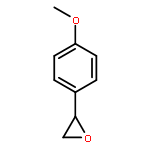
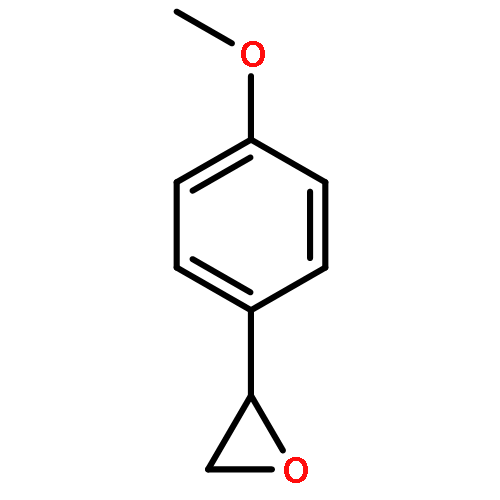
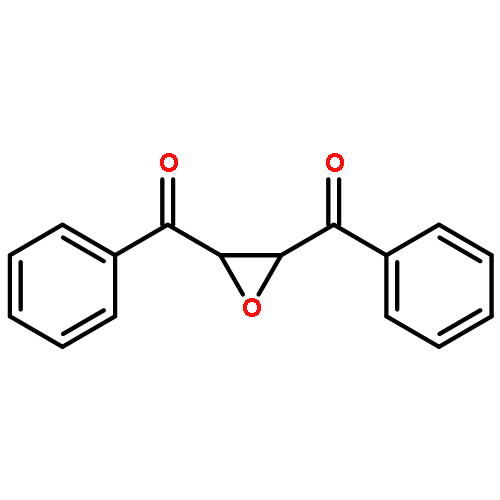
Co-reporter:Ekaterina V. Bakhmutova-Albert, Huirong Yao, Daniel E. Denevan, and David E. Richardson
Inorganic Chemistry 2010 Volume 49(Issue 24) pp:11287-11296
Publication Date(Web):November 15, 2010
DOI:10.1021/ic1007389
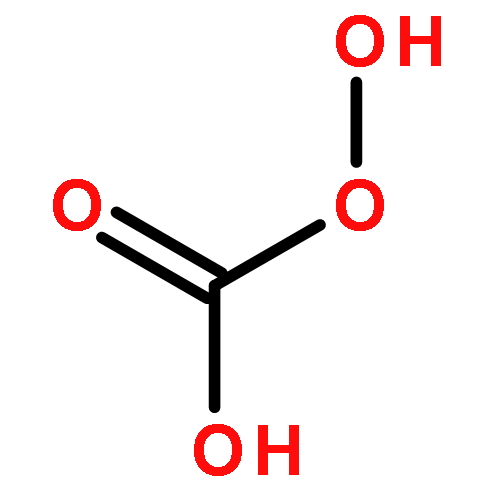
The kinetics and mechanism of peroxymonocarbonate (HCO4−) formation in the reaction of hydrogen peroxide with bicarbonate have been investigated for the pH 6−9 range. A double pH jump method was used in which 13C-labeled bicarbonate solutions are first acidified to produce 13CO2 and then brought to higher pH values by addition of base in the presence of hydrogen peroxide. The time evolution of the 13C NMR spectrum was used to establish the competitive formation and subsequent equilibration of bicarbonate and peroxymonocarbonate following the second pH jump. Kinetic simulations are consistent with a mechanism for the bicarbonate reaction with peroxide in which the initial formation of CO2 via dehydration of bicarbonate is followed by reaction of CO2 with H2O2 (perhydration) and its conjugate base HOO− (base-catalyzed perhydration). The rate of peroxymonocarbonate formation from bicarbonate increases with decreasing pH because of the increased availability of CO2 as an intermediate. The selectivity for formation of HCO4− relative to the hydration product HCO3− increases with increasing pH as a consequence of the HOO− pathway and the slower overall equilibration rate, and this pH dependence allows estimation of rate constants for the reaction of CO2 with H2O2 and HOO− at 25 °C (2 × 10−2 M−1 s−1 and 280 M−1 s−1, respectively). The contributions of the HOO− and H2O2 pathways are comparable at pH 8. In contrast to the perhydration of many other common inorganic and organic acids, the facile nature of the CO2/HCO3− equilibrium and relatively high equilibrium availability of the acid anhydride (CO2) at neutral pH allows for rapid formation of the peroxymonocarbonate ion without strong acid catalysis. Formation of peroxymonocarbonate by the reaction of HCO3− with H2O2 is significantly accelerated by carbonic anhydrase and the model complex [Zn(II)L(H2O)]2+ (L = 1,4,7,10-tetraazacyclododecane).
Co-reporter:Ana Ison, Cheng Xu, G. Ken Weakley, David E. Richardson
Journal of Molecular Catalysis A: Chemical 2008 Volume 293(1–2) pp:1-7
Publication Date(Web):1 October 2008
DOI:10.1016/j.molcata.2008.06.010
The hydrocarbon-soluble coordination complex [Fe(4,7-diphenyl-1,10-phenanthroline)3](SO3CF3)2 (1) is an active catalyst for the autoxidation of cumene and cyclohexane. The activity of 1 in the autoxidation of cumene at 60 °C is comparable to that of tetra(pentafluorophenyl)porphyrin iron(III) chloride (2), a halogenated iron porphyrin with high autoxidation activity. The kinetic data have been fit by a mechanism in which the iron catalyst is activated by reaction with peroxide and the resulting active complex acts as a peroxide decomposition catalyst producing chain-carrying radicals. The activity of 1 is also comparable to that of 2 in the autoxidation of cyclohexane at 135 °C. The utility of catalyst 1 is enhanced by its solubility in pure hydrocarbon substrates.The hydrocarbon-soluble coordination complex [Fe(4,7-diphenyl-1,10-phenanthroline)3](SO3CF3)2 (1) is an active catalyst for the autoxidation of cumene and cyclohexane. The activity of 1 in the autoxidation of cumene at 60 °C is comparable to that of tetra(pentafluorophenyl)porphyrin iron(III) chloride (2), a halogenated iron porphyrin with high autoxidation activity. The kinetic data have been fit by a mechanism in which the iron catalyst is activated by reaction with peroxide and the resulting active complex acts as a peroxide decomposition catalyst producing chain-carrying radicals. The activity of 1 is also comparable to that of 2 in the autoxidation of cyclohexane at 135 °C. The utility of catalyst 1 is enhanced by its solubility in pure hydrocarbon substrates.
Co-reporter:Celeste Aida S. Regino, David E. Richardson
Inorganica Chimica Acta 2007 Volume 360(Issue 14) pp:3971-3977
Publication Date(Web):1 November 2007
DOI:10.1016/j.ica.2007.05.020
The effect of bicarbonate on the rates of the H2O2 oxidation of cysteine, gluthathione, and N-acetylcysteine to the corresponding disulfides was investigated. The relative oxidation rates at pH 8 for the different thiols are inversely related to the pKa values of the thiol groups, and the reactive nucleophiles are identified as the thiolate anions or their kinetic equivalents. The second-order rate constants at 25 °C for the reaction of the thiolate anions with hydrogen peroxide are 17 ± 2 M−1 s−1 for all three substrates. In the presence of bicarbonate (>25 mM), the observed rate of thiolate oxidation is increased by a factor of two or more, and the catalysis is proposed to be associated with the formation of peroxymonocarbonate from the equilibrium reaction of hydrogen peroxide with bicarbonate (via CO2). The calculated second-order rate constants for the direct reaction of the three thiolate anions with peroxymonocarbonate fall within the range of 900–2000 M−1 s−1. Further oxidation of disulfides by peroxymonocarbonate results in the formation of thiosulfonate and sulfonate products. These results strongly suggest that peroxymonocarbonate should be considered as a reactive oxygen species in aerobic metabolism with relevance in thiol oxidations.The effect of bicarbonate on the rates of the H2O2 oxidation of cysteine, gluthathione, and N-acetylcysteine to the corresponding disulfides was investigated. Bicarbonate catalysis is associated with the formation of peroxymonocarbonate (via peroxide reaction with CO2). Calculated second-order rate constants for the reaction of the three thiolate anions with peroxymonocarbonate are 900–2000 M−1 s−1.
Co-reporter:David E. Richardson, G.H.Lisa Lang, Elisa Crestoni, Matthew F. Ryan, John R. Eyler
International Journal of Mass Spectrometry 2001 Volume 204(1–3) pp:255-266
Publication Date(Web):6 February 2001
DOI:10.1016/S1387-3806(00)00359-6
Fourier transform ion cyclotron resonance mass spectrometry was used to study the gas-phase reactions of three zirconium(IV) hydroxide ions, Cp2ZrOH+ (Cp = η5-cyclopentadienyl), Cp2ZrOD+, and Cp2Zr18OH+. Product distributions were determined for reactions with alcohols, amines, ethers, esters, and amides. Reactions with alcohols lead to O–H bond activation with formation of alkoxide complexes Cp2ZrOR+ and elimination of water. Equilibrium constants for the reactions were used to determine the relative energetics of Zr–OR bonds, and the affinities of hydroxide and alkoxides toward the zirconium(IV) center in Cp2Zr2+ decrease (OH− > MeO− > EtO− ≈ iso-PrO− ≈ sec-BuO− ≈ tert-BuO−) in the same order as the proton affinities of RO−. Adducts of the alkoxide complex ion with alcohols are formed at long reaction times. Reaction with acetic acid leads to formation of the carboxylate complex ion Cp2ZrO2CCH3+ and elimination of water. Amines, ethers, esters, and amides all form adducts with Cp2ZrOH+, but no other reaction pathways, such as hydrolysis, are observed. The unsolvated zirconium(IV) hydroxide species can coordinate substrates and activate O–H bonds, but it does not efficiently cleave esters and amides despite its reactive, bound hydroxide and coordinative unsaturation. The relationship of these results to solution reactions of metal–hydroxides is discussed.


![7-OXABICYCLO[4.1.0]HEPTANE-3,3-DIMETHANOL](http://img.cochemist.com/ccimg/54100/54061-77-7.png)
![7-OXABICYCLO[4.1.0]HEPTANE-3,3-DIMETHANOL](http://img.cochemist.com/ccimg/54100/54061-77-7_b.png)