10.1007/s11244-014-0340-2
Recent electrochemical studies have reported aqueous CO2 reduction to formic acid, formaldehyde and methanol at potentials of ca. −600 mV versus SCE, when using a Pt working electrode in acidic pyridine solutions. In those experiments, pyridinium is thought to function as a one-electron shuttle for the underlying multielectron reduction of CO2. DFT studies proposed that the critical step of the underlying reaction mechanism is the one-electron reduction of pyridinium at the Pt surface through proton coupled electron transfer. Such reaction forms a H adsorbate that is subsequently transferred to CO2 as a hydride, through a proton coupled hydride transfer mechanism where pyridinium functions as a Brønsted acid. Here, we find that imidazolium exhibits an electrochemical behavior analogous to pyridinium, as characterized by the experimental and theoretical analysis of the initial reduction on Pt. A cathodic wave, with a cyclic voltammetric half wave potential of ca. −680 mV versus SCE, is consistent with the theoretical prediction based on the recently proposed reaction mechanism suggesting that positively charged Brønsted acids could serve as electrocatalytic one-electron shuttle species for multielectron CO2 reduction.
Co-reporter:Brian T. Psciuk, Mirabelle Prémont-Schwarz, Benjamin Koeppe, Sharon Keinan, Dequan Xiao, Erik T. J. Nibbering, and Victor S. Batista
The Journal of Physical Chemistry A 2015 Volume 119(Issue 20) pp:4800-4812
Publication Date(Web):April 16, 2015
DOI:10.1021/acs.jpca.5b01530
To assess the potential use of O–H stretching modes of aromatic alcohols as ultrafast local probes of transient structures and photoacidity, we analyze the response of the O–H stretching mode in the 2-naphthol-acetonitrile (2N–CH3CN) 1:1 complex after UV photoexcitation. We combine femtosecond UV-infrared pump–probe spectroscopy and a theoretical treatment of vibrational solvatochromic effects based on the Pullin perturbative approach, parametrized at the density functional theory (DFT) level. We analyze the effect of hydrogen bonding on the vibrational properties of the photoacid–base complex in the S0 state, as compared to O–H stretching vibrations in a wide range of substituted phenols and naphthols covering the 3000–3650 cm–1 frequency range. Ground state vibrational properties of these phenols and naphthols with various substituent functional groups are analyzed in solvents of different polarity and compared to the vibrational frequency shift of 2N induced by UV photoexcitation to the 1Lb electronic excited state. We find that the O–H stretching frequency shifts follow a linear relationship with the solvent polarity function F0 = (2ε0 – 2)/(2ε0 + 1), where ε0 is the static dielectric constant of the solvent. These changes are directly correlated with photoacidity trends determined by reported pKa values and with structural changes in the O···N and O–H hydrogen-bond distances induced by solvation or photoexcitation of the hydrogen-bonded complexes.
Co-reporter:Eric Block;Seogjoo Jang;Hiroaki Matsunami;Hanyi Zhuang
PNAS 2015 Volume 112 (Issue 25 ) pp:E3155
Publication Date(Web):2015-06-23
DOI:10.1073/pnas.1508443112
Co-reporter:Christian F. A. Negre, Karin J. Young, Ma. Belén Oviedo, Laura J. Allen, Cristián G. Sánchez, Katarzyna N. Jarzembska, Jason B. Benedict, Robert H. Crabtree, Philip Coppens, Gary W. Brudvig, and Victor S. Batista
Journal of the American Chemical Society 2014 Volume 136(Issue 46) pp:16420-16429
Publication Date(Web):October 22, 2014
DOI:10.1021/ja509270f
We find that crystallographically resolved Ti17O24(OPri)20 nanoparticles, functionalized by covalent attachment of 4-nitrophenyl-acetylacetonate or coumarin 343 adsorbates, exhibit hole injection into surface states when photoexcited with visible light (λ = 400–680 nm). Our findings are supported by photoelectrochemical measurements, EPR spectroscopy, and quantum dynamics simulations of interfacial charge transfer. The underlying mechanism is consistent with measurements of photocathodic currents generated with visible light for thin layers of functionalized polyoxotitanate nanocrystals deposited on FTO working electrodes. The reported experimental and theoretical analysis demonstrates for the first time the feasibility of p-type sensitization of TiO2 solely based on covalent binding of organic adsorbates.
Co-reporter:Victoria Mooney; Sivakumar Sekharan; Jian Liu; Ying Guo; Victor S. Batista;Elsa C. Y. Yan
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:307-313
Publication Date(Web):December 16, 2014
DOI:10.1021/ja510553f
Visual pigments can be thermally activated via isomerization of the retinyl chromophore and hydrolysis of the Schiff base (SB) through which the retinyl chromophore is bound to the opsin protein. Here, we present the first combined experimental and theoretical study of the thermal activation of a Siberian hamster ultraviolet (SHUV) pigment. We measured the rates of thermal isomerization and hydrolysis in the SHUV pigment and bovine rhodopsin. We found that these rates were significantly faster in the UV pigment than in rhodopsin due to the difference in the structural and electrostatic effects surrounding the unprotonated Schiff base (USB) retinyl chromophore in the UV pigment. Theoretical (DFT-QM/MM) calculations of the cis–trans thermal isomerization revealed a barrier of ∼23 kcal/mol for the USB retinyl chromophore in SHUV compared to ∼40 kcal/mol for protonated Schiff base (PSB) chromophore in rhodopsin. The lower barrier for thermal isomerization in the SHUV pigment is attributed to the (i) lessening of the steric restraints near the β-ionone ring and SB ends of the chromophore, (ii) displacement of the transmembrane helix 6 (TM6) away from the binding pocket toward TM5 due to absence of the salt bridge between the USB and the protonated E113 residue, and (iii) change in orientation of the hydrogen-bonding networks (HBNs) in the extracellular loop 2 (EII). The results in comparing thermal stability of UV cone pigment and rhodopsin provide insight into molecular evolution of vertebrate visual pigments in achieving low discrete dark noise and high photosensitivity in rod pigments for dim-light vision.
Co-reporter:Wendu Ding, Christian F. A. Negre, Leslie Vogt, and Victor S. Batista
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 8) pp:3393-3400
Publication Date(Web):July 15, 2014
DOI:10.1021/ct5004687
A mechanism for electronic rectification under low bias potentials is elucidated for the prototype molecule HS-phenyl-amide-phenyl-SH. We apply density functional theory (DFT) combined with the nonequilibrium Green’s function formalism (NEGF), as implemented in the TranSIESTA computational code to calculate transport properties. We find that a single frontier orbital, the closest to the Fermi level, provides the dominant contribution to the overall transmission and determines the current. The asymmetric distribution of electron density in that orbital leads to rectification in charge transport due to its asymmetric response, shifting toward (or away from) the Fermi level under forward (or reverse) applied bias voltage. These findings provide a simple design principle to suppress recombination in molecular assemblies of dye-sensitized solar cells (DSSCs) where interfacial electron transfer is mediated by frontier orbitals with asymmetric character.
Co-reporter:Junming Ho, Michael B. Newcomer, Christina M. Ragain, Jose A. Gascon, Enrique R. Batista, J. Patrick Loria, and Victor S. Batista
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 11) pp:5125-5135
Publication Date(Web):September 24, 2014
DOI:10.1021/ct500571k
A generalization of the Moving-Domain Quantum Mechanics/Molecular Mechanics (MoD-QM/MM) hybrid method [Gascon, J. A.; Leung, S. S. F.; Batista, E. R.; Batista, V. S. J. Chem. Theory Comput. 2006, 2, 175–186] is introduced to provide a self-consistent computational protocol for structural refinement of extended systems. The method partitions the system into molecular domains that are iteratively optimized as quantum mechanical (QM) layers embedded in their surrounding molecular environment to obtain an ab initio quality description of the geometry and the molecular electrostatic potential of the extended system composed of those constituent fragments. The resulting methodology is benchmarked as applied to model systems that allow for full QM optimization as well as through refinement of the hydrogen bonding geometry in Oxytricha nova guanine quadruplex for which several studies have been reported, including the X-ray structure and NMR data. Calculations of 1H NMR chemical shifts based on the gauge independent atomic orbital (GIAO) method and direct comparisons with experiments show that solvated MoD-QM/MM structures, sampled from explicit solvent molecular dynamics simulations, allow for NMR simulations in much improved agreement with experimental data than models based on the X-ray structure or those optimized using classical molecular mechanics force fields.
Co-reporter:C. Koenigsmann, T. S. Ripolles, B. J. Brennan, C. F. A. Negre, M. Koepf, A. C. Durrell, R. L. Milot, J. A. Torre, R. H. Crabtree, V. S. Batista, G. W. Brudvig, J. Bisquert and C. A. Schmuttenmaer
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 31) pp:16629-16641
Publication Date(Web):04 Jul 2014
DOI:10.1039/C4CP02405B
An efficient synthetic protocol to functionalize the cyanoacrylic acid anchoring group of commercially available MK-2 dye with a highly water-stable hydroxamate anchoring group is described. Extensive characterization of this hydroxamate-modified dye (MK-2HA) reveals that the modification does not affect its favorable optoelectronic properties. Dye-sensitized solar cells (DSSCs) prepared with the MK-2HA dye attain improved efficiency (6.9%), relative to analogously prepared devices with commercial MK-2 and N719 dyes. The hydroxamate anchoring group also contributes to significantly increased water stability, with a decrease in the rate constant for dye desorption of MK-2HA relative to MK-2 in the presence of water by as much as 37.5%. In addition, the hydroxamate-anchored dye undergoes essentially no loss in DSSC efficiency and the external quantum efficiency improves when up to 20% water is purposefully added to the electrolyte. In contrast, devices prepared with the commercial dye suffer a 50% decline in efficiency under identical conditions, with a concomitant decrease in external quantum efficiency. Collectively, our results indicate that covalent functionalization of organic dyes with hydroxamate anchoring groups is a simple and efficient approach to improving the water stability of the dye–semiconductor interface and overall device durability.
Co-reporter:Sandra Luber, Sophie Leung, Carmen Herrmann, Wenge Han Du, Louis Noodleman and Victor S. Batista
Dalton Transactions 2014 vol. 43(Issue 2) pp:576-583
Publication Date(Web):16 Oct 2013
DOI:10.1039/C3DT51563J
Ribonucleotide reductases (RNRs) catalyze the reduction of ribonucleotides into deoxyribonucleotides necessary for DNA biosynthesis. Unlike the conventional class Ia RNRs which use a diiron cofactor in their subunit R2, the active site of the RNR-R2 from Chlamydia trachomatis (Ct) contains a Mn/Fe cofactor. The detailed structure of the Mn/Fe core has yet to be established. In this paper we evaluate six different structural models of the Ct RNR active site in the Mn(IV)/Fe(III) state by using Mössbauer parameter calculations and simulations of Mn/Fe extended X-ray absorption fine structure (EXAFS) spectroscopy, and we identify a structure similar to a previously proposed DFT-optimized model that shows quantitative agreement with both EXAFS and Mössbauer spectroscopic data.
Co-reporter:Mikhail Askerka, Jimin Wang, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2014 Volume 53(Issue 44) pp:
Publication Date(Web):October 27, 2014
DOI:10.1021/bi5011915
The S1 → S2 transition of the oxygen-evolving complex (OEC) of photosystem II does not involve the transfer of a proton to the lumen and occurs at cryogenic temperatures. Therefore, it is commonly thought to involve only Mn oxidation without any significant change in the structure of the OEC. Here, we analyze structural changes upon the S1 → S2 transition, as revealed by quantum mechanics/molecular mechanics methods and the isomorphous difference Fourier method applied to serial femtosecond X-ray diffraction data. We find that the main structural change in the OEC is in the position of the dangling Mn and its coordination environment.
Co-reporter:Omar F. Mohammed, Dequan Xiao, Victor S. Batista, and Erik T. J. Nibbering
The Journal of Physical Chemistry A 2014 Volume 118(Issue 17) pp:3090-3099
Publication Date(Web):March 31, 2014
DOI:10.1021/jp501612f
We combine ultrafast electronic and vibrational spectroscopy and computational modeling to investigate the photoinduced excited-state intramolecular hydrogen-transfer dynamics in 1,8-dihydroxy-9,10-anthraquinone (DHAQ) in tetrachloroethene, acetonitrile, dimethyl sulfoxide, and methanol. We analyze the electronic excited states of DHAQ with various possible hydrogen-bonding schemes and provide a general description of the electronic excited-state dynamics based on a systematic analysis of femtosecond UV/vis and UV/IR pump–probe spectroscopic data. Upon photoabsorption at 400 nm, the S2 electronic excited state is initially populated, followed by a rapid equilibration within 150 fs through population transfer to the S1 state where DHAQ exhibits ESIHT dynamics. In this equilibration process, the excited-state population is distributed between the 9,10-quinone (S2) and 1,10-quinone (S1) states while undergoing vibrational energy redistribution, vibrational cooling, and solvation dynamics on the 0.1–50 ps time scale. Transient UV/vis pump–probe data in methanol also suggest additional relaxation dynamics on the subnanosecond time scale, which we tentatively ascribe to hydrogen bond dynamics of DHAQ with the protic solvent, affecting the equilibrium population dynamics within the S2 and S1 electronic excited states. Ultimately, the two excited singlet states decay with a solvent-dependent time constant ranging from 139 to 210 ps. The concomitant electronic ground-state recovery is, however, only partial because a large fraction of the population relaxes to the first triplet state. From the similarity of the time scales involved, we conjecture that the solvent plays a crucial role in breaking the intramolecular hydrogen bond of DHAQ during the S2/S1 relaxation to either the ground or triplet state.
Co-reporter:Wendu Ding;Dr. Christian F. A. Negre;Dr. Julio L. Palma;Dr. Alec C. Durrell;Dr. Laura J. Allen;Dr. Karin J. Young;Rebecca L. Milot; Charles A. Schmuttenmaer; Gary W. Brudvig; Robert H. Crabtree; Victor S. Batista
ChemPhysChem 2014 Volume 15( Issue 6) pp:1138-1147
Publication Date(Web):
DOI:10.1002/cphc.201400063
Abstract
Linkers that favor rectification of interfacial electron transfer are likely to be required for efficient photo-driven catalysis of multi-electron reactions at electrode surfaces. Design principles are discussed, together with the synthesis and characterization of a specific pair of molecular linkers, related by inversion of the direction of an amide bond in the heart of the molecule. The linkers have a terpyridyl group that can covalently bind Mn as in a well-known water oxidation catalyst and an acetylacetonate group that allows attachment to TiO2 surfaces. The appropriate choice of the sense of the amide linkage yields directionality of interfacial electron transfer, essential to enhance electron injection and slow back-electron transfer. Support comes from electron paramagnetic resonance and terahertz spectroscopic measurements, as well as computational modeling characterizing the asymmetry of electron transfer properties.
Co-reporter:Wendu Ding ; Christian F. A. Negre ; Leslie Vogt
The Journal of Physical Chemistry C 2014 Volume 118(Issue 16) pp:8316-8321
Publication Date(Web):April 4, 2014
DOI:10.1021/jp503193m
Understanding charge transport across single molecular junctions is essential for the rational design and optimization of molecular device components. However, the correlation between calculated and experimental transport properties of single molecules probed by current–voltage (I–V) characteristics is often uncertain. Part of the challenge is that molecular conductance is sensitive to several factors that are difficult to control, including molecular orientation, conformation, aggregation, and chemical stability. Other challenges include the limitations of computational methodologies. Here, we implement the Σ-Extended Hückel (EH) nonequilibrium Green’s function (NEGF) method to analyze the histogram of I–V curves of 4,4′-diaminostilbene probed by break-junction experiments. We elucidate the nature of the molecular conformations with a widespread distribution of I–V curves, typically probed under experimental conditions. We find maximum conductance for molecules that are not at the minimum energy configuration but rather are aligned almost parallel to the transport direction. The increased conductance is due to the more favorable electronic coupling between the transport channel state and the electronic states in the contacts, as indicated by the broadening of bands in the transmission function near the Fermi level. These findings provide valuable guidelines for the design of anchoring groups that stabilize conformations of molecular assemblies with optimal charge transport properties.
Co-reporter:Sivakumar Sekharan ; Victoria L. Mooney ; Ivan Rivalta ; Manija A. Kazmi ; Maureen Neitz ; Jay Neitz ; Thomas P. Sakmar ; Elsa C. Y. Yan
Journal of the American Chemical Society 2013 Volume 135(Issue 51) pp:19064-19067
Publication Date(Web):December 2, 2013
DOI:10.1021/ja409896y
Ultraviolet (UV) cone pigments can provide insights into the molecular evolution of vertebrate vision since they are nearer to ancestral pigments than the dim-light rod photoreceptor rhodopsin. While visible-absorbing pigments contain an 11-cis retinyl chromophore with a protonated Schiff-base (PSB11), UV pigments uniquely contain an unprotonated Schiff-base (USB11). Upon F86Y mutation in model UV pigments, both the USB11 and PSB11 forms of the chromophore are found to coexist at physiological pH. The origin of this intriguing equilibrium remains to be understood at the molecular level. Here, we address this phenomenon and the role of the USB11 environment in spectral tuning by combining mutagenesis studies with spectroscopic (UV–vis) and theoretical [DFT-QM/MM (SORCI+Q//B3LYP/6-31G(d): Amber96)] analysis. We compare structural models of the wild-type (WT), F86Y, S90A and S90C mutants of Siberian hamster ultraviolet (SHUV) cone pigment to explore structural rearrangements that stabilize USB11 over PSB11. We find that the PSB11 forms upon F86Y mutation and is stabilized by an “inter-helical lock” (IHL) established by hydrogen-bonding networks between transmembrane (TM) helices TM6, TM2, and TM3 (including water w2c and amino acid residues Y265, F86Y, G117, S118, A114, and E113). The findings implicate the involvement of the IHL in constraining the displacement of TM6, an essential component of the activation of rhodopsin, in the spectral tuning of UV pigments.
Co-reporter:Li Fu ; Dequan Xiao ; Zhuguang Wang ; Victor S. Batista ;Elsa C. Y. Yan
Journal of the American Chemical Society 2013 Volume 135(Issue 9) pp:3592-3598
Publication Date(Web):February 8, 2013
DOI:10.1021/ja3119527
Studying hydrogen/deuterium (H/D) exchange in proteins can provide valuable insight on protein structure and dynamics. Several techniques are available for probing H/D exchange in the bulk solution, including NMR, mass spectroscopy, and Fourier transform infrared spectroscopy. However, probing H/D exchange at interfaces is challenging because it requires surface-selective methods. Here, we introduce the combination of in situ chiral sum frequency generation (cSFG) spectroscopy and ab initio simulations of cSFG spectra as a powerful methodology to probe the dynamics of H/D exchange at interfaces. This method is applied to characterize H/D exchange in the antiparallel β-sheet peptide LK7β. We report here for the first time that the rate of D-to-H exchange is about 1 order of magnitude faster than H-to-D exchange in the antiparallel structure at the air/water interface, which is consistent with the existing knowledge that O–H/D dissociation in water is the rate-limiting step, and breaking the O–D bond is slower than breaking the O–H bond. The reported analysis also provides fundamental understanding of several vibrational modes and their couplings in peptide backbones that have been difficult to characterize by conventional methods, including Fermi resonances of various combinations of peptide vibrational modes such as amide I and amide II, C–N stretch, and N–H/N–D bending. These results demonstrate cSFG as a sensitive technique for probing the kinetics of H/D exchange in proteins at interfaces, with high signal-to-noise N–H/N–D stretch bands that are free of background from the water O–H/O–D stretch.
Co-reporter:Rhitankar Pal ; Sivakumar Sekharan
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9624-9627
Publication Date(Web):June 18, 2013
DOI:10.1021/ja404600z
The spectral tuning of halorhodopsin from Halobacterium salinarum (shR) during anion transport was analyzed at the molecular level using DFT-QM/MM [SORCI+Q//B3LYP/6-31G(d):Amber96] hybrid methods. Insights into the influence of Cl– depletion, Cl– substitution by N3– or NO3–, and mutation of key amino acid residues along the ion translocation pathway (H95A, H95R, Q105E, R108H, R108I, R108K, R108Q, T111V, R200A, R200H, R200K, R200Q, and T203V) were analyzed for the first time in a fully atomistic model of the shR photoreceptor. We found evidence that structural rearrangements mediated by specific hydrogen bonds of internal water molecules and counterions (D238 and Cl–) in the active site induce changes in the bond-length alternation of the all-trans retinyl chromophore and affect the wavelength of maximal absorption in shR.
Co-reporter:James D. Blakemore, Michael W. Mara, Maxwell N. Kushner-Lenhoff, Nathan D. Schley, Steven J. Konezny, Ivan Rivalta, Christian F. A. Negre, Robert C. Snoeberger, Oleksandr Kokhan, Jier Huang, Andrew Stickrath, Lan Anh Tran, Maria L. Parr, Lin X. Chen, David M. Tiede, Victor S. Batista, Robert H. Crabtree, and Gary W. Brudvig
Inorganic Chemistry 2013 Volume 52(Issue 4) pp:1860-1871
Publication Date(Web):February 5, 2013
DOI:10.1021/ic301968j
Upon electrochemical oxidation of the precursor complexes [Cp*Ir(H2O)3]SO4 (1) or [(Cp*Ir)2(OH)3]OH (2) (Cp* = pentamethylcyclopentadienyl), a blue layer of amorphous iridium oxide containing a carbon admixture (BL) is deposited onto the anode. The solid-state, amorphous iridium oxide material that is formed from the molecular precursors is significantly more active for water-oxidation catalysis than crystalline IrO2 and functions as a remarkably robust catalyst, capable of catalyzing water oxidation without deactivation or significant corrosion for at least 70 h. Elemental analysis reveals that BL contains carbon that is derived from the Cp* ligand (∼ 3% by mass after prolonged electrolysis). Because the electrodeposition of precursors 1 or 2 gives a highly active catalyst material, and electrochemical oxidation of other iridium complexes seems not to result in immediate conversion to iridium oxide materials, we investigate here the nature of the deposited material. The steps leading to the formation of BL and its structure have been investigated by a combination of spectroscopic and theoretical methods. IR spectroscopy shows that the carbon content of BL, while containing some C–H bonds intact at short times, is composed primarily of components with C═O fragments at longer times. X-ray absorption and X-ray absorption fine structure show that, on average, the six ligands to iridium in BL are likely oxygen atoms, consistent with formation of iridium oxide under the oxidizing conditions. High-energy X-ray scattering (HEXS) and pair distribution function (PDF) analysis (obtained ex situ on powder samples) show that BL is largely free of the molecular precursors and is composed of small, <7 Å, iridium oxide domains. Density functional theory (DFT) modeling of the X-ray data suggests a limited set of final components in BL; ketomalonate has been chosen as a model fragment because it gives a good fit to the HEXS-PDF data and is a potential decomposition product of Cp*.
Co-reporter:Timothy P. Brewster, Steven J. Konezny, Stafford W. Sheehan, Lauren A. Martini, Charles A. Schmuttenmaer, Victor S. Batista, and Robert H. Crabtree
Inorganic Chemistry 2013 Volume 52(Issue 11) pp:6752-6764
Publication Date(Web):May 21, 2013
DOI:10.1021/ic4010856
We present the first analysis of performance of hydroxamate linkers as compared to carboxylate and phosphonate groups when anchoring ruthenium-polypyridyl dyes to TiO2 surfaces in dye-sensitized solar cells (DSSCs). The study provides fundamental insight into structure/function relationships that are critical for cell performance. Our DSSCs have been produced by using newly synthesized dye molecules and characterized by combining measurements and simulations of experimental current density–voltage (J-V) characteristic curves. We show that the choice of anchoring group has a direct effect on the overall sunlight-to-electricity conversion efficiency (η), with hydroxamate anchors showing the best performance. Solar cells based on the pyridyl-hydroxamate complex exhibit higher efficiency since they suppress electron transfer from the photoanode to the electrolyte and have superior photoinjection characteristics. These findings suggest that hydroxamate anchoring groups should be particularly valuable in DSSCs and photocatalytic applications based on molecular adsorbates covalently bound to semiconductor surfaces. In contrast, analogous acetylacetonate anchors might undergo decomposition under similar conditions suggesting limited potential in future applications.
Co-reporter:Oana R. Luca, Steven J. Konezny, Eric K. Paulson, Fatemah Habib, Kurt M. Luthy, Muralee Murugesu, Robert H. Crabtree and Victor S. Batista
Dalton Transactions 2013 vol. 42(Issue 24) pp:8802-8807
Publication Date(Web):03 May 2013
DOI:10.1039/C3DT50528F
A tridentate NNN NiII complex, shown to be an electrocatalyst for aqueous H2 production at low overpotentials, is studied by using temperature-dependent paramagnetic 1H NMR. The NMR T1 relaxation rates, temperature dependence of the chemical shifts, and dc SQUID magnetic susceptibility are correlated to DFT chemical shifts and compared with the properties of a diamagnetic Zn analogue complex. The resulting characterization provides an unambiguous assignment of the six proton environments in the meridionally coordinating tridentate NNN ligand. The demonstrated NMR/DFT methodology should be valuable in the search for appropriate ligands to optimize the reactivity of 3d metal complexes bound to attract increasing attention in catalytic applications.
Co-reporter:Rhitankar Pal, Christian F. A. Negre, Leslie Vogt, Ravi Pokhrel, Mehmed Z. Ertem, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2013 Volume 52(Issue 44) pp:
Publication Date(Web):October 15, 2013
DOI:10.1021/bi401214v
The S0 → S1 transition of the oxygen-evolving complex (OEC) of photosystem II is one of the least understood steps in the Kok cycle of water splitting. We introduce a quantum mechanics/molecular mechanics (QM/MM) model of the S0 state that is consistent with extended X-ray absorption fine structure spectroscopy and X-ray diffraction data. In conjunction with the QM/MM model of the S1 state, we address the proton-coupled electron-transfer (PCET) process that occurs during the S0 → S1 transition, where oxidation of a Mn center and deprotonation of a μ-oxo bridge lead to a significant rearrangement in the OEC. A hydrogen bonding network, linking the D1-D61 residue to a Mn-bound water molecule, is proposed to facilitate the PCET mechanism.
Co-reporter:Christian F. A. Negre, Rebecca L. Milot, Lauren A. Martini, Wendu Ding, Robert H. Crabtree, Charles A. Schmuttenmaer, and Victor S. Batista
The Journal of Physical Chemistry C 2013 Volume 117(Issue 46) pp:24462-24470
Publication Date(Web):October 23, 2013
DOI:10.1021/jp408738b
High performance dye-sensitized solar cells (DSSCs) rely upon molecular linkers that allow efficient electron transport from the photoexcited dye into the conduction band of the semiconductor host substrate. We have studied photoinduced electron injection efficiencies from modular assemblies of a Zn-porphyrin dye and a series of linker molecules which are axially bound to the Zn-porphyrin complex and covalently bound to TiO2 nanoparticles. Experimental measurements based on terahertz spectroscopy are compared to the calculated molecular conductance of the linker molecules. We find a linear relationship between measured electron injection efficiency and calculated single-molecule conductance of the linker employed. Since the same chromophore is used in all cases, variations in the absorptivities of the adsorbate complexes are quite small and cannot account for the large variations in observed injection efficiencies. These results suggest that the linker single-molecule conductance is a key factor that should be optimized for maximum electron injection efficiencies in DSSCs. In addition, our findings demonstrate for the first time the possibility of inferring values of single molecule conductance for bridging molecules at semiconductor interfaces by using time-resolved THz spectroscopy.
Co-reporter:Sandra Luber, Katrin Adamczyk, Erik T. J. Nibbering, and Victor S. Batista
The Journal of Physical Chemistry A 2013 Volume 117(Issue 25) pp:5269-5279
Publication Date(Web):May 29, 2013
DOI:10.1021/jp403342w
We characterize the structural and electronic changes during the photoinduced enol–keto tautomerization of 2-(2′-hydroxyphenyl)-benzothiazole (HBT) in a nonpolar solvent (tetrachloroethene). We quantify the redistribution of electronic charge and intramolecular proton translocation in real time by combining UV-pump/IR-probe spectroscopy and quantum chemical modeling. We find that the photophysics of this prototypical molecule involves proton coupled electron transfer (PCET), from the hydroxyphenyl to the benzothiazole rings, resulting from excited state intramolecular proton transfer (ESIPT) coupled to electron transfer through the conjugated double bond linking the two rings. The combination of polarization-resolved mid-infrared spectroscopy of marker modes and time-dependent density functional theory (TD-DFT) provides key insights into the transient structures of the molecular chromophore during ultrafast isomerization dynamics.
Co-reporter:Muhamed Amin, Leslie Vogt, Serguei Vassiliev, Ivan Rivalta, Mohammad M. Sultan, Doug Bruce, Gary W. Brudvig, Victor S. Batista, and M. R. Gunner
The Journal of Physical Chemistry B 2013 Volume 117(Issue 20) pp:6217-6226
Publication Date(Web):April 9, 2013
DOI:10.1021/jp403321b
The influence of electrostatic interactions on the free energy of proton coupled electron transfer in biomimetic oxomanganese complexes inspired by the oxygen-evolving complex (OEC) of photosystem II (PSII) are investigated. The reported study introduces an enhanced multiconformer continuum electrostatics (MCCE) model, parametrized at the density functional theory (DFT) level with a classical valence model for the oxomanganese core. The calculated pKa’s and oxidation midpoint potentials (Em’s) match experimental values for eight complexes, indicating that purely electrostatic contributions account for most of the observed couplings between deprotonation and oxidation state transitions. We focus on pKa’s of terminal water ligands in [Mn(II/III)(H2O)6]2+/3+ (1), [Mn(III)(P)(H2O)2]3– (2, P = 5,10,15,20-tetrakis(2,6-dichloro-3-sulfonatophenyl)porphyrinato), [Mn2(IV,IV)(μ-O)2(terpy)2(H2O)2]4+ (3, terpy = 2,2′:6′,2″-terpyridine), and [Mn3(IV,IV,IV)(μ-O)4(phen)4(H2O)2]4+ (4, phen = 1,10-phenanthroline) and the pKa’s of μ-oxo bridges and Mn Em’s in [Mn2(μ-O)2(bpy)4] (5, bpy = 2,2′-bipyridyl), [Mn2(μ-O)2(salpn)2] (6, salpn = N,N′-bis(salicylidene)-1,3-propanediamine), [Mn2(μ-O)2(3,5-di(Cl)-salpn)2] (7), and [Mn2(μ-O)2(3,5-di(NO2)-salpn)2] (8). The analysis of complexes 6–8 highlights the strong coupling between electron and proton transfers, with any Mn oxidation lowering the pKa of an oxo bridge by 10.5 ± 0.9 pH units. The model also accounts for changes in the Em’s by ligand substituents, such as found in complexes 6–8, due to the electron withdrawing Cl (7) and NO2 (8). The reported study provides the foundation for analysis of electrostatic effects in other oxomanganese complexes and metalloenzymes, where proton coupled electron transfer plays a fundamental role in redox-leveling mechanisms.
Co-reporter:C. Moyses Araujo, Davide L. Simone, Steven J. Konezny, Aaron Shim, Robert H. Crabtree, Grigorii L. Soloveichik and Victor S. Batista
Energy & Environmental Science 2012 vol. 5(Issue 11) pp:9534-9542
Publication Date(Web):29 Aug 2012
DOI:10.1039/C2EE22749E
Our work focuses on the feasibility of utilizing organic fuels for virtual hydrogen flow cell battery systems, based on thermodynamic considerations of fuel hydrogenation/dehydrogenation reactions. An assessment of the energy density and open circuit potentials (OCPs) as determined by the structure of carbocyclic and heterocyclic saturated hydrocarbons and their dehydrogenation products has been pursued and we identified promising organic carriers that could yield theoretical OCPs higher than that for the hydrogen fuel cell.
Co-reporter:Karin J. Young, Lauren A. Martini, Rebecca L. Milot, Robert C. Snoeberger III, Victor S. Batista, Charles A. Schmuttenmaer, Robert H. Crabtree, Gary W. Brudvig
Coordination Chemistry Reviews 2012 Volume 256(21–22) pp:2503-2520
Publication Date(Web):November 2012
DOI:10.1016/j.ccr.2012.03.031
Light-driven water oxidation is an essential step for conversion of sunlight into storable chemical fuels. Fujishima and Honda reported the first example of photoelectrochemical water oxidation in 1972. In their system, TiO2 was irradiated with ultraviolet light, producing oxygen at the anode and hydrogen at a platinum cathode. Inspired by this system, more recent work has focused on functionalizing nanoporous TiO2 or other semiconductor surfaces with molecular adsorbates, including chromophores and catalysts that absorb visible light and generate electricity (i.e., dye-sensitized solar cells) or trigger water oxidation at low overpotentials (i.e., photocatalytic cells). The physics involved in harnessing multiple photochemical events for multi-electron reactions, as required in the four-electron water-oxidation process, has been the subject of much experimental and computational study. In spite of significant advances with regard to individual components, the development of highly efficient photocatalytic cells for solar water splitting remains an outstanding challenge. This article reviews recent progress in the field with emphasis on water-oxidation photoanodes inspired by the design of functionalized thin-film semiconductors of typical dye-sensitized solar cells.Graphical abstract.Highlights► Anodes for light-driven water oxidation. ► Design includes semiconductor, light-harvesting molecule, and catalyst. ► Integration of components is greatest challenge.
Co-reporter:Sivakumar Sekharan ; Jennifer N. Wei
Journal of the American Chemical Society 2012 Volume 134(Issue 48) pp:19536-19539
Publication Date(Web):November 12, 2012
DOI:10.1021/ja308763b
The nonvisual ocular photoreceptor melanopsin, found in the neurons of vertebrate inner retina, absorbs blue light and triggers the “biological clock” of mammals by activating the suprachiasmatic nuclei (a small region of the brain that regulates the circadian rhythms of neuronal and hormonal activities over 24 h cycles). The structure of melanopsin, however, has yet to be established. Here, we propose for the first time a structural model of the active site of mouse melanopsin. The homology model is based on the crystal structure of squid rhodopsin (λmax = 490 nm) and shows a maximal absorbance (λmax = 447 nm) consistent with the observed absorption of the photoreceptor. The 43 nm spectral shift is due to an increased bond-length alternation of the protonated Schiff base of 11-cis-retinal chromophore, induced by N87Q mutation and water-mediated H-bonding interactions with the Schiff base linkage. These findings, analogous to spectral changes observed in the G89Q bovine rhodopsin mutant, suggest that single site mutations can convert photopigments into visual light sensors or nonvisual sensory photoreceptors.
Co-reporter:Robert C. Snoeberger ; III; Karin J. Young ; Jiji Tang ; Laura J. Allen ; Robert H. Crabtree ; Gary W. Brudvig ; Philip Coppens ; Victor; S. Batista ;Jason B. Benedict
Journal of the American Chemical Society 2012 Volume 134(Issue 21) pp:8911-8917
Publication Date(Web):May 1, 2012
DOI:10.1021/ja301238t
Interfacial electron transfer (IET) between a chromophore and a semiconductor nanoparticle is one of the key processes in a dye-sensitized solar cell. Theoretical simulations of the electron transfer in polyoxotitanate nanoclusters Ti17O24(OPri)20 (Ti17) functionalized with four p-nitrophenyl acetylacetone (NPA-H) adsorbates, of which the atomic structure has been fully established by X-ray diffraction measurements, are presented. Complementary experimental information showing IET has been obtained by EPR spectroscopy. Evolution of the time-dependent photoexcited electron during the initial 5 fs after instantaneous excitation to the NPA LUMO + 1 has been evaluated. Evidence for delocalization of the excitation over multiple chromophores after excitation to the NPA LUMO + 2 state on a 15 fs time scale is also obtained. While chromophores are generally considered electronically isolated with respect to neighboring sensitizers, our calculations show that this is not necessarily the case. The present work is the most comprehensive study to date of a sensitized semiconductor nanoparticle in which the structure of the surface and the mode of molecular adsorption are precisely defined.
Co-reporter:Andreas Markmann, Frank Graziani, and Victor S. Batista
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 1) pp:24-35
Publication Date(Web):November 8, 2011
DOI:10.1021/ct200452h
An accurate and efficient algorithm for dynamics simulations of particles with attractive 1/r singular potentials is introduced. The method is applied to semiclassical dynamics simulations of electron–proton scattering processes in the Wigner-transform time-dependent picture, showing excellent agreement with full quantum dynamics calculations. Rather than avoiding the singularity problem by using a pseudopotential, the algorithm predicts the outcome of close-encounter two-body collisions for the true 1/r potential by solving the Kepler problem analytically and corrects the trajectory for multiscattering with other particles in the system by using standard numerical techniques (e.g., velocity Verlet, or Gear Predictor corrector algorithms). The resulting integration is time-reversal symmetric and can be applied to the general multibody dynamics problem featuring close encounters as occur in electron–ion scattering events, in particle–antiparticle dynamics, as well as in classical simulations of charged interstellar gas dynamics and gravitational celestial mechanics.
Co-reporter:Oana R. Luca, James D. Blakemore, Steven J. Konezny, Jeremy M. Praetorius, Timothy J. Schmeier, Glendon B. Hunsinger, Victor S. Batista, Gary W. Brudvig, Nilay Hazari, and Robert H. Crabtree
Inorganic Chemistry 2012 Volume 51(Issue 16) pp:8704-8709
Publication Date(Web):July 31, 2012
DOI:10.1021/ic300009a
Nonplatinum metals are needed to perform cost-effective water reduction electrocatalysis to enable technological implementation of a proposed hydrogen economy. We describe electrocatalytic proton reduction and H2 production by two organometallic nickel complexes with tridentate pincer ligands. The kinetics of H2 production from voltammetry is consistent with an overall third order rate law: the reaction is second order in acid and first order in catalyst. Hydrogen production with 90–95% Faradaic yields was confirmed by gas analysis, and UV–vis spectroscopy suggests that the ligand remains bound to the catalyst over the course of the reaction. A computational study provides mechanistic insights into the proposed catalytic cycle. Furthermore, two proposed intermediates in the proton reduction cycle were isolated in a representative system and show a catalytic response akin to the parent compound.
Co-reporter:Louise M. Guard, Julio L. Palma, William P. Stratton, Laura J. Allen, Gary W. Brudvig, Robert H. Crabtree, Victor S. Batista and Nilay Hazari
Dalton Transactions 2012 vol. 41(Issue 26) pp:8098-8110
Publication Date(Web):13 Feb 2012
DOI:10.1039/C2DT12426B
The reactions of the substituted 2,2′:6,2′′-terpyridine ligands, 4′-mesityl-2,2′:6′,2′′-terpyridine (mesitylterpy) (1a), 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine (tri-tButerpy) (1b) and 4′-phenyl-2,2′:6′,2′′-terpyridine (phenylterpy) (1c) with Grignard reagents were investigated. When half an equivalent of mesitylterpy or tri-tButerpy were treated with MeMgBr in diethyl ether, the only products were (R-terpy)MgBr2 (R = mesityl (5a), or tri-tBu (5b)) and Me2Mg and a similar reaction was observed in THF. Compounds 5a and 5b were characterized by X-ray crystallography. Changing the Grignard reagent to PhMgBr also generated 5a and 5b along with Ph2Mg, while the reaction between MeMgCl or PhMgCl and 1a or 1b generated (R-terpy)MgCl2 (R = mesityl (6a), or tri-tBu (6b)) and either Me2Mg or Ph2Mg, respectively. The products from reactions between phenylterpy (1c) and Grignard reagents were highly insoluble and could not be fully characterized but appeared to be the same as those from reactions with 1a and 1b. In contrast to other studies using tridentate nitrogen ligands, which formed either mixed halide alkyl species or dihalide and bis(alkyl) species depending on whether the Grignard reagent was reacted with the ligand in diethyl ether or THF, the formation of mixed halide, alkyl complexes of the type (R-terpy)MgR′X (R′ = Me or Ph; X = Cl or Br) or dialkyl species such as (R-terpy)MgR′2 (R′ = Me or Ph) was not observed here, regardless of the reaction conditions. DFT studies were performed to complement the experimental studies. The experimental results could not be accurately reproduced unless π-stacking effects associated with free terpyridine were included in the model. When these effects were included, the calculations were consistent with the experimental results which indicated that the formation of the terpy Mg dihalide species and R′2Mg (R′ = Me or Ph) is thermodynamically preferred over the formation of mixed alkyl halide Mg species. This is proposed to be due to the increased steric bulk of the terpy ligand in the coordination plane, compared with other tridentate nitrogen donors.
Co-reporter:C. Moyses Araujo, Mark D. Doherty, Steven J. Konezny, Oana R. Luca, Alex Usyatinsky, Hans Grade, Emil Lobkovsky, Grigorii L. Soloveichik, Robert H. Crabtree and Victor S. Batista
Dalton Transactions 2012 vol. 41(Issue 12) pp:3562-3573
Publication Date(Web):08 Feb 2012
DOI:10.1039/C2DT12195F
The structure and electrochemical properties of a series of bis(imino)pyridine CoII complexes (NNN)CoX2 and [(NNN)2Co][PF6]2 (NNN = 2,6-bis[1-(4-R-phenylimino)ethyl]pyridine, with R = CN, CF3, H, CH3, OCH3, N(CH3)2; NNN = 2,6-bis[1-(2,6-(iPr)2-phenylimino)ethyl]pyridine and X = Cl, Br) were studied using a combination of electrochemical and theoretical methods. Cyclic voltammetry measurements and DFT/B3LYP calculations suggest that in solution (NNN)CoCl2 complexes exist in equilibrium with disproportionation products [(NNN)2Co]2+ [CoCl4]2− with the position of the equilibrium heavily influenced by both the solvent polarity and the steric and electronic properties of the bis(imino)pyridine ligands. In strong polar solvents (e.g., CH3CN or H2O) or with electron donating substituents (R = OCH3 or N(CH3)2) the equilibrium is shifted and only oxidation of the charged products [(NNN)2Co]2+ and [CoCl4]2− is observed. Conversely, in nonpolar organic solvents such as CH2Cl2 or with electron withdrawing substituents (R = CN or CF3), disproportionation is suppressed and oxidation of the (NNN)CoCl2 complexes leads to 18e− CoIII complexes stabilized by coordination of a solvent moiety. In addition, the [(NNN)2Co][PF6]2 complexes exhibit reversible CoII/III oxidation potentials that are strongly dependent on the electron withdrawing/donating nature of the N-aryl substituents, spanning nearly 750 mV in acetonitrile. The resulting insight on the regulation of redox properties of a series of bis(imino)pyridine cobalt(II) complexes should be particularly valuable to tune suitable conditions for reactivity.
Co-reporter:Oana R. Luca, Steven J. Konezny, James D. Blakemore, Dominic M. Colosi, Shubhro Saha, Gary W. Brudvig, Victor S. Batista and Robert H. Crabtree
New Journal of Chemistry 2012 vol. 36(Issue 5) pp:1149-1152
Publication Date(Web):19 Mar 2012
DOI:10.1039/C2NJ20912H
A NiII complex with a redox-active pincer ligand reduces protons at a low overpotential in aqueous acidic conditions. A combined experimental and computational study provides mechanistic insights into a putative catalytic cycle.
Co-reporter:Victor S. Batista, Robert H. Crabtree, Steven J. Konezny, Oana R. Luca and Jeremy M. Praetorius
New Journal of Chemistry 2012 vol. 36(Issue 5) pp:1141-1144
Publication Date(Web):08 Mar 2012
DOI:10.1039/C2NJ40021A
C–H activation of the methyl group of toluene and related ArCH3 derivatives by 2,3-dichloro-4,5-dicyano-1,4-benzoquinone (DDQ) gives insertion products, ArCH2O[C6Cl2(CN)2]OH via a rate-determining hydride abstraction by DDQ. The resulting benzylic ether can undergo reactions with phosphines to give benzylic phosphonium salts (Wittig reagents) and with phosphites to give phosphonate esters (Horner–Wadsworth–Emmons reagents).
Co-reporter:Dequan Xiao, Mirabelle Prémont-Schwarz, Erik T. J. Nibbering, and Victor S. Batista
The Journal of Physical Chemistry A 2012 Volume 116(Issue 11) pp:2775-2790
Publication Date(Web):November 1, 2011
DOI:10.1021/jp208426v
We study the solvent-induced frequency shifts of the OH-stretching mode of 1-naphthol and 2-naphthol in nonpolar/weakly polar solvents, subject to electronic excitation, with ultrafast UV/mid-infrared pump–probe spectroscopy and theoretical modeling based on Pullin’s perturbative treatment of vibrational solvatochromic effects. The model is parametrized at the density functional theory (DFT) level, including the B3LYP/TZVP and TD-B3LYP/TZVP descriptions, for the naphthol chromophores in the S0- and 1Lb-states and accounts for both the static and the optical dielectric response of the solvent on time scales comparable to that of the OH-stretching vibrational motions. The favorable comparison between experimental and theoretical values of the solvent-induced vibrational frequency shifts suggests that the ultrafast dielectric response of the solvent contributes predominantly to the solvatochromic shifts in solvents of moderate polarity where specific solute–solvent interactions are absent.
Co-reporter:Ivan Rivalta;Mohammad M. Sultan;Ning-Shiuan Lee;Gregory A. Manley;J. Patrick Loria
PNAS 2012 Volume 109 (Issue 22 ) pp:
Publication Date(Web):2012-05-29
DOI:10.1073/pnas.1120536109
Protein allosteric pathways are investigated in the imidazole glycerol phosphate synthase heterodimer in an effort to elucidate
how the effector (PRFAR, N′-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide) activates glutaminase
catalysis at a distance of 25 Å from the glutamine-binding site. We apply solution NMR techniques and community analysis of
dynamical networks, based on mutual information of correlated protein motions in the active and inactive enzymes. We find
evidence that the allosteric pathways in the PRFAR bound enzyme involve conserved residues that correlate motion of the PRFAR
binding loop to motion at the protein-protein interface, and ultimately at the glutaminase active site. The imidazole glycerol
phosphate synthase bienzyme is an important branch point for the histidine and nucleotide biosynthetic pathways and represents
a potential therapeutic target against microbes. The proposed allosteric mechanism and the underlying allosteric pathways
provide fundamental insights for the design of new allosteric drugs and/or alternative herbicides.
Co-reporter:Gary F. Moore ; Steven J. Konezny ; Hee-eun Song ; Rebecca L. Milot ; James D. Blakemore ; Minjoo L. Lee ; Victor S. Batista ; Charles A. Schmuttenmaer ; Robert H. Crabtree ;Gary W. Brudvig
The Journal of Physical Chemistry C 2012 Volume 116(Issue 7) pp:4892-4902
Publication Date(Web):January 23, 2012
DOI:10.1021/jp210096m
We report a selection of high-potential porphyrin photoanodes (HPPPs) for use in photoelectrochemical cells (PECs). The anodes consist of bispentafluorophenyl free-base and metallo-porphyrin sensitizers bearing anchoring groups for attachment to metal-oxide surfaces including TiO2 and SnO2 nanoparticles. The term “high potential” refers to the relatively large and positive value of the electrochemical reduction potential for the bispentafluorophenyl porphyrin radical cation (P•+ + e– → P) as compared with more conventional nonfluorinated analogues. Photoelectrochemical measurements demonstrate the sensitizers used in these HPPPs extend the absorption of the bare anode well into the visible region. Terahertz spectroscopic studies show the photoexcited dyes are capable of injecting electrons into the conduction band of an underlying metal-oxide with appropriate energetics. The reduction potentials of the resulting photogenerated porphyrin radical cations are relatively high (ranging from ∼1.35 to 1.65 V vs NHE depending on the sensitizer). This is demonstrated by the ability of dye-sensitized solar cells, containing our HPPPs, to use the Br3–/Br– redox couple as a regenerative electron mediator with superior performance in comparison to results obtained using the lower-potential I3–/I– relay. Computational modeling of the structures and equivalent circuits assists in a molecular-based understanding of these systems. Further, the oxidation power of the porphyrin radical cations generated in these bioinspired constructs is similar to that found in the reaction centers of their natural counterpart (photosystem II); thus, HPPPs are promising as components in artificial systems for photochemical water spitting applications.
Co-reporter:Steven J. Konezny, Mark D. Doherty, Oana R. Luca, Robert H. Crabtree, Grigorii L. Soloveichik, and Victor S. Batista
The Journal of Physical Chemistry C 2012 Volume 116(Issue 10) pp:6349-6356
Publication Date(Web):February 21, 2012
DOI:10.1021/jp300485t
Reliable calculations of redox potentials could provide valuable insight into catalytic mechanisms of electrochemically active transition-metal complexes as well as guidelines for the design of new electrocatalysts. However, the correlation between theoretical and experimental data is often uncertain, since redox properties depend strongly on experimental conditions of electrochemical measurements, including the nature of the solvent, electrolyte, and working electrode. Here, we show that the use of internal references allows for quantitative theoretical predictions of redox potentials with standard deviations σ comparable to typical experimental errors of cyclic voltammetry measurements. Agreement for first-, second-, and third-row transition-metal complexes is demonstrated even at a rather modest level of density functional theory (σ = 64 mV for the UB3LYP/6-311G* level). This is shown for a series of benchmark redox couples, including ([MCp2]0/+ (Cp = η5-cyclopentadienyl), [MCp*2]0/+ (Cp* = η5-1,2,3,4,5-pentamethylcyclopentadienyl), [M(bpy)3]2+/3+ (bpy =2,2′-bipyridine), and [Ir(acac)3]0/+ (acac = acetylacetonate), with M = Fe, Co, Ni, Ru, Os, or Ir) in various nonaqueous solvents [acetonitrile (MeCN), dimethyl sulfoxide (DMSO), and dichloromethane (DCM)].
Co-reporter:Chantelle L. Anfuso ; Robert C. Snoeberger ; III; Allen M. Ricks ; Weimin Liu ; Dequan Xiao ; Victor S. Batista ;Tianquan Lian
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6922-6925
Publication Date(Web):April 19, 2011
DOI:10.1021/ja2013664
We have characterized the covalent binding of the CO2 reduction electrocatalyst ReC0A (Re(CO)3Cl(dcbpy) (dcbpy =4,4′-dicarboxy-2,2′-bipyridine)) to the TiO2 rutile (001) surface. The analysis based on sum frequency generation (SFG) spectroscopy and density functional theory (DFT) calculations indicates that ReC0A binds to TiO2 through the carboxylate groups in bidentate or tridentate linkage motifs. The adsorbed complex has the dcbpy moiety nearly perpendicular to the TiO2 surface and the Re exposed to the solution in a configuration suitable for catalysis.
Co-reporter:Dequan Xiao ; Lauren A. Martini ; Robert C. Snoeberger ; III; Robert H. Crabtree
Journal of the American Chemical Society 2011 Volume 133(Issue 23) pp:9014-9022
Publication Date(Web):May 9, 2011
DOI:10.1021/ja2020313
An inverse design methodology suitable to assist the synthesis and optimization of molecular sensitizers for dye-sensitized solar cells is introduced. The method searches for molecular adsorbates with suitable photoabsorption properties through continuous optimization of “alchemical” structures in the vicinity of a reference molecular framework. The approach is illustrated as applied to the design and optimization of linker chromophores for TiO2 sensitization, using the recently developed phenyl-acetylacetonate (i.e., phenyl-acac) anchor [McNamara et al. J. Am. Chem. Soc.2008, 130, 14329–14338] as a reference framework. A novel anchor (3-acac-pyran-2-one) is found to be a local optimum, with improved sensitization properties when compared to phenyl-acac. Its molecular structure is related to known coumarin dyes that could be used as lead chromophore anchors for practical applications in dye-sensitized solar cells. Synthesis and spectroscopic characterization confirms that the linker provides robust attachment to TiO2, even in aqueous conditions, yielding improved sensitization to solar light and ultrafast interfacial electron injection. The findings are particularly relevant to the design of sensitizers for dye-sensitized solar cells because of the wide variety of structures that are possible but they should be equally useful for other applications such as ligand design for homogeneous catalysis.
Co-reporter:Timothy P. Brewster ; Wendu Ding ; Nathan D. Schley ; Nilay Hazari ; Victor S. Batista ;Robert H. Crabtree
Inorganic Chemistry 2011 Volume 50(Issue 23) pp:11938-11946
Publication Date(Web):November 8, 2011
DOI:10.1021/ic200950e
Ruthenium polypyridyl complexes have seen extensive use in solar energy applications. One of the most efficient dye-sensitized solar cells produced to date employs the dye-sensitizer N719, a ruthenium polypyridyl thiocyanate complex. Thiocyanate complexes are typically present as an inseparable mixture of N-bound and S-bound linkage isomers. Here we report the synthesis of a new complex, [Ru(terpy)(tbbpy)SCN][SbF6] (terpy = 2,2′;6′,2″-terpyridine, tbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine), as a mixture of N-bound and S-bound thiocyanate linkage isomers that can be separated based on their relative solubility in ethanol. Both isomers have been characterized spectroscopically and by X-ray crystallography. At elevated temperatures the isomers equilibrate, the product being significantly enriched in the more thermodynamically stable N-bound form. Density functional theory analysis supports our experimental observation that the N-bound isomer is thermodynamically preferred, and provides insight into the isomerization mechanism.
Co-reporter:Oana R. Luca, Ting Wang, Steven J. Konezny, Victor S. Batista and Robert H. Crabtree
New Journal of Chemistry 2011 vol. 35(Issue 5) pp:998-999
Publication Date(Web):07 Apr 2011
DOI:10.1039/C0NJ01011A
2,3-Dichloro-5,6-dicyanobenzoquinone (DDQ) is an electrochemical oxidation catalyst for a secondary amine, a model system for virtual hydrogen storage by removal of a hydrogen equivalent from an amine; a computational study provides mechanistic information.
Co-reporter:Ivan Rivalta, Muhamed Amin, Sandra Luber, Serguei Vassiliev, Ravi Pokhrel, Yasufumi Umena, Keisuke Kawakami, Jian-Ren Shen, Nobuo Kamiya, Doug Bruce, Gary W. Brudvig, M. R. Gunner, and Victor S. Batista
Biochemistry 2011 Volume 50(Issue 29) pp:
Publication Date(Web):June 16, 2011
DOI:10.1021/bi200685w
Chloride binding in photosystem II (PSII) is essential for photosynthetic water oxidation. However, the functional roles of chloride and possible binding sites, during oxygen evolution, remain controversial. This paper examines the functions of chloride based on its binding site revealed in the X-ray crystal structure of PSII at 1.9 Å resolution. We find that chloride depletion induces formation of a salt bridge between D2-K317 and D1-D61 that could suppress the transfer of protons to the lumen.
Co-reporter:Sandra Luber, Ivan Rivalta, Yasufumi Umena, Keisuke Kawakami, Jian-Ren Shen, Nobuo Kamiya, Gary W. Brudvig, and Victor S. Batista
Biochemistry 2011 Volume 50(Issue 29) pp:
Publication Date(Web):June 16, 2011
DOI:10.1021/bi200681q
We introduce a quantum mechanics/molecular mechanics model of the oxygen-evolving complex of photosystem II in the S1 Mn4(IV,III,IV,III) state, where Ca2+ is bridged to manganese centers by the carboxylate moieties of D170 and A344 on the basis of the new X-ray diffraction (XRD) model recently reported at 1.9 Å resolution. The model is also consistent with high-resolution spectroscopic data, including polarized extended X-ray absorption fine structure data of oriented single crystals. Our results provide refined intermetallic distances within the Mn cluster and suggest that the XRD model most likely corresponds to a mixture of oxidation states, including species more reduced than those observed in the catalytic cycle of water splitting.
Co-reporter: Dr. Victor S. Batista; Dr. Stefan Grimme; Dr. Markus Reiher
ChemPhysChem 2011 Volume 12( Issue 17) pp:3043-3044
Publication Date(Web):
DOI:10.1002/cphc.201100862
No abstract is available for this article.
Co-reporter:Mirabelle Prémont-Schwarz, Dequan Xiao, Victor S. Batista, and Erik T. J. Nibbering
The Journal of Physical Chemistry A 2011 Volume 115(Issue 38) pp:10511-10516
Publication Date(Web):August 18, 2011
DOI:10.1021/jp207642k
We investigate the OH stretch vibrational frequency shifts of a prototype photoacid, 2-naphthol (2N), when dissolved in solvents of low polarity. We combine femtosecond mid-infrared spectroscopy and a theoretical model based on the Pullin–van der Zwan–Hynes perturbative approach to explore vibrational solvatochromic effects in the ground S0 and the first electronically excited 1Lb states. The model is parametrized using density functional theory (DFT), at the B3LYP/TZVP and TD-B3LYP/TZVP levels for the 2N chromophore in the S0 and 1Lb states, respectively. From the agreement between experiment and theory we conclude that vibrational solvatochromic effects are dominated by the instantaneous dielectric response of the solvent, while time-dependent nuclear rearrangements are of secondary importance.
Co-reporter:Steven J. Konezny, Christiaan Richter, Robert C. Snoeberger III, Alexander R. Parent, Gary W. Brudvig, Charles A. Schmuttenmaer, and Victor S. Batista
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 15) pp:1931-1936
Publication Date(Web):July 14, 2011
DOI:10.1021/jz200853v
The electronic mechanisms responsible for dark conductivity in nanoporous TiO2 thin films remain only partially understood, although they control the efficiency of charge transport in a wide range of technological applications. Measurements in the 78–335 K temperature range show DC conductivity values spanning over 4 orders of magnitude, with a high-temperature Arrhenius dependence that gradually changes into a temperature-independent plateau at low temperatures. We show evidence that a fluctuation-induced tunneling conductivity (FITC) mechanism is fully consistent with the experimental data. Quantitative agreement is demonstrated for the entire temperature range (T = 78–335 K) with a FITC model parametrized according to atomistic models of nanoporous TiO2 and the characterization of the films by X-ray diffraction and scanning electron microscopy measurements. These findings suggest that dark DC conductivity in nanoporous TiO2 films depends strongly on the properties of the junctions linking the constituent nanoparticles.Keywords: charge transport; chemical sensors; DC conductivity; nanoporous TiO2; photocatalytic cells; solar cells; tunneling;
Co-reporter:Rajdeep Saha and Victor S. Batista
The Journal of Physical Chemistry B 2011 Volume 115(Issue 18) pp:5234-5242
Publication Date(Web):March 9, 2011
DOI:10.1021/jp108331x
A general coherent control scenario to suppress or accelerate tunneling of quantum states decaying into a continuum is investigated. The method is based on deterministic, or stochastic, sequences of unitary pulses that affect the underlying interference phenomena responsible for quantum dynamics, without inducing decoherence, or collapsing the coherent evolution of the system. The influence of control sequences on the ensuing quantum dynamics is analyzed by using perturbation theory to first order in the control pulse fields and compared to dynamical decoupling protocols and to sequences of pulses that collapse the coherent evolution and induce quantum Zeno (QZE) or quantum anti-Zeno effects (AZE). The analysis reveals a subtle interplay between coherent and incoherent phenomena and demonstrates that dynamics analogous to the evolution due to QZE or AZE can be generated from stochastic sequences of unitary pulses when averaged over all possible realizations.
Co-reporter:William R. McNamara, Rebecca L. Milot, Hee-eun Song, Robert C. Snoeberger III, Victor S. Batista, Charles A. Schmuttenmaer, Gary W. Brudvig and Robert H. Crabtree
Energy & Environmental Science 2010 vol. 3(Issue 7) pp:917-923
Publication Date(Web):26 Apr 2010
DOI:10.1039/C001065K
A novel class of derivatized hydroxamic acid linkages for robust sensitization of TiO2 nanoparticles (NPs) under various aqueous conditions is described. The stability of linkages bound to metal oxides under various conditions is important in developing photocatalytic cells which incorporate transition metal complexes for solar energy conversion. In order to compare the standard carboxylate anchor to hydroxamates, two organic dyes differing only in anchoring groups were synthesized and attached to TiO2 NPs. At acidic, basic, and close to neutral pH, hydroxamic acid linkages resist detachment compared to the labile carboxylic acids. THz spectroscopy was used to compare ultrafast interfacial electron transfer (IET) into the conduction band of TiO2 for both linkages and found similar IET characteristics. Observable electron injection and stronger binding suggest that hydroxamates are a suitable class of anchors for designing water stable molecules for functionalizing TiO2.
Co-reporter:Ting Wang, Gary W. Brudvig and Victor S. Batista
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 8) pp:2395-2401
Publication Date(Web):July 15, 2010
DOI:10.1021/ct1002658
The oxomanganese complex [H2O(terpy)MnIII(μ-O)2MnIV(terpy)H2O]3+ (1, terpy = 2,2′:6-2′′-terpyridine) is a biomimetic model of the oxygen-evolving complex of photosystem II with terminal water ligands. When bound to TiO2 surfaces, 1 is activated by primary oxidants (e.g., Ce4+(aq) or oxone in acetate buffers) to catalyze the oxidation of water yielding O2 evolution [G. Li et al. Energy Environ. Sci. 2009, 2, 230−238]. The activation is thought to involve oxidation of the inorganic core [MnIII(μ-O)2MnIV]3+ to generate the [MnIV(μ-O)2MnIV]4+ state 1ox first and then the highly reactive Mn oxyl species MnIVO• through proton coupled electron transfer (PCET). Here, we investigate the step 1 → 1ox as compared to the analogous conversion in an oxomanganese complex without terminal water ligands, the [(bpy)2 MnIII(μ-O)2 MnIV (bpy)2]3+ complex (2, bpy = 2,2′-bipyridyl). We characterize the oxidation in terms of free energy calculations of redox potentials and pKa’s as directly compared to cyclic voltammogram measurements. We find that the pKa’s of terminal water ligands depend strongly on the oxidation states of the Mn centers, changing by ∼13 pH units (i.e., from 14 to 1) during the III,IV → IV,IV transition. Furthermore, we find that the oxidation potential of 1 is strongly dependent on pH (in contrast to the pH-independent redox potential of 2) as well as by coordination of Lewis base moieties (e.g., carboxylate groups) that competitively bind to Mn by exchange with terminal water ligands. The reported analysis of ligand binding free energies, pKa’s, and redox potentials indicates that the III,IV → IV,IV oxidation of 1 in the presence of acetate (AcO−) involves the following PCET: [H2O(terpy)MnIII(μ-O)2MnIV(terpy)AcO]2+ → [HO(terpy)MnIV(μ-O)2MnIV(terpy)AcO]2+ + H+ + e−.
Co-reporter:Ting Wang, Gary Brudvig and Victor S. Batista
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 3) pp:755-760
Publication Date(Web):January 29, 2010
DOI:10.1021/ct900615b
The capabilities and limitations of the Becke-3-Lee−Yang−Parr (B3LYP) density functional theory (DFT) for modeling proton coupled electron transfer (PCET) in the mixed-valence oxomanganese complex [(bpy)2MnIII(μ-O)2MnIV(bpy)2]3+ (1; bpy = 2,2′-bipyridyl) are analyzed. Complex 1 serves as a prototypical synthetic model for studies of redox processes analogous to those responsible for water oxidation in the oxygen-evolving complex (OEC) of photosystem II (PSII). DFT B3LYP free energy calculations of redox potentials and pKa’s are obtained according to the thermodynamic cycle formalism applied in conjunction with a continuum solvation model. We find that the pKa’s of the oxo-ligands depend strongly on the oxidation states of the complex, changing by approximately 10 pH units (i.e., from pH ∼ 2 to pH ∼ 12) upon III,IV → III,III reduction of complex 1. These computational results are consistent with the experimental pKa’s determined by solution magnetic susceptibility and near-IR spectroscopy as well as with the pH dependence of the redox potential reported by cyclic voltammogram measurements, suggesting that the III,IV → III,III reduction of complex 1 is coupled to protonation of the di-μ-oxo bridge as follows: [(bpy)2MnIII(μ-O)2MnIV(bpy)2]3+ + H+ + e− → [(bpy)2MnIII(μ-O)(μ-OH)MnIII(bpy)2]3+. It is thus natural to expect that analogous redox processes might strongly modulate the pKa’s of oxo and hydroxo/water ligands in the OEC of PSII, leading to deprotonation of the OEC upon oxidation state transitions.
Co-reporter:Michael D. Kurland, Michael B. Newcomer, Zita Peterlin, Kevin Ryan, Stuart Firestein and Victor S. Batista
Biochemistry 2010 Volume 49(Issue 30) pp:
Publication Date(Web):July 7, 2010
DOI:10.1021/bi100976w
The discrimination of n-alkyl-saturated aldehydes during the early stage of odorant recognition by the rat I7 olfactory receptor (OR-I7) is investigated. The concentrations of odorants necessary for 50% activation (or inhibition) of the OR-I7 are measured by calcium imaging recordings of dissociated rat olfactory sensory neurons, expressing the recombinant OR-I7 from an adenoviral vector. These are correlated with the corresponding binding free energies computed for a homology structural model of the OR-I7 built from the crystal structure of bovine visual rhodopsin at 2.2 Å resolution.
Co-reporter:Shengye Jin, Robert C. Snoeberger III, Abey Issac, David Stockwell, Victor S. Batista, and Tianquan Lian
The Journal of Physical Chemistry B 2010 Volume 114(Issue 45) pp:14309-14319
Publication Date(Web):March 12, 2010
DOI:10.1021/jp911662g
Photoinduced interfacial electron transfer (IET) in sulforhodamine B (SRhB)-aminosilane-Tin oxide (SnO2) nanoparticle donor−bridge−acceptor complexes has been studied on a single molecule and ensemble average level. On both SnO2 and ZrO2, the sum of single molecule fluorescence decays agree with the ensemble average results, suggesting complete sampling of molecules under single molecule conditions. Shorter fluorescence lifetime on SnO2 than on ZrO2 is observed and attributed to IET from SRhB to SnO2. Single molecule lifetimes fluctuate with time and vary among different molecules, suggesting both static and dynamic IET heterogeneity in this system. Computational modeling of the complexes shows a distribution of molecular conformation, leading to a distribution of electronic coupling strengths and ET rates. It is likely that the conversion between these conformations led to the fluctuation of ET rate and fluorescence lifetime on the single molecule level.
Co-reporter:Gonghu Li, Eduardo M. Sproviero, William R. McNamara, Robert C. Snoeberger III, Robert H. Crabtree, Gary W. Brudvig, and Victor S. Batista
The Journal of Physical Chemistry B 2010 Volume 114(Issue 45) pp:14214-14222
Publication Date(Web):November 19, 2009
DOI:10.1021/jp908925z
Several polynuclear transition-metal complexes, including our own dinuclear di-μ-oxo manganese compound [H2O(terpy)MnIII(μ-O)2MnIV(terpy)H2O](NO3)3 (1, terpy = 2,2′:6′,2′′-terpyridine), have been reported to be homogeneous catalysts for water oxidation. This paper reports the covalent attachment of 1 onto nanoparticulate TiO2 surfaces using a robust chromophoric linker L. L, a phenylterpy ligand attached to a 3-phenyl-acetylacetonate anchoring moiety via an amide bond, absorbs visible light and leads to photoinduced interfacial electron transfer into the TiO2 conduction band. We characterize the electronic and structural properties of the 1−L−TiO2 assemblies by using a combination of methods, including computational modeling and UV−visible, IR, and EPR spectroscopies. We show that the Mn(III,IV) state of 1 can be reversibly advanced to the Mn(IV,IV) state by visible-light photoexcitation of 1−L−TiO2 nanoparticles (NPs) and recombines back to the Mn(III,IV) state in the dark, in the absence of electron scavengers. Our findings also indicate that a high degree of crystallinity of the TiO2 NPs is essential for promoting photooxidation of the adsorbates by photoinduced charge separation when the TiO2 NPs serve as electron acceptors in artificial photosynthetic assemblies. The reported results are particularly relevant to the development of photocatalytic devices for oxidation chemistry based on inexpensive materials (e.g., TiO2 and Mn complexes) that are robust under aqueous and oxidative conditions.
Co-reporter:William R. McNamara, Robert C. Snoeberger III, Gonghu Li, Christiaan Richter, Laura J. Allen, Rebecca L. Milot, Charles A. Schmuttenmaer, Robert H. Crabtree, Gary W. Brudvig and Victor S. Batista
Energy & Environmental Science 2009 vol. 2(Issue 11) pp:1173-1175
Publication Date(Web):07 Aug 2009
DOI:10.1039/B910241H
A graphical abstract is available for this content
Co-reporter:Gonghu Li, Eduardo M. Sproviero, Robert C. Snoeberger III, Nobuhito Iguchi, James D. Blakemore, Robert H. Crabtree, Gary W. Brudvig and Victor S. Batista
Energy & Environmental Science 2009 vol. 2(Issue 2) pp:230-238
Publication Date(Web):12 Jan 2009
DOI:10.1039/B818708H
Inexpensive water oxidation catalysts are needed to develop photocatalytic solar cells that mimic photosynthesis and produce fuel from sunlight and water. This paper reports the successful attachment of a dinuclear di-µ-oxo manganese water oxidation catalyst [H2O(terpy)MnIII(µ-O)2 MnIV(terpy)H2O](NO3)3 (1, terpy = 2,2′:6′2″-terpyridine) onto TiO2 nanoparticles (NPs) via direct adsorption, or in situ synthesis. The resulting surface complexes are characterized by EPR and UV-visible spectroscopy, electrochemical measurements and computational modeling. We conclude that the mixed-valence (III,IV) state of 1 attaches to near-amorphous TiO2 NPs by substituting one of its water ligands by the TiO2 NP, as suggested by low-temperature (7 K) EPR data. In contrast, the analogous attachment onto well-crystallized TiO2 NPs leads to dimerization of 1 forming Mn(IV) tetramers on the TiO2 surface as suggested by EPR spectroscopy and electrochemical studies.
Co-reporter:Gonghu Li, Christiaan P. Richter, Rebecca L. Milot, Lawrence Cai, Charles A. Schmuttenmaer, Robert H. Crabtree, Gary W. Brudvig and Victor S. Batista
Dalton Transactions 2009 (Issue 45) pp:10078-10085
Publication Date(Web):02 Sep 2009
DOI:10.1039/B908686B
A synergistic effect between anatase and rutile TiO2 is known, in which the addition of rutile can remarkably enhance the photocatalytic activity of anatase in the degradation of organic contaminants. In this study, mixed-phase TiO2 nanocomposites consisting of anatase and rutile nanoparticles (NPs) were prepared for use as photoanodes in dye-sensitized solar cells (DSSCs) and were characterized by using UV-vis spectroscopy, powder X-ray diffraction and scanning electron microscopy. The addition of 10–15% rutile significantly improved light harvesting and the overall solar conversion efficiency of anatase NPs in DSSCs. The underlying mechanism for the synergistic effect in DSSCs is now explored by using time-resolved terahertz spectroscopy. It is clearly demonstrated that photo-excited electrons injected into the rutile NPs can migrate to the conduction band of anatase NPs, enhancing the photocurrent and efficiency. Interfacial electron transfer from rutile to anatase, similar to that in heterogeneous photocatalysis, is proposed to account for the synergistic effect in DSSCs. Our results further suggest that the synergistic effect can be used to explain the beneficial effect of TiCl4 treatment on DSSC efficiency.
Co-reporter:Justin Kim;Yinghua Wu;Jean-Luc Brédas
Israel Journal of Chemistry 2009 Volume 49( Issue 2) pp:187-197
Publication Date(Web):
DOI:10.1560/IJC.49.2.187
Abstract
The excited-state intramolecular proton-transfer dynamics and photoabsorption associated with the ketoenolic tautomerization reaction in 2-(2′-hydr oxyphenyl)benzothiazole are simulated according to a numerically exact quantum-dynamics propagation method and a full-dimensional excited-state potential energy surface based on an ab initio reaction surface Hamiltonian. The simulations involve the propagation of 69-dimensional wave packets according to the matching-pursuit/split-operator Fourier transform (MP/SOFT) method (Wu, Y.; Batista, V.S. J. Chem. Phys.2004, 121, 1676–1686). The underlying propagation scheme recursively applies the time-evolution operator as defined by the Trotter expansion to second-order accuracy in dynamically adaptive coherent-state expansions. Computations of time-dependent survival amplitudes, the time-dependent product population, and photoabsorption linewidths are compared to experimental data. The reported results provide fundamental insight on the nature of the excited-state reaction dynamics and demonstrate the capabilities of the MP/SOFT method as a powerful computational tool to study ultrafast reaction dynamics in polyatomic systems.
Co-reporter:Eduardo M. Sproviero;James P. McEvoy;José A. Gascón
Photosynthesis Research 2008 Volume 97( Issue 1) pp:91-114
Publication Date(Web):2008 July
DOI:10.1007/s11120-008-9307-0
Mechanistic investigations of the water-splitting reaction of the oxygen-evolving complex (OEC) of photosystem II (PSII) are fundamentally informed by structural studies. Many physical techniques have provided important insights into the OEC structure and function, including X-ray diffraction (XRD) and extended X-ray absorption fine structure (EXAFS) spectroscopy as well as mass spectrometry (MS), electron paramagnetic resonance (EPR) spectroscopy, and Fourier transform infrared spectroscopy applied in conjunction with mutagenesis studies. However, experimental studies have yet to yield consensus as to the exact configuration of the catalytic metal cluster and its ligation scheme. Computational modeling studies, including density functional (DFT) theory combined with quantum mechanics/molecular mechanics (QM/MM) hybrid methods for explicitly including the influence of the surrounding protein, have proposed chemically satisfactory models of the fully ligated OEC within PSII that are maximally consistent with experimental results. The inorganic core of these models is similar to the crystallographic model upon which they were based, but comprises important modifications due to structural refinement, hydration, and proteinaceous ligation which improve agreement with a wide range of experimental data. The computational models are useful for rationalizing spectroscopic and crystallographic results and for building a complete structure-based mechanism of water-splitting in PSII as described by the intermediate oxidation states of the OEC. This review summarizes these recent advances in QM/MM modeling of PSII within the context of recent experimental studies.
Co-reporter:George P. Lisi, Gregory A. Manley, Heidi Hendrickson, Ivan Rivalta, ... J. Patrick Loria
Structure (6 July 2016) Volume 24(Issue 7) pp:1155-1166
Publication Date(Web):6 July 2016
DOI:10.1016/j.str.2016.04.010
•Allosteric ligand binding is correlated to structural dynamics in a model enzyme•A widely dispersed allosteric network has been identified•Catalytic rate enhancement varies with the degree of millisecond flexibility•Allosteric ligand binding is communicated over a 25-Å distanceThe allosteric mechanism of the heterodimeric enzyme imidazole glycerol phosphate synthase was studied in detail with solution nuclear magnetic resonance spectroscopy and molecular dynamics simulations. We studied IGPS in complex with a series of allosteric activators corresponding to a large range of catalytic rate enhancements (26- to 4,900-fold), in which ligand binding is entropically driven. Conformational flexibility on the millisecond timescale plays a crucial role in intersubunit communication. Carr-Purcell-Meiboom-Gill relaxation dispersion experiments probing Ile, Leu, and Val methyl groups reveal that the apo- and glutamine-mimicked complexes are static on the millisecond timescale. Domain-wide motions are stimulated in the presence of the allosteric activators. These studies, in conjunction with ligand titrations, demonstrate that the allosteric network is widely dispersed and varies with the identity of the effector. Furthermore, we find that stronger allosteric ligands create more conformational flexibility on the millisecond timescale throughout HisF. This domain-wide loosening leads to maximum catalytic activity.
Co-reporter:Dequan Xiao, Li Fu, Jian Liu, Victor S. Batista, Elsa C.Y. Yan
Journal of Molecular Biology (24 August 2012) Volume 421(Issues 4–5) pp:537-547
Publication Date(Web):24 August 2012
DOI:10.1016/j.jmb.2011.12.035
Many amyloid proteins misfold into β-sheet aggregates upon interacting with biomembranes at the onset of diseases, such as Parkinson's disease and type II diabetes. The molecular mechanisms triggering aggregation depend on the orientation of β-sheets at the cell membranes. However, understanding how β-sheets adsorb onto lipid/aqueous interfaces is challenging. Here, we combine chiral sum frequency generation (SFG) spectroscopy and ab initio quantum chemistry calculations based on a divide-and-conquer strategy to characterize the orientation of human islet amyloid polypeptides (hIAPPs) at lipid/aqueous interfaces. We show that the aggregates bind with β-strands oriented at 48° relative to the interface. This orientation reflects the amphiphilic properties of hIAPP β-sheet aggregates and suggests the potential disruptive effect on membrane integrity.Download high-res image (228KB)Download full-size imageHighlights► We obtain a high-resolution chiral SFG spectrum of hIAPP aggregates. ► In theory, we relate the ratio of two SFG peaks to the aggregates' orientation. ► We find four possible orientations for the hIAPP by the theory. ► We simulate the four possible SFG spectra by a new “divide-and-conquer” approach. ► By comparison, we find that the hIAPP orients at an angle of 48° at interfaces.
Co-reporter:Sivakumar Sekharan, Mehmed Z. Ertem, Hanyi Zhuang, Eric Block, Hiroaki Matsunami, Ruina Zhang, Jennifer N. Wei, Yi Pan, Victor S. Batista
Biophysical Journal (2 September 2014) Volume 107(Issue 5) pp:
Publication Date(Web):2 September 2014
DOI:10.1016/j.bpj.2014.07.031
Understanding structure/function relationships of olfactory receptors is challenging due to the lack of x-ray structural models. Here, we introduce a QM/MM model of the mouse olfactory receptor MOR244-3, responsive to organosulfur odorants such as (methylthio)methanethiol. The binding site consists of a copper ion bound to the heteroatoms of amino-acid residues H105, C109, and N202. The model is consistent with site-directed mutagenesis experiments and biochemical measurements of the receptor activation, and thus provides a valuable framework for further studies of the sense of smell at the molecular level.
Co-reporter:Chetan Poojari, Dequan Xiao, Victor S. Batista, Birgit Strodel
Biophysical Journal (19 November 2013) Volume 105(Issue 10) pp:
Publication Date(Web):19 November 2013
DOI:10.1016/j.bpj.2013.09.045
Several neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases as well as nonneuropathic diseases such as type II diabetes and atrial amyloidosis are associated with aggregation of amyloid polypeptides into fibrillar structures, or plaques. In this study, we use molecular dynamics simulations to test the stability and orientation of membrane-embedded aggregates of the human islet amyloid polypeptide (hIAPP) implicated in type II diabetes. We find that in both monolayers and bilayers of dipalmitoylphosphatidylglycerol (DPPG) hIAPP trimers and tetramers remain inside the membranes and preserve their β-sheet secondary structure. Lipid bilayer-inserted hIAPP trimers and tetramers orient inside DPPG at 60° relative to the membrane/water interface and lead to water permeation and Na+ intrusion, consistent with ion-toxicity in islet β-cells. In particular, hIAPP trimers form a water-filled β-sandwich that induce water permeability comparable with channel-forming proteins, such as aquaporins and gramicidin-A. The predicted disruptive orientation is consistent with the amphiphilic properties of the hIAPP aggregates and could be probed by chiral sum frequency generation (SFG) spectroscopy, as predicted by the simulated SFG spectra.
Co-reporter:Sandra Luber, Sophie Leung, Carmen Herrmann, Wenge Han Du, Louis Noodleman and Victor S. Batista
Dalton Transactions 2014 - vol. 43(Issue 2) pp:NaN583-583
Publication Date(Web):2013/10/16
DOI:10.1039/C3DT51563J
Ribonucleotide reductases (RNRs) catalyze the reduction of ribonucleotides into deoxyribonucleotides necessary for DNA biosynthesis. Unlike the conventional class Ia RNRs which use a diiron cofactor in their subunit R2, the active site of the RNR-R2 from Chlamydia trachomatis (Ct) contains a Mn/Fe cofactor. The detailed structure of the Mn/Fe core has yet to be established. In this paper we evaluate six different structural models of the Ct RNR active site in the Mn(IV)/Fe(III) state by using Mössbauer parameter calculations and simulations of Mn/Fe extended X-ray absorption fine structure (EXAFS) spectroscopy, and we identify a structure similar to a previously proposed DFT-optimized model that shows quantitative agreement with both EXAFS and Mössbauer spectroscopic data.
Co-reporter:Jianbing Jiang, John R. Swierk, Svante Hedström, Adam J. Matula, Robert H. Crabtree, Victor S. Batista, Charles A. Schmuttenmaer and Gary W. Brudvig
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 28) pp:NaN18682-18682
Publication Date(Web):2016/06/30
DOI:10.1039/C6CP04377A
Interfacial electron transfer dynamics of a series of photosensitizers bound to TiO2via linkers of varying conjugation strength are explored by spectroscopic and computational techniques. Injection and recombination depend on the extent of conjugation in the linker, where the LUMO delocalization determines the injection dynamics but both the HOMO and HOMO−1 are involved in recombination.
Co-reporter:Gonghu Li, Christiaan P. Richter, Rebecca L. Milot, Lawrence Cai, Charles A. Schmuttenmaer, Robert H. Crabtree, Gary W. Brudvig and Victor S. Batista
Dalton Transactions 2009(Issue 45) pp:NaN10085-10085
Publication Date(Web):2009/09/02
DOI:10.1039/B908686B
A synergistic effect between anatase and rutile TiO2 is known, in which the addition of rutile can remarkably enhance the photocatalytic activity of anatase in the degradation of organic contaminants. In this study, mixed-phase TiO2 nanocomposites consisting of anatase and rutile nanoparticles (NPs) were prepared for use as photoanodes in dye-sensitized solar cells (DSSCs) and were characterized by using UV-vis spectroscopy, powder X-ray diffraction and scanning electron microscopy. The addition of 10–15% rutile significantly improved light harvesting and the overall solar conversion efficiency of anatase NPs in DSSCs. The underlying mechanism for the synergistic effect in DSSCs is now explored by using time-resolved terahertz spectroscopy. It is clearly demonstrated that photo-excited electrons injected into the rutile NPs can migrate to the conduction band of anatase NPs, enhancing the photocurrent and efficiency. Interfacial electron transfer from rutile to anatase, similar to that in heterogeneous photocatalysis, is proposed to account for the synergistic effect in DSSCs. Our results further suggest that the synergistic effect can be used to explain the beneficial effect of TiCl4 treatment on DSSC efficiency.
Co-reporter:C. Moyses Araujo, Mark D. Doherty, Steven J. Konezny, Oana R. Luca, Alex Usyatinsky, Hans Grade, Emil Lobkovsky, Grigorii L. Soloveichik, Robert H. Crabtree and Victor S. Batista
Dalton Transactions 2012 - vol. 41(Issue 12) pp:NaN3573-3573
Publication Date(Web):2012/02/08
DOI:10.1039/C2DT12195F
The structure and electrochemical properties of a series of bis(imino)pyridine CoII complexes (NNN)CoX2 and [(NNN)2Co][PF6]2 (NNN = 2,6-bis[1-(4-R-phenylimino)ethyl]pyridine, with R = CN, CF3, H, CH3, OCH3, N(CH3)2; NNN = 2,6-bis[1-(2,6-(iPr)2-phenylimino)ethyl]pyridine and X = Cl, Br) were studied using a combination of electrochemical and theoretical methods. Cyclic voltammetry measurements and DFT/B3LYP calculations suggest that in solution (NNN)CoCl2 complexes exist in equilibrium with disproportionation products [(NNN)2Co]2+ [CoCl4]2− with the position of the equilibrium heavily influenced by both the solvent polarity and the steric and electronic properties of the bis(imino)pyridine ligands. In strong polar solvents (e.g., CH3CN or H2O) or with electron donating substituents (R = OCH3 or N(CH3)2) the equilibrium is shifted and only oxidation of the charged products [(NNN)2Co]2+ and [CoCl4]2− is observed. Conversely, in nonpolar organic solvents such as CH2Cl2 or with electron withdrawing substituents (R = CN or CF3), disproportionation is suppressed and oxidation of the (NNN)CoCl2 complexes leads to 18e− CoIII complexes stabilized by coordination of a solvent moiety. In addition, the [(NNN)2Co][PF6]2 complexes exhibit reversible CoII/III oxidation potentials that are strongly dependent on the electron withdrawing/donating nature of the N-aryl substituents, spanning nearly 750 mV in acetonitrile. The resulting insight on the regulation of redox properties of a series of bis(imino)pyridine cobalt(II) complexes should be particularly valuable to tune suitable conditions for reactivity.
Co-reporter:Oana R. Luca, Steven J. Konezny, Eric K. Paulson, Fatemah Habib, Kurt M. Luthy, Muralee Murugesu, Robert H. Crabtree and Victor S. Batista
Dalton Transactions 2013 - vol. 42(Issue 24) pp:NaN8807-8807
Publication Date(Web):2013/05/03
DOI:10.1039/C3DT50528F
A tridentate NNN NiII complex, shown to be an electrocatalyst for aqueous H2 production at low overpotentials, is studied by using temperature-dependent paramagnetic 1H NMR. The NMR T1 relaxation rates, temperature dependence of the chemical shifts, and dc SQUID magnetic susceptibility are correlated to DFT chemical shifts and compared with the properties of a diamagnetic Zn analogue complex. The resulting characterization provides an unambiguous assignment of the six proton environments in the meridionally coordinating tridentate NNN ligand. The demonstrated NMR/DFT methodology should be valuable in the search for appropriate ligands to optimize the reactivity of 3d metal complexes bound to attract increasing attention in catalytic applications.
Co-reporter:Bradley J. Brennan, Jeffrey Chen, Benjamin Rudshteyn, Subhajyoti Chaudhuri, Brandon Q. Mercado, Victor S. Batista, Robert H. Crabtree and Gary W. Brudvig
Chemical Communications 2016 - vol. 52(Issue 14) pp:NaN2975-2975
Publication Date(Web):2016/01/12
DOI:10.1039/C5CC09857B
Hydroxamate binding modes and protonation states have yet to be conclusively determined. Molecular titanium(IV) phenylhydroxamate complexes were synthesized as structural and spectroscopic models, and compared to functionalized TiO2 nanoparticles. In a combined experimental–theoretical study, we find that the predominant binding form is monodeprotonated, with evidence for the chelate mode.
Co-reporter:C. Koenigsmann, T. S. Ripolles, B. J. Brennan, C. F. A. Negre, M. Koepf, A. C. Durrell, R. L. Milot, J. A. Torre, R. H. Crabtree, V. S. Batista, G. W. Brudvig, J. Bisquert and C. A. Schmuttenmaer
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 31) pp:NaN16641-16641
Publication Date(Web):2014/07/04
DOI:10.1039/C4CP02405B
An efficient synthetic protocol to functionalize the cyanoacrylic acid anchoring group of commercially available MK-2 dye with a highly water-stable hydroxamate anchoring group is described. Extensive characterization of this hydroxamate-modified dye (MK-2HA) reveals that the modification does not affect its favorable optoelectronic properties. Dye-sensitized solar cells (DSSCs) prepared with the MK-2HA dye attain improved efficiency (6.9%), relative to analogously prepared devices with commercial MK-2 and N719 dyes. The hydroxamate anchoring group also contributes to significantly increased water stability, with a decrease in the rate constant for dye desorption of MK-2HA relative to MK-2 in the presence of water by as much as 37.5%. In addition, the hydroxamate-anchored dye undergoes essentially no loss in DSSC efficiency and the external quantum efficiency improves when up to 20% water is purposefully added to the electrolyte. In contrast, devices prepared with the commercial dye suffer a 50% decline in efficiency under identical conditions, with a concomitant decrease in external quantum efficiency. Collectively, our results indicate that covalent functionalization of organic dyes with hydroxamate anchoring groups is a simple and efficient approach to improving the water stability of the dye–semiconductor interface and overall device durability.
Co-reporter:Louise M. Guard, Julio L. Palma, William P. Stratton, Laura J. Allen, Gary W. Brudvig, Robert H. Crabtree, Victor S. Batista and Nilay Hazari
Dalton Transactions 2012 - vol. 41(Issue 26) pp:NaN8110-8110
Publication Date(Web):2012/02/13
DOI:10.1039/C2DT12426B
The reactions of the substituted 2,2′:6,2′′-terpyridine ligands, 4′-mesityl-2,2′:6′,2′′-terpyridine (mesitylterpy) (1a), 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine (tri-tButerpy) (1b) and 4′-phenyl-2,2′:6′,2′′-terpyridine (phenylterpy) (1c) with Grignard reagents were investigated. When half an equivalent of mesitylterpy or tri-tButerpy were treated with MeMgBr in diethyl ether, the only products were (R-terpy)MgBr2 (R = mesityl (5a), or tri-tBu (5b)) and Me2Mg and a similar reaction was observed in THF. Compounds 5a and 5b were characterized by X-ray crystallography. Changing the Grignard reagent to PhMgBr also generated 5a and 5b along with Ph2Mg, while the reaction between MeMgCl or PhMgCl and 1a or 1b generated (R-terpy)MgCl2 (R = mesityl (6a), or tri-tBu (6b)) and either Me2Mg or Ph2Mg, respectively. The products from reactions between phenylterpy (1c) and Grignard reagents were highly insoluble and could not be fully characterized but appeared to be the same as those from reactions with 1a and 1b. In contrast to other studies using tridentate nitrogen ligands, which formed either mixed halide alkyl species or dihalide and bis(alkyl) species depending on whether the Grignard reagent was reacted with the ligand in diethyl ether or THF, the formation of mixed halide, alkyl complexes of the type (R-terpy)MgR′X (R′ = Me or Ph; X = Cl or Br) or dialkyl species such as (R-terpy)MgR′2 (R′ = Me or Ph) was not observed here, regardless of the reaction conditions. DFT studies were performed to complement the experimental studies. The experimental results could not be accurately reproduced unless π-stacking effects associated with free terpyridine were included in the model. When these effects were included, the calculations were consistent with the experimental results which indicated that the formation of the terpy Mg dihalide species and R′2Mg (R′ = Me or Ph) is thermodynamically preferred over the formation of mixed alkyl halide Mg species. This is proposed to be due to the increased steric bulk of the terpy ligand in the coordination plane, compared with other tridentate nitrogen donors.
.jpg)


![21H,23H-porphine, 5,10,15,20-tetrakis[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/127300/127286-86-6.png)
![21H,23H-porphine, 5,10,15,20-tetrakis[3,5-bis(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/127300/127286-86-6_b.png)
















![[2,2'-Bipyridine]-4,4'-diamine](http://img.cochemist.com/ccimg/18600/18511-69-8.png)
![[2,2'-Bipyridine]-4,4'-diamine](http://img.cochemist.com/ccimg/18600/18511-69-8_b.png)



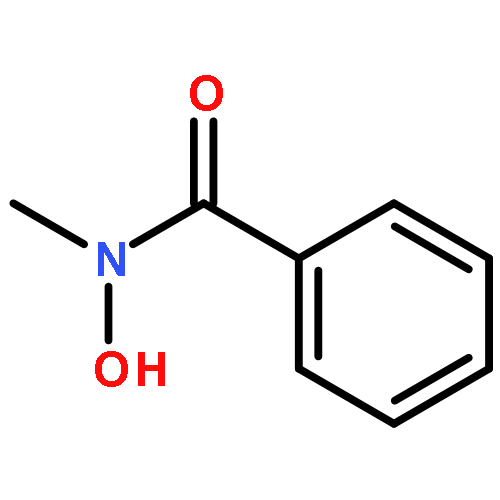
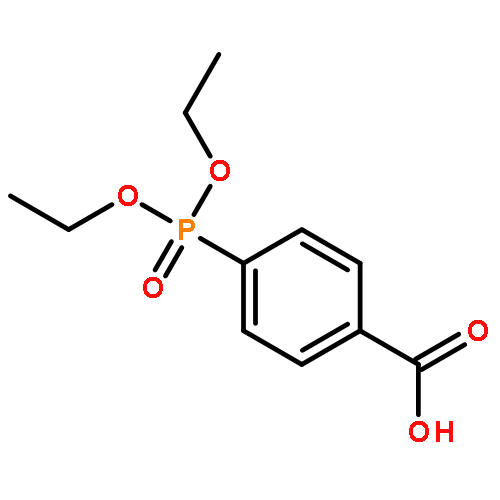





![4-Pyridinecarboxamide, N-[(tetrahydro-2H-pyran-2-yl)oxy]-](/data/chemimg/101300/1437782-69-8.png)
![4-Pyridinecarboxamide, N-[(tetrahydro-2H-pyran-2-yl)oxy]-](/data/chemimg/101300/1437782-69-8_b.png)
![2,4-Pentanedione, 3-[4-(1H-imidazol-1-yl)phenyl]-](/data/chemimg/101300/1437782-64-3.png)
![2,4-Pentanedione, 3-[4-(1H-imidazol-1-yl)phenyl]-](/data/chemimg/101300/1437782-64-3_b.png)


![Phosphonic acid, P-[4-(1H-imidazol-1-yl)phenyl]-, diethyl ester](/data/chemimg/101300/1437782-56-3.png)
![Phosphonic acid, P-[4-(1H-imidazol-1-yl)phenyl]-, diethyl ester](/data/chemimg/101300/1437782-56-3_b.png)
![Benzamide, 4-(1H-imidazol-1-yl)-N-[(tetrahydro-2H-pyran-2-yl)oxy]-](/data/chemimg/101300/1437782-51-8.png)
![Benzamide, 4-(1H-imidazol-1-yl)-N-[(tetrahydro-2H-pyran-2-yl)oxy]-](/data/chemimg/101300/1437782-51-8_b.png)




















![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9.png)
![2,2'-[(2,3,4,5,6-pentafluorophenyl)methylene]bis-1H-Pyrrole](http://img.cochemist.com/ccimg/167500/167482-91-9_b.png)
![4-([2,2':6',2''-Terpyridin]-4'-yl)benzoic acid](/data/chemimg/104300/158014-74-5.png)
![4-([2,2':6',2''-Terpyridin]-4'-yl)benzoic acid](/data/chemimg/104300/158014-74-5_b.png)
















![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4_b.png)
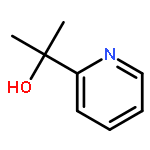
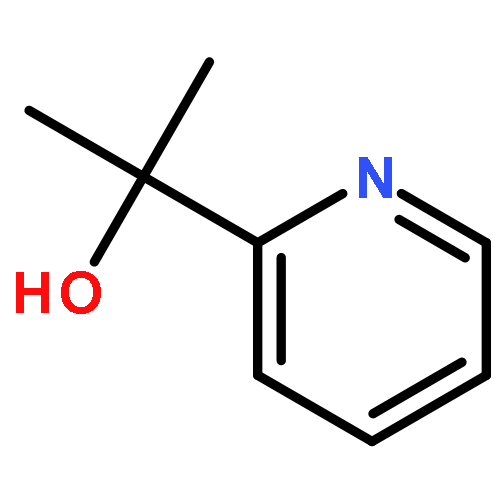



![Benzoic acid, 4-[[4-(dimethylamino)phenyl]azo]-, methyl ester](http://img.cochemist.com/ccimg/24700/24630-00-0.png)
![Benzoic acid, 4-[[4-(dimethylamino)phenyl]azo]-, methyl ester](http://img.cochemist.com/ccimg/24700/24630-00-0_b.png)








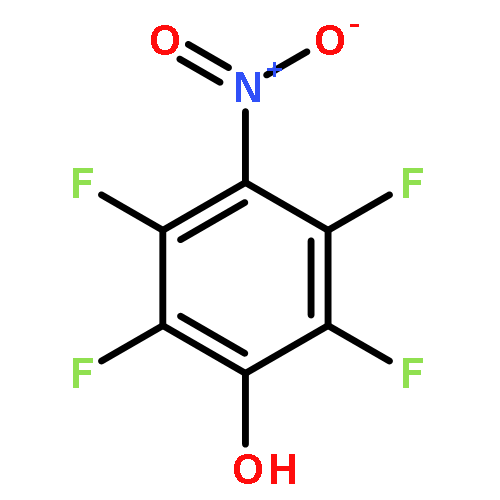


































![[(2R,3S,4R,5R)-5-[4-carbamoyl-5-[[(E)-[(3R,4R)-3,4-dihydroxy-2-oxo-5-phosphonooxy-pentyl]iminomethyl]amino]imidazol-1-yl]-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl dihydrogen phosphate](http://img.cochemist.com/ccimg/36300/36244-86-7.png)
![[(2R,3S,4R,5R)-5-[4-carbamoyl-5-[[(E)-[(3R,4R)-3,4-dihydroxy-2-oxo-5-phosphonooxy-pentyl]iminomethyl]amino]imidazol-1-yl]-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl dihydrogen phosphate](http://img.cochemist.com/ccimg/36300/36244-86-7_b.png)