Co-reporter:Dennis W. Szymanski;Malvina Papanastasiou;Katja Melchior;Richard W. Mercier;Ganesh A. Thakur;Nikolai Zvonok;Sangwon Cha;Billy Wu;David R. Janero;Barry Karger
Journal of Proteome Research October 7, 2011 Volume 10(Issue 10) pp:4789-4798
Publication Date(Web):Publication Date (Web): August 24, 2011
DOI:10.1021/pr2005583
The lack of experimental characterization of the structures and ligand-binding motifs of therapeutic G-protein coupled receptors (GPCRs) hampers rational drug discovery. The human cannabinoid receptor 2 (hCB2R) is a class-A GPCR and promising therapeutic target for small-molecule cannabinergic agonists as medicines. Prior mutational and modeling data constitute provisional evidence that AM-841, a high-affinity classical cannabinoid, interacts with cysteine C6.47(257) in hCB2R transmembrane helix 6 (TMH6) to afford improved hCB2R selectivity and unprecedented agonist potency. We now apply bottom-up mass spectrometry (MS)-based proteomics to define directly the hCB2R-AM-841 interaction at the amino-acid level. Recombinant hCB2R, overexpressed as an N-terminal FLAG-tagged/C-terminal 6His-tagged protein (FLAG-hCB2R-6His) with a baculovirus system, was solubilized and purified by immunochromatography as functional receptor. A multiplex multiple reaction monitoring (MRM)-MS method was developed that allowed us to observe unambiguously all seven discrete TMH peptides in the tryptic digest of purified FLAG-hCB2R-6His and demonstrate that AM-841 modifies hCB2R TMH6 exclusively. High-resolution mass spectra of the TMH6 tryptic peptide obtained by Q-TOF MS/MS analysis demonstrated that AM-841 covalently and selectively modifies hCB2R at TMH6 cysteine C6.47(257). These data demonstrate how integration of MS-based proteomics into a ligand-assisted protein structure (LAPS) experimental paradigm can offer guidance to structure-enabled GPCR agonist design.Keywords: agonist; covalent probe; drug discovery; electrospray ionization; GPCR; ligand binding domain; multiple reaction monitoring; protein structural biology;
Co-reporter:Han Zhou, Yan Peng, Aneetha Halikhedkar, Pusheng Fan, David R. Janero, Ganesh A. Thakur, Richard W. Mercier, Xin Sun, Xiaoyu Ma, and Alexandros Makriyannis
ACS Chemical Neuroscience June 21, 2017 Volume 8(Issue 6) pp:1338-1338
Publication Date(Web):February 21, 2017
DOI:10.1021/acschemneuro.7b00003
Cannabinoid receptor 2 (CB2R)-dependent signaling is implicated in neuronal physiology and immune surveillance by brain microglia. Selective CB2R agonists hold therapeutic promise for inflammatory and other neurological disorders. Information on human CB2R (hCB2R) ligand-binding and functional domains is needed to inform the rational design and optimization of candidate druglike hCB2R agonists. Prior demonstration that hCB2R transmembrane helix 2 (TMH2) cysteine C2.59(89) reacts with small-molecule methanethiosulfonates showed that this cysteine residue is accessible to sulfhydryl derivatization reagents. We now report the design and application of two novel, pharmacologically active, high-affinity molecular probes, AM4073 and AM4099, as chemical reporters to interrogate directly the interaction of classical cannabinoid agonists with hCB2R cysteine residues. AM4073 has one electrophilic isothiocyanate (NCS) functionality at the C9 position of its cyclohexenyl C-ring, whereas AM4099 has NCS groups at that position and at the terminus of its aromatic A-ring C3 side chain. Pretreatment of wild-type hCB2R with either probe reduced subsequent [3H]CP55,940 specific binding by ∼60%. Conservative serine substitution of any hCB2R TMH cysteine residue except C2.59(89) did not affect the reduction of [3H]CP55,940 specific binding by either probe, suggesting that AM4073 and AM4099 interact irreversibly with this TMH2 cysteine. In contrast, AM841, an exceptionally potent hCB2R megagonist and direct AM4073/4099 congener bearing a single electrophilic NCS group at the terminus of its C3 side chain, had been demonstrated to bind covalently to TMH6 cysteine C6.47(257) and not C2.59(89). Molecular modeling indicates that the AM4073–hCB2R* interaction at C2.59(89) orients this classical cannabinoid away from TMH6 and toward the TMH2–TMH3 interface in the receptor’s hydrophobic binding pocket, whereas the AM841–hCB2R* interaction at C6.47(257) favors agonist orientation toward TMH6/7. These data constitute initial evidence that TMH2 cysteine C2.59(89) is a component of the hCB2R binding pocket for classical cannabinoids. The results further demonstrate how interactions between classical cannabinoids and specific amino acids within the hCB2R* ligand-binding domain act as determinants of agonist pharmacological properties and the architecture of the agonist-hCB2R* conformational ensemble, allowing the receptor to adopt distinct activity states, such that interaction of classical cannabinoids with TMH6 cysteine C6.47(257) favors a binding pose more advantageous for agonist potency than does their interaction with TMH2 cysteine C2.59(89).Keywords: Binding motif; cannabinoid receptor 2; covalent chemical probe; cysteine; G-protein coupled receptor; isothiocyanate; ligand-binding domain; molecular probe; signal transduction; transmembrane helix;
Co-reporter:Thanh C. Ho, Naoyuki Shimada, Marcus A. Tius, Spyros P. Nikas, Wen Zhang, and Alexandros Makriyannis
The Journal of Organic Chemistry August 4, 2017 Volume 82(Issue 15) pp:7839-7839
Publication Date(Web):July 5, 2017
DOI:10.1021/acs.joc.7b00988
We report the design, synthesis, and biological evaluation of a novel class of cannabinergic ligands, namely C1′-azacycloalkyl hexahydrocannabinols. Our synthetic approaches utilize an advanced common chiral intermediate triflate from which all analogues could be derived. Key synthetic steps involve microwave-assisted Liebeskind–Srogl C–C cross-coupling and palladium-catalyzed decarboxylative coupling reactions. The C1′-N-methylazetidinyl and C1′-N-methylpyrrolidinyl analogues were found to be high affinity ligands for the CB1 and CB2 cannabinoid receptors.
Co-reporter:Nikolai Zvonok;Wei Xu;John Williams;David R. Janero;Srinivasan C. Krishnan
Journal of Proteome Research April 5, 2010 Volume 9(Issue 4) pp:1746-1753
Publication Date(Web):2017-2-22
DOI:10.1021/pr900870p
The human cannabinoid 1 receptor (hCB1), a ubiquitous G protein-coupled receptor (GPCR), transmits cannabinergic signals that participate in diverse (patho)physiological processes. Pharmacotherapeutic hCB1 targeting is considered a tractable approach for treating such prevalent diseases as obesity, mood disorders, and drug addiction. The hydrophobic nature of the transmembrane helices of hCB1 presents a formidable difficulty to its direct structural analysis. Comprehensive experimental characterization of functional hCB1 by mass spectrometry (MS) is essential to the targeting of affinity probes that can be used to define directly hCB1 binding domains using a ligand-assisted experimental approach. Such information would greatly facilitate the rational design of hCB1-selective agonists/antagonists with therapeutic potential. We report the first high-coverage MS analysis of the primary sequence of the functional hCB1 receptor, one of the few such comprehensive MS-based analyses of any GPCR. Recombinant C-terminal hexa-histidine-tagged hCB1 (His6-hCB1) was expressed in cultured insect (Spodoptera frugiperda) cells, solubilized by a procedure devised to enhance receptor purity following metal-affinity chromatography, desalted by buffer exchange, and digested in solution with (chymo)trypsin. “Bottom-up” nanoLC−MS/MS of the (chymo)tryptic digests afforded a degree of overall hCB1 coverage (>94%) thus far reported for only two other GPCRs. This MS-compatible procedure devised for His6-hCB1 sample preparation, incorporating in-solution (chymo)trypsin digestion in the presence of a low concentration of CYMAL-5 detergent, may be applicable to the MS-based proteomic characterization of other GPCRs. This work should help enable future ligand-assisted structural characterization of hCB1 binding motifs at the amino-acid level using rationally designed and targeted covalent cannabinergic probes.Keywords: affinity purification; binding motifs; drug design; endocannabinoid signaling system; G protein-coupled receptor; ligand targeting; ligand-assisted protein analysis; nanoLC−MS/MS; protein expression; proteomic analysis; signal transduction; transmembrane protein;
Co-reporter:Srikrishnan Mallipeddi, David R. Janero, Nikolai Zvonok, Alexandros Makriyannis
Biochemical Pharmacology 2017 Volume 128(Volume 128) pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.bcp.2016.11.014
The phenomenon of functional selectivity, whereby a ligand preferentially directs the information output of a G-protein coupled receptor (GPCR) along (a) particular effector pathway(s) and away from others, has redefined traditional GPCR signaling paradigms to provide a new approach to structure-based drug design. The two principal cannabinoid receptors (CBRs) 1 and 2 belong to the class-A GPCR subfamily and are considered tenable therapeutic targets for several indications. Yet conventional orthosteric ligands (agonists, antagonists/inverse agonists) for these receptors have had very limited clinical utility due to their propensity to incite on-target adverse events. Chemically distinct classes of cannabinergic ligands exhibit signaling bias at CBRs towards individual subsets of signal transduction pathways. In this review, we discuss the known signaling pathways regulated by CBRs and examine the current evidence for functional selectivity at CBRs in response to endogenous and exogenous cannabinergic ligands as biased agonists. We further discuss the receptor and ligand structural features allowing for selective activation of CBR-dependent functional responses. The design and development of biased ligands may offer a pathway to therapeutic success for novel CBR-targeted drugs.Download high-res image (175KB)Download full-size image
Co-reporter:Shashank Kulkarni; Spyros P. Nikas; Rishi Sharma; Shan Jiang; Carol A. Paronis; Michael Z. Leonard; Bin Zhang; Chandrashekhar Honrao; Srikrishnan Mallipeddi; Jimit Girish Raghav; Othman Benchama; Torbjörn U. C. Järbe; Jack Bergman
Journal of Medicinal Chemistry 2016 Volume 59(Issue 14) pp:6903-6919
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.jmedchem.6b00717
In pursuit of safer controlled-deactivation cannabinoids with high potency and short duration of action, we report the design, synthesis, and pharmacological evaluation of novel C9- and C11-hydroxy-substituted hexahydrocannabinol (HHC) and tetrahydrocannabinol (THC) analogues in which a seven atom long side chain, with or without 1′-substituents, carries a metabolically labile 2′,3′-ester group. Importantly, in vivo studies validated our controlled deactivation approach in rodents and non-human primates. The lead molecule identified here, namely, butyl-2-[(6aR,9R,10aR)-1-hydroxy-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromen-3-yl]-2-methylpropanoate (AM7499), was found to exhibit remarkably high in vitro and in vivo potency with shorter duration of action than the currently existing classical cannabinoid agonists.
Co-reporter:Spyros P. Nikas; Rishi Sharma; Carol A. Paronis; Shashank Kulkarni; Ganesh A. Thakur; Dow Hurst; JodiAnne T. Wood; Roger S. Gifford; Girija Rajarshi; Yingpeng Liu; Jimit Girish Raghav; Jason Jianxin Guo; Torbjörn U.C. Järbe; Patricia H. Reggio; Jack Bergman
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:665-681
Publication Date(Web):December 3, 2014
DOI:10.1021/jm501165d
We recently reported on a controlled deactivation/detoxification approach for obtaining cannabinoids with improved druggability. Our design incorporates a metabolically labile ester group at strategic positions within the THC structure. We have now synthesized a series of (−)-Δ8-THC analogues encompassing a carboxyester group within the 3-alkyl chain in an effort to explore this novel cannabinergic chemotype for CB receptor binding affinity, in vitro and in vivo potency and efficacy, as well as controlled deactivation by plasma esterases. We have also probed the chain’s polar characteristics with regard to fast onset and short duration of action. Our lead molecule, namely 2-[(6aR,10aR)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic acid 3-cyano-propyl ester (AM7438), showed picomolar affinity for CB receptors and is deactivated by plasma esterases while the respective acid metabolite is inactive. In further in vitro and in vivo experiments, the compound was found to be a remarkably potent and efficacious CB1 receptor agonist with relatively fast onset/offset of action.
Co-reporter:Go Ogawa; Marcus A. Tius; Han Zhou; Spyros P. Nikas; Aneetha Halikhedkar; Srikrishnan Mallipeddi
Journal of Medicinal Chemistry 2015 Volume 58(Issue 7) pp:3104-3116
Publication Date(Web):March 11, 2015
DOI:10.1021/jm501960u
The aliphatic side chain plays a pivotal role in determining the cannabinergic potency of tricyclic classical cannabinoids, and we have previously shown that this chain could be substituted successfully by adamantyl or other polycyclic groups. In an effort to explore the pharmacophoric features of these conformationally fixed groups, we have synthesized a series of analogues in which the C3 position is substituted directly with an adamantyl group bearing functionality at one of the tertiary carbon atoms. These substituents included the electrophilic isothiocyanate and photoactivatable azido groups, both of which are capable of covalent attachment with the target protein. Our results show that substitution at the 3′-adamantyl position can lead to ligands with improved affinities and CB1/CB2 selectivities. Our work has also led to the development of two successful covalent probes with high affinities for both cannabinoid receptors, namely, the electrophilic isothiocyanate AM994 and the photoactivatable aliphatic azido AM993 analogues.
Co-reporter:David R. Janero, Suma Yaddanapudi, Nikolai Zvonok, Kumar V. Subramanian, Vidyanand G. Shukla, Edward Stahl, Lei Zhou, Dow Hurst, James Wager-Miller, Laura M. Bohn, Patricia H. Reggio, Ken Mackie, and Alexandros Makriyannis
ACS Chemical Neuroscience 2015 Volume 6(Issue 8) pp:1400
Publication Date(Web):May 15, 2015
DOI:10.1021/acschemneuro.5b00090
The cannabinoid 1 receptor (CB1R) is one of the most abundant G protein-coupled receptors (GPCRs) in the central nervous system. CB1R involvement in multiple physiological processes, especially neurotransmitter release and synaptic function, has made this GPCR a prime drug discovery target, and pharmacological CB1R activation has been demonstrated to be a tenable therapeutic modality. Accordingly, the design and profiling of novel, drug-like CB1R modulators to inform the receptor’s ligand-interaction landscape and molecular pharmacology constitute a prime contemporary research focus. For this purpose, we report utilization of AM3677, a designer endocannabinoid (anandamide) analogue derivatized with a reactive electrophilic isothiocyanate functionality, as a covalent, CB1R-selective chemical probe. The data demonstrate that reaction of AM3677 with a cysteine residue in transmembrane helix 6 of human CB1R (hCB1R), C6.47(355), is a key feature of AM3677’s ligand-binding motif. Pharmacologically, AM3677 acts as a high-affinity, low-efficacy CB1R agonist that inhibits forskolin-stimulated cellular cAMP formation and stimulates CB1R coupling to G protein. AM3677 also induces CB1R endocytosis and irreversible receptor internalization. Computational docking suggests the importance of discrete hydrogen bonding and aromatic interactions as determinants of AM3677’s topology within the ligand-binding pocket of active-state hCB1R. These results constitute the initial identification and characterization of a potent, high-affinity, hCB1R-selective covalent agonist with utility as a pharmacologically active, orthosteric-site probe for providing insight into structure–function correlates of ligand-induced CB1R activation and the molecular features of that activation by the native ligand, anandamide.Keywords: 7-transmembrane receptor; Amino acid; binding domain; central nervous system; chemical probe; cysteine; G protein-coupled receptor; homology modeling; isothiocyanate; ligand-binding motif; receptor activation; signal transduction
Co-reporter:Erin L. Shelnut, Spyros P. Nikas, David F. Finnegan, Nan Chiang, Charles N. Serhan, Alexandros Makriyannis
Tetrahedron Letters 2015 Volume 56(Issue 11) pp:1411-1415
Publication Date(Web):11 March 2015
DOI:10.1016/j.tetlet.2015.01.164
Novel prostaglandin-ethanolamide (PGE2-EA) and glycerol ester (2-PGE2-G) analogs were designed and synthesized to aid in the characterization of a putative prostamide receptor. Our design incorporates the electrophilic isothiocyanato and the photoactivatable azido groups at the terminal tail position of the prototype. Stereoselective Wittig and Horner–Wadsworth–Emmons reactions install the head and the tail moieties of the PGE2 skeleton. The synthesis is completed using Mitsunobu azidation and peptide coupling as the key steps. A chemoenzymatic synthesis for the 2-PGE2-G is described for first time.
Co-reporter:Alexandros Makriyannis
Journal of Medicinal Chemistry 2014 Volume 57(Issue 10) pp:3891-3911
Publication Date(Web):April 7, 2014
DOI:10.1021/jm500220s
My involvement with the field of cannabinoids spans close to 3 decades and covers a major part of my scientific career. It also reflects the robust progress in this initially largely unexplored area of biology. During this period of time, I have witnessed the growth of modern cannabinoid biology, starting from the discovery of its two receptors and followed by the characterization of its endogenous ligands and the identification of the enzyme systems involved in their biosynthesis and biotransformation. I was fortunate enough to start at the beginning of this new era and participate in a number of the new discoveries. It has been a very exciting journey. With coverage of some key aspects of my work during this period of “modern cannabinoid research,” this Award Address, in part historical, intends to give an account of how the field grew, the key discoveries, and the most promising directions for the future.
Co-reporter:David R. Janero and Alexandros Makriyannis
ACS Chemical Neuroscience 2014 Volume 5(Issue 11) pp:1097
Publication Date(Web):May 28, 2014
DOI:10.1021/cn5000875
Endocananbnoid-system G-protein coupled receptors (GPCRs) and transient receptor potential (TRP) cation channels are critical components of cellular biosignaling networks. These plasma-membrane proteins are pleiotropic in their ability to interact with and engage structurally diverse ligands. The endocannabinoid and TRP signaling systems overlap in their recognition properties with respect to select naturally occurring plant-derived ligands that belong to the terpene and lipid chemical classes, the overlap establishing a physiological connectivity between these two ubiquitous cell-signaling systems. Identification and pharmacological profiling of phytochemicals engaged by cannabinoid GPCRs and/or TRP channels has inspired the synthesis of novel designer ligands that interact with cannabinoid receptors and/or TRP channels as xenobiotics. Functional interplay between the endocannabinoid and TRP-channel signaling systems is responsible for the antinocifensive action of some synthetic cananbinoids (WIN55,212-2 and AM1241), vasorelaxation by the endocannabinoid N-arachidonylethanolamide (anandamide), and the pain-relief afforded by the synthetic anandamide analogue N-arachidonoylaminophenol (AM404), the active metabolite of the widely used nonprescription analgesic and antipyretic acetaminophen (paracetamol). The biological actions of some plant-derived cannabinoid-receptor (e.g., Δ9-tetrahydrocannabinol) or TRP-channel (e.g,, menthol) ligands either carry abuse potential themselves or promote the use of other addictive substances, suggesting the therapeutic potential for modulating these signaling systems for abuse-related disorders. The pleiotropic nature of and therapeutically relevant interactions between cananbinergic and TRP-channel signaling suggest the possibility of dual-acting ligands as drugs.Keywords: Drug discovery; endocananbinoid; G-protein coupled receptors; ion channels; ligands; phytocannabinoid; phytochemicals; signal transduction
Co-reporter:Rishi Sharma, Spyros P. Nikas, Jason Jianxin Guo, Srikrishnan Mallipeddi, JodiAnne T. Wood, and Alexandros Makriyannis
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 4) pp:400-404
Publication Date(Web):January 14, 2014
DOI:10.1021/ml4005304
As a part of our controlled-deactivation ligand development project, we recently disclosed a series of (−)-Δ8-tetrahydrocannabinols (THCs) with a metabolically labile ester group at the 2′-position of the side chain. Now, we have replaced the C-ring in the classical THC structure with a hydrolyzable seven-membered lactone. One of the synthesized analogues binds with high affinity to the CB1 receptor (Ki = 4.6 nM) and exhibits much lower affinities for the mCB2 and the hCB2. Also, in vitro functional characterization found the compound to be an agonist at rCB1. Consistent with our rational design, the lead cannabinergic lactone identified here is susceptible to metabolic inactivation by plasma esterases, while the respective acid metabolite is inactive at CB receptors. These results are highlighted with molecular modeling of the two regiosomeric lactones.Keywords: Baeyer−Villiger rearrangement; Cannabinoids; lactones;
Co-reporter:Ganesh A. Thakur ; Shama Bajaj ; Carol Paronis ; Yan Peng ; Anna L. Bowman ; Lawrence S. Barak ; Marc G. Caron ; Demon Parrish ; Jeffrey R. Deschamps
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:3904-3921
Publication Date(Web):April 26, 2013
DOI:10.1021/jm4000775
In previous studies, compound 1 (AM411), a 3-(1-adamantyl) analogue of the phytocannabinoid (−)-Δ8-tetrahydrocannabinol (Δ8-THC), was shown to have improved affinity and selectivity for the CB1 receptor. In this work, we further explored the role of the 1-adamantyl group at the C-3 position in a series of tricyclic cannabinoid analogues modified at the 9-northern aliphatic hydroxyl (NAH) position. Of these, 9-hydroxymethyl hexahydrocannabinol 11 (AM4054) exhibited high CB1 affinity and full agonist profile. In the cAMP assay, the 9-hydroxymethyl cannabinol analogue 24 (AM4089) had a partial agonist profile, with high affinity and moderate selectivity for rCB1 over hCB2. In vivo results in rat models of hypothermia and analgesia were congruent with in vitro data. Our in vivo data indicate that 3-(1-adamantyl) substitution, within NAH cannabinergics, imparts improved pharmacological profiles when compared to the corresponding, traditionally used 3-dimethylheptyl analogues and identifies 11 and 24 as potentially useful in vivo CB1 cannabinergic probes.
Co-reporter:Rishi Sharma ; Spyros P. Nikas ; Carol A. Paronis ; JodiAnne T. Wood ; Aneetha Halikhedkar ; Jason Jianxin Guo ; Ganesh A. Thakur ; Shashank Kulkarni ; Othman Benchama ; Jimit Girish Raghav ; Roger S. Gifford ; Torbjörn U. C. Järbe ; Jack Bergman
Journal of Medicinal Chemistry 2013 Volume 56(Issue 24) pp:10142-10157
Publication Date(Web):November 28, 2013
DOI:10.1021/jm4016075
We report an approach for obtaining novel cannabinoid analogues with controllable deactivation and improved druggability. Our design involves the incorporation of a metabolically labile ester group at the 2′-position on a series of (−)-Δ8-THC analogues. We have sought to introduce benzylic substituents α to the ester group which affect the half-lives of deactivation through enzymatic activity while enhancing the affinities and efficacies of individual ligands for the CB1 and CB2 receptors. The 1′-(S)-methyl, 1′-gem-dimethyl, and 1′-cyclobutyl analogues exhibit remarkably high affinities for both CB receptors. The novel ligands are susceptible to enzymatic hydrolysis by plasma esterases in a controllable manner, while their metabolites are inactive at the CB receptors. In further in vitro and in vivo experiments key analogues were shown to be potent CB1 receptor agonists and to exhibit CB1-mediated hypothermic and analgesic effects.
Co-reporter:Ioannis Karageorgos, Thomas E. Wales, David R. Janero, Nikolai Zvonok, V. Kiran Vemuri, John R. Engen, and Alexandros Makriyannis
Biochemistry 2013 Volume 52(Issue 29) pp:
Publication Date(Web):June 24, 2013
DOI:10.1021/bi400430k
Human monoacylglycerol lipase (hMGL) regulates endocannabinoid signaling primarily by deactivating the lipid messenger 2-arachidonoylglycerol. Agents that carbamylate hMGLs catalytic Ser122 constitute a leading class of therapeutically promising hMGL inhibitors. We have applied peptide-level hydrogen/deuterium exchange mass spectrometry to characterize hMGL’s conformational responses to two potent carbamylating inhibitors, AM6580 (irreversible) and AM6701 (slowly reversible). A dynamic, solvent-exposed lid domain is characteristic of hMGL’s solution conformation. Both hMGL inhibitors restricted backbone enzyme motility in the active-site region and increased substrate binding-pocket solvent exposure. Covalent reaction of AM6580 with hMGL generates a bulkier carbamylated Ser122 residue as compared to the more discrete Ser122 modification by AM6701, a difference reflected in AM6580’s more pronounced effect upon hMGL conformation. We demonstrate that structurally distinct carbamylating hMGL inhibitors generate particular conformational ensembles characterized by region-specific hMGL dynamics. By demonstrating the distinctive influences of two hMGL inhibitors on enzyme conformation, this study furthers our understanding at the molecular level of the dynamic features of hMGL interaction with small-molecule ligands.
Co-reporter:Jianqin Zhuang;De-Ping Yang;Spyros P. Nikas;Jianhong Zhao
The AAPS Journal 2013 Volume 15( Issue 2) pp:477-482
Publication Date(Web):2013 April
DOI:10.1208/s12248-013-9455-9
It has been reported that the endocannabinoid anandamide (AEA) binds to a class of fatty acid-binding proteins and serum albumin which can serve as carrier proteins and potentiate the cellular uptake of AEA and its intracellular translocation. Here, we employed 19F nuclear magnetic resonance spectroscopy to study the interactions of serum albumin with two inhibitors of fatty acid amide hydrolase (FAAH), the enzyme involved in the deactivation of anandamide. We found that, for both inhibitors AM5206 and AM5207, the primary binding site on serum albumin is drug site 1 located at subdomain IIA. Neither inhibitor binds to drug site 2. While AM5207 binds exclusively to drug site 1, AM5206 also interacts with other fatty acid-binding sites on serum albumin. Additionally, AM5206 has an affinity for serum albumin approximately one order of magnitude higher than that of AM5207. The data suggest that interactions of FAAH inhibitors with albumin may provide added advantages for their ability to modulate endocannabinoid levels for a range of applications including analgesia, antiemesis, and neuroprotection.
Co-reporter:Shakiru O. Alapafuja ; Spyros P. Nikas ; Indu T. Bharathan ; Vidyanand G. Shukla ; Mahmoud L. Nasr ; Anna L. Bowman ; Nikolai Zvonok ; Jing Li ; Xiaomeng Shi ; John R. Engen
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:10074-10089
Publication Date(Web):October 19, 2012
DOI:10.1021/jm301205j
Sulfonyl fluorides are known to inhibit esterases. Early work from our laboratory has identified hexadecyl sulfonylfluoride (AM374) as a potent in vitro and in vivo inhibitor of fatty acid amide hydrolase (FAAH). We now report on later generation sulfonyl fluoride analogs that exhibit potent and selective inhibition of FAAH. Using recombinant rat and human FAAH, we show that 5-(4-hydroxyphenyl)pentanesulfonyl fluoride (AM3506) has similar inhibitory activity for both the rat and the human enzyme, while rapid dilution assays and mass spectrometry analysis suggest that the compound is a covalent modifier for FAAH and inhibits its action in an irreversible manner. Our SAR results are highlighted by molecular docking of key analogs.
Co-reporter:Jay M. West, Nikolai Zvonok, Kyle M. Whitten, JodiAnne T. Wood, and Alexandros Makriyannis
Journal of Proteome Research 2012 Volume 11(Issue 2) pp:972-981
Publication Date(Web):2017-2-22
DOI:10.1021/pr200735a
N-Acylethanolamine-hydrolyzing acid amidase (NAAA) is a lysosomal enzyme that primarily degrades palmitoylethanolamine (PEA), a lipid amide that inhibits inflammatory responses. We developed a HEK293 cell line stably expressing the NAAA pro-enzyme (zymogen) and a single step chromatographic purification of the protein from the media. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry MALDI-TOF MS analysis of the zymogen (47.7 kDa) treated with peptide-N-glycosidase F (PNGase F) identified 4 glycosylation sites, and acid cleavage of the zymogen into α- and β-subunits (14.6 and 33.3 kDa) activated the enzyme. Size exclusion chromatography estimated the mass of the active enzyme as 45 ± 3 kDa, suggesting formation of an α/β heterodimer. MALDI-TOF MS fingerprinting covered more than 80% of the amino acid sequence, including the N-terminal peptides, and evidence for the lack of a disulfide bond between subunits. The significance of the cysteine residues was established by their selective alkylation resulting in almost complete loss of activity. The purified enzyme was kinetically characterized with PEA and a novel fluorogenic substrate, N-(4-methyl coumarin) palmitamide (PAMCA). The production of sufficient quantities of NAAA and a high throughput assay could be useful in discovering novel inhibitors and determining the structure and function of this enzyme.
Co-reporter:Ioannis Karageorgos, Nikolai Zvonok, David R. Janero, V. Kiran Vemuri, Vidyanand Shukla, Thomas E. Wales, John R. Engen, and Alexandros Makriyannis
ACS Chemical Neuroscience 2012 Volume 3(Issue 5) pp:393
Publication Date(Web):March 20, 2012
DOI:10.1021/cn3000263
In the mammalian central nervous system, monoacylglycerol lipase (MGL) is principally responsible for inactivating the endocannabinoid signaling lipid 2-arachidonoylglycerol (2-AG) and modulates cannabinoid-1 receptor (CB1R) desensitization and signal intensity. MGL is also a drug target for diseases in which CB1R stimulation may be therapeutic. To inform the design of human MGL (hMGL) inhibitors, we have engineered a Leu(Leu169;Leu176)-to-Ser(Ser169;Ser176) double hMGL mutant (sol-hMGL) which exhibited enhanced solubility properties, and we further mutated this variant by substituting its catalytic-triad Ser122 with Cys (sol-S-hMGL). The hMGL variants hydrolyzed both 2-AG and a fluorogenic reporter substrate with comparable affinities. Our results suggest that the hMGL cysteine mutant maintains the same overall architecture as wild-type hMGL. The results also underscore the superior nucleophilic nature of the reactive catalytic Ser122 residue as compared to that of Cys122 in the sol-S-hMGL mutant and suggest that the nucleophilic character of the Cys122 residue is not commensurately enhanced within the three dimensional architecture of hMGL. The interaction of the sol-hMGL variants with the irreversible inhibitors AM6580 and N-arachidonylmaleimide (NAM) and the reversible inhibitor AM10212 was profiled. LC/MS analysis of tryptic digests from sol-S-hMGL directly demonstrate covalent modification of this variant by NAM and AM6580, consistent with enzyme thiol alkylation and carbamoylation, respectively. These data provide insight into hMGL catalysis, the key role of the nucleophilic character of Ser122, and the mechanisms underlying hMGL inhibition by different classes of small molecules.Keywords: Active site; catalytic mechanism; drug design; enzyme inhibition; serine hydrolase
Co-reporter:Meghan Johnston, Shachi R. Bhatt, Surina Sikka, Richard W. Mercier, Jay M. West, Alexandros Makriyannis, S. John Gatley, Richard I. Duclos Jr.
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4585-4592
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.101
A series of N-formyl-α-amino acid esters of β-lactone derivatives structurally related to tetrahydrolipstatin (THL) and O-3841 were synthesized that inhibit human and murine diacylglycerol lipase (DAGL) activities. New ether lipid reporter compounds were developed for an in vitro assay to efficiently screen inhibitors of 1,2-diacyl-sn-glycerol hydrolysis and related lipase activities using fluorescence resonance energy transfer (FRET). A standardized thin layer chromatography (TLC) radioassay of diacylglycerol lipase activity utilizing the labeled endogenous substrate [1″-14C]1-stearoyl-2-arachidonoyl-sn-glycerol with phosphorimaging detection was used to quantify inhibition by following formation of the initial product [1″-14C]2-arachidonoylglycerol and further hydrolysis under the assay conditions to [1-14C]arachidonic acid.New N-formyl-α-amino acid analogs of tetrahydrolipstatin (THL) were synthesized that inhibit human and murine diacylglycerol lipases. New reporter compounds were developed to screen inhibitors of 1,2-diacyl-sn-glycerol hydrolysis activity using fluorescence resonance energy transfer (FRET). Diacylglycerol lipase (DAGL) activities were quantified using [1″-14C]1-stearoyl-2-arachidonoyl-sn-glycerol substrate in a thin layer chromatography (TLC) assay using phosphorimaging analysis.
Co-reporter:Dai Lu, Spyros P. Nikas, Xiu-Wen Han, Damon A. Parrish, Alexandros Makriyannis
Tetrahedron Letters 2012 Volume 53(Issue 35) pp:4636-4638
Publication Date(Web):29 August 2012
DOI:10.1016/j.tetlet.2012.05.165
Resorcinol derivatives are important building blocks in the synthesis of natural products and pharmaceutical compounds including cannabinoids. Here we describe the synthesis and the structural characterization of a key resorcinol which carries a fully restricted bridged bicyclic group. We also report a potential mechanism for the acid catalyzed condensation of (+)- or (−)-3-pinanol with 2,6-dimethoxyphenol. The synthesized resorcinol facilitates the development of novel conformationally restricted cannabinoid analogs.
Co-reporter:Spyros P. Nikas, Marsha D'Souza, Alexandros Makriyannis
Tetrahedron 2012 68(31) pp: 6329-6337
Publication Date(Web):
DOI:10.1016/j.tet.2012.05.010
Co-reporter:Alexandros Makriyannis, Spyros P. Nikas
Chemistry & Biology 2011 Volume 18(Issue 10) pp:1208-1209
Publication Date(Web):28 October 2011
DOI:10.1016/j.chembiol.2011.10.005
Aspirin triggers the biosynthesis of oxygenated metabolites from arachidonic, eicosapentaenoic, and docosahexaenoic (DHA) acids. In a preceding issue, Serhan et al. (2011) describe a novel aspirin-triggered DHA pathway for the biosynthesis of a potent anti-inflammatory and proresolving molecule.
Co-reporter:Elvis K. Tiburu;Sergiy Tyukhtenko;Han Zhou;David R. Janero
The AAPS Journal 2011 Volume 13( Issue 1) pp:92-98
Publication Date(Web):2011 March
DOI:10.1208/s12248-010-9244-7
G protein-coupled receptors (GPCRs) play critical physiological and therapeutic roles. The human cannabinoid 1 GPCR (hCB1) is a prime pharmacotherapeutic target for addiction and cardiometabolic disease. Our prior biophysical studies on the structural biology of a synthetic peptide representing the functionally significant hCB1 transmembrane helix 7 (TMH7) and its cytoplasmic extension, helix 8 (H8), [hCB1(TMH7/H8)] demonstrated that the helices are oriented virtually perpendicular to each other in membrane-mimetic environments. We identified several hCB1(TMH7/H8) structure-function determinants, including multiple electrostatic amino-acid interactions and a proline kink involving the highly conserved NPXXY motif. In phospholipid bicelles, TMH7 structure, orientation, and topology relative to H8 are dynamically modulated by the surrounding membrane phospholipid bilayer. These data provide a contextual basis for the present solid-state NMR study to investigate whether intermolecular interactions between hCB1(TMH7/H8) and its phospholipid environment may affect membrane-bilayer structure. For this purpose, we measured 1H–13C heteronuclear dipolar couplings for the choline, glycerol, and acyl-chain regions of dimyristoylphosphocholine in a magnetically aligned hCB1(TMH7/H8) bicelle sample. The results identify discrete regional interactions between hCB1(TMH7/H8) and membrane lipid molecules that increase phospholipid motion and decrease phospholipid order, indicating that the peptide’s partial traversal of the bilayer alters membrane structure. These data offer new insight into hCB1(TMH7/H8) properties and support the concept that the membrane bilayer itself may serve as a mechanochemical mediator of hCB1/GPCR signal transduction. Since interaction with its membrane environment has been implicated in hCB1 function and its modulation by small-molecule therapeutics, our work should help inform hCB1 pharmacology and the design of hCB1-targeted drugs.
Co-reporter:Heidi Teng, Ganesh A. Thakur, Alexandros Makriyannis
Bioorganic & Medicinal Chemistry Letters 2011 21(19) pp: 5999-6002
Publication Date(Web):
DOI:10.1016/j.bmcl.2011.07.017
Co-reporter:Grzegorz Godlewski, Shakiru O. Alapafuja, Sándor Bátkai, Spyros P. Nikas, Resat Cinar, László Offertáler, Douglas Osei-Hyiaman, Jie Liu, Bani Mukhopadhyay, Judith Harvey-White, Joseph Tam, Karel Pacak, Jacqueline L. Blankman, Benjamin F. Cravatt, Alexandros Makriyannis, George Kunos
Chemistry & Biology 2010 Volume 17(Issue 11) pp:1256-1266
Publication Date(Web):24 November 2010
DOI:10.1016/j.chembiol.2010.08.013
The enzyme fatty acid amide hydrolase (FAAH) catalyzes the in vivo degradation of the endocannabinoid anandamide, thus controlling its action at receptors. A novel FAAH inhibitor, AM3506, normalizes the elevated blood pressure and cardiac contractility of spontaneously hypertensive rats (SHR) without affecting these parameters in normotensive rats. These effects are due to blockade of FAAH and a corresponding rise in brain anandamide levels, resulting in CB1 receptor-mediated decrease in sympathetic tone. The supersensitivity of SHR to CB1 receptor-mediated cardiovascular depression is related to increased G protein coupling of CB1 receptors. Importantly, AM3506 does not elicit hyperglycemia and insulin resistance seen with other FAAH inhibitors or in FAAH−/− mice, which is related to its inability to inhibit FAAH in the liver due to rapid hepatic uptake and metabolism. This unique activity profile offers improved therapeutic value in hypertension.Highlights► Unique pharmacological profile of novel fatty acid amide hydrolase inhibitor ► Selectivity of AM3506 in FAAH inhibition in brain over liver in vivo ► AM3506 as centrally acting antihypertensive agent with minimal cardiovascular effects under normotensive conditions and without adverse metabolic effects
Co-reporter:Richard W. Mercier, Ying Pei, Lakshmipathi Pandarinathan, David R. Janero, Jing Zhang, Alexandros Makriyannis
Chemistry & Biology 2010 Volume 17(Issue 10) pp:1132-1142
Publication Date(Web):29 October 2010
DOI:10.1016/j.chembiol.2010.08.010
The human cannabinoid 2 GPCR (hCB2) is a prime therapeutic target. To define potential cysteine-related binding motifs critical to hCB2-ligand interaction, a library of hCB2 cysteine-substitution mutants and a novel, high-affinity biarylpyrazole hCB2 antagonist/inverse agonist (AM1336) functionalized to serve as a covalent affinity probe to target cysteine residues within (or in the microenvironment of) its hCB2 binding pocket were generated. The data provide direct experimental demonstration that both hCB2 TMH7 cysteines [i.e., C7.38(284) and C7.42(288)] are critical to optimal hCB2-AM1336 binding interaction and AM1336 pharmacological activity in a cell-based functional assay (cAMP formation). Elongating the AM1336 aliphatic side chain generated another novel hCB2 inverse agonist that binds covalently and selectively to C7.42(288) only. Identification of specific cysteine residues critical to hCB2 ligand interaction and function informs the structure-based design of hCB2-targeted medicines.Highlights► A novel hCB2 inverse agonist/covalent affinity probe has been generated ► Cysteine residues critical to hCB2 ligand interaction and function are identified ► The data inform the structure-based design of hCB2-targeted medicines
Co-reporter:Darryl D. Dixon ; Divakaramenon Sethumadhavan ; Tore Benneche ; April R. Banaag ; Marcus A. Tius ; Ganesh A. Thakur ; Anna Bowman ; JodiAnne T. Wood
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5656-5666
Publication Date(Web):July 1, 2010
DOI:10.1021/jm100390h
The aliphatic side chain plays a pivotal role in determining the cannabinergic potency of tricyclic classical cannabinoids. We have synthesized a series of analogues in which the C3 position is substituted either directly or through a one-carbon atom linker with an adamantylamine or with an oxa- or an oxazaadamantane. The oxaadamantane pharmacophore in analogue 16 showed the best binding profile for both receptors.
Co-reporter:Spyros P. Nikas ; Shakiru O. Alapafuja ; Ioannis Papanastasiou ; Carol A. Paronis ; Vidyanand G. Shukla ; Demetris P. Papahatjis ; Anna L. Bowman ; Aneetha Halikhedkar ; Xiuwen Han
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:6996-7010
Publication Date(Web):September 9, 2010
DOI:10.1021/jm100641g
In pursuit of a more detailed understanding of the structural requirements for the key side chain cannabinoid pharmacophore, we have extended our SAR to cover a variety of conformationally modified side chains within the 9-keto and 9-hydroxyl tricyclic structures. Of the compounds described here, those with a seven-atom long side chain substituted with a cyclopentyl ring at C1′ position have very high affinities for both CB1 and CB2 (0.97 nM < Ki < 5.25 nM), with no preference for either of the two receptors. However, presence of the smaller cyclobutyl group at the C1′ position leads to an optimal affinity and selectivity interaction with CB1. Thus, two of the C1′-cyclobutyl analogues, namely, (6aR,10aR)-3-(1-hexyl-cyclobut-1-yl)-6,6a,7,8,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo[b,d]pyran-9-one and (6aR,9R,10aR)-3-(1-hexyl-cyclobut-1-yl)-6a,7,8,9,10,10a-hexahydro-6,6-dimethyl-6H-dibenzo[b,d]pyran-1,9 diol (7e-β, AM2389), exhibited remarkably high affinities (0.84 and 0.16 nM, respectively) and significant selectivities (16- and 26-fold, respectively) for CB1. Compound 7e-β was found to exhibit exceptionally high in vitro and in vivo potency with a relatively long duration of action.
Co-reporter:NL Cluny;VK Vemuri;AP Chambers;CL Limebeer;H Bedard;JT Wood;B Lutz;A Zimmer;LA Parker;A Makriyannis;KA Sharkey
British Journal of Pharmacology 2010 Volume 161( Issue 3) pp:629-642
Publication Date(Web):
DOI:10.1111/j.1476-5381.2010.00908.x
BACKGROUND AND PURPOSE Cannabinoid CB1 receptor antagonists reduce food intake and body weight, but clinical use in humans is limited by effects on the CNS. We have evaluated a novel cannabinoid antagonist (AM6545) designed to have limited CNS penetration, to see if it would inhibit food intake in rodents, without aversive effects.
EXPERIMENTAL APPROACH Cannabinoid receptor binding studies, cAMP assays, brain penetration studies and gastrointestinal motility studies were carried out to assess the activity profile of AM6545. The potential for AM6545 to induce malaise in rats and the actions of AM6545 on food intake and body weight were also investigated.
KEY RESULTS AM6545 binds to CB1 receptors with a Ki of 1.7 nM and CB2 receptors with a Ki of 523 nM. AM6545 is a neutral antagonist, having no effect on cAMP levels in transfected cells and was less centrally penetrant than AM4113, a comparable CB1 receptor antagonist. AM6545 reversed the effects of WIN55212-2 in an assay of colonic motility. In contrast to AM251, AM6545 did not produce conditioned gaping or conditioned taste avoidance in rats. In rats and mice, AM6545 dose-dependently reduced food intake and induced a sustained reduction in body weight. The effect on food intake was maintained in rats with a complete subdiaphragmatic vagotomy. AM6545 inhibited food intake in CB1 receptor gene-deficient mice, but not in CB1/CB2 receptor double knockout mice.
CONCLUSIONS AND IMPLICATIONS Peripherally active, cannabinoid receptor antagonists with limited brain penetration may be useful agents for the treatment of obesity and its complications.
Co-reporter:Ioannis Karageorgos, Sergiy Tyukhtenko, Nikolai Zvonok, David R. Janero, Christine Sallum and Alexandros Makriyannis
Molecular BioSystems 2010 vol. 6(Issue 8) pp:1381-1388
Publication Date(Web):12 May 2010
DOI:10.1039/C004515B
Intramolecular hydrogen bonding is an important determinant of enzyme structure, catalysis, and inhibitor action. Monoacylglycerol lipase (MGL) modulates cannabinergic signaling as the main enzyme responsible for deactivating 2-arachidonoylglycerol (2-AG), a primary endocannabinoid lipid messenger. By enhancing tissue-protective 2-AG tone, targeted MGL inhibitors hold therapeutic promise for managing pain and treating inflammatory and neurodegenerative diseases. We report study of purified, solubilized human MGL (hMGL) to explore the details of hMGL catalysis by using two known covalent hMGL inhibitors, the carbamoyl tetrazole AM6701 and N-arachidonoylmaleimide (NAM), that act through distinct mechanisms. Using proton nuclear magnetic resonance spectroscopy (NMR) with purified wild-type and mutant hMGLs, we have directly observed a strong hydrogen-bond network involving Asp239 and His269 of the catalytic triad and neighboring Leu241 and Cys242 residues. hMGL inhibition by AM6701 alters this hydrogen-bonding pattern through subtle active-site structural rearrangements without influencing hydrogen-bond occupancies. Rapid carbamoylation of hMGL Ser122 by AM6701 and elimination of the leaving group is followed by a slow hydrolysis of the carbamate group, ultimately regenerating catalytically competent hMGL. In contrast, hMGL titration with NAM, which leads to cysteine alkylation, stoichiometrically decreases the population of the active-site hydrogen bonds. NAM prevents reformation of this network, and in this manner inhibits hMGL irreversibly. These data provide detailed molecular insight into the distinctive mechanisms of two covalent hMGL inhibitors and implicate a hydrogen-bond network as a structural feature of hMGL catalytic function.
Co-reporter:Jianxin Guo, Richard I. Duclos Jr., V. Kiran Vemuri, Alexandros Makriyannis
Tetrahedron Letters 2010 Volume 51(Issue 27) pp:3465-3469
Publication Date(Web):7 July 2010
DOI:10.1016/j.tetlet.2010.04.077
The conformational structures of the hormone 17β-estradiol (E2) and the epimeric 17α-estradiol determined by solution NMR spectroscopy and restrained molecular dynamics calculations found a single low energy conformation.
Co-reporter:Elvis K. Tiburu, Stefano V. Gulla, Mark Tiburu, David R. Janero, David E. Budil and Alexandros Makriyannis
Biochemistry 2009 Volume 48(Issue 22) pp:
Publication Date(Web):May 4, 2009
DOI:10.1021/bi802235w
The influence of membrane environment on human cannabinoid 1 (hCB1) receptor transmembrane helix (TMH) conformational dynamics was investigated by solid-state NMR and site-directed spin labeling/EPR with a synthetic peptide, hCB1(T377-E416), corresponding to the receptor’s C-terminal component, i.e., TMH7 and its intracellular α-helical extension (H8) (TMH7/H8). Solid-state NMR experiments with mechanically aligned hCB1(T377-E416) specifically 2H- or 15N-labeled at Ala380 and reconstituted in membrane-mimetic dimyristoylphosphocholine (DMPC) or 1-palmitoyl-2-oleoyl-sn-glycerophosphocholine (POPC) bilayers demonstrate that the conformation of the TMH7/H8 peptide is more heterogeneous in the thinner DMPC bilayer than in the thicker POPC bilayer. As revealed by EPR studies on hCB1(T377-E416) spin-labeled at Cys382 and reconstituted into the phospholipid bilayers, the spin label partitions actively between hydrophobic and hydrophilic environments. In the DMPC bilayer, the hydrophobic component dominates, regardless of temperature. Mobility parameters (ΔH0−1) are 0.3 and 0.73 G for the peptide in the DMPC or POPC bilayer environment, respectively. Interspin distances of doubly labeled hCB1(T377-E416) peptide reconstituted into a TFE/H2O mixture or a POPC or DMPC bilayer were estimated to be 10.6 ± 0.5, 16.8 ± 1, and 11.6 ± 0.8 Å, respectively. The extent of coupling (≥50%) between spin labels located at i and i + 4 in a TFE/H2O mixture or a POPC bilayer is indicative of an α-helical TMH conformation, whereas the much lower coupling (14%) when the peptide is in a DMPC bilayer suggests a high degree of peptide conformational heterogeneity. These data demonstrate that hCB1(T377-E416) backbone dynamics as well as spin-label rotameric freedom are sensitive to and altered by the peptide’s phospholipid bilayer environment, which exerts a dynamic influence on the conformation of a TMH critical to signal transmission by the hCB1 receptor.
Co-reporter:Anna L. Bowman
Journal of Computer-Aided Molecular Design 2009 Volume 23( Issue 11) pp:
Publication Date(Web):2009 November
DOI:10.1007/s10822-009-9289-9
Monoacylglycerol lipase (MGL) is primarily responsible for the hydrolysis of 2-arachidonoylglycerol (2-AG), an endocannabinoid with full agonist activity at both cannabinoid receptors. Increased tissue 2-AG levels consequent to MGL inhibition are considered therapeutic against pain, inflammation, and neurodegenerative disorders. However, the lack of MGL structural information has hindered the development of MGL-selective inhibitors. Here, we detail a fully refined homology model of MGL which preferentially identifies MGL inhibitors over druglike noninhibitors. We include for the first time insight into the active-site geometry and potential hydrogen-bonding interactions along with molecular dynamics simulations describing the opening and closing of the MGL helical-domain lid. Docked poses of both the natural substrate and known inhibitors are detailed. A comparison of the MGL active-site to that of the other principal endocannabinoid metabolizing enzyme, fatty acid amide hydrolase, demonstrates key differences which provide crucial insight toward the design of selective MGL inhibitors as potential drugs.
Co-reporter:Shakiru O. Alapafuja, Spyros P. Nikas, Vidyanand G. Shukla, Ioannis Papanastasiou, Alexandros Makriyannis
Tetrahedron Letters 2009 50(50) pp: 7028-7031
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.09.167
Co-reporter:Rishi Sharma, Subramanian K. Vadivel, Richard I. Duclos Jr., Alexandros Makriyannis
Tetrahedron Letters 2009 50(42) pp: 5780-5782
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.07.079
Co-reporter:Dai Lu ; Jianxin Guo ; Richard I. Duclos ; Jr.; Anna L. Bowman
Journal of Medicinal Chemistry 2008 Volume 51(Issue 20) pp:6393-6399
Publication Date(Web):October 1, 2008
DOI:10.1021/jm8005299
Structure−activity relationship studies of classical cannabinoid analogues have established that the C3 aliphatic side chain plays a pivotal role in determining cannabinergic potency. In earlier work, we provided evidence for the presence of subsites within the CB1 and CB2 cannabinoid receptor binding domains that can accommodate bulky conformationally defined substituents at the C3 alkyl side chain pharmacophore of classical cannabinoids. We have now extended this work with the synthesis of a series of Δ8-THC analogues in which bornyl substituents are introduced at the C3 position. Our results indicate that, for optimal interactions with both CB1 and CB2 receptors, the bornyl substituents need to be within close proximity of the tricyclic core of Δ8-THC and that the conformational space occupied by the C3 substituents influences CB1/CB2 receptor subtype selectivity.
Co-reporter:Jianxin Guo ; De-Ping Yang ; Ravi Chari ; Xiaoyu Tian ; Spiro Pavlopoulos ; Dai Lu
Journal of Medicinal Chemistry 2008 Volume 51(Issue 21) pp:6793-6799
Publication Date(Web):October 4, 2008
DOI:10.1021/jm800766x
Magnetically aligned bicelles were used as a model membrane to study the orientation and dynamic properties of two cannabinoids (Δ8-THC and Me-Δ8-THC) using 31P and 2H NMR. The uniform alignment of the bicelles allowed us to obtain well resolved deuterium spectra from a solution NMR spectrometer. The preferred orientations of Δ8-THC and Me-Δ8-THC were calculated on the basis of the measurements of individual quadrupolar splittings. Our results agree with previous experiments using multilamellar membranes as well as with molecular dynamics simulation data described here. In conjunction with our earlier report using small and fast tumbling bicelles, the present work of well aligned bicelles shows that bicelle preparations can provide either pseudoisotropic or anisotropic NMR spectra to study the conformation, orientation, and dynamic properties of ligands in membrane bilayers. Such data are of critical value for understanding the interactions of lipophilic drug molecules with membrane proteins.
Co-reporter:Ying Pei, Richard W. Mercier, Jenine K. Anday, Ganesh A. Thakur, Alexander M. Zvonok, Dow Hurst, Patricia H. Reggio, David R. Janero, Alexandros Makriyannis
Chemistry & Biology 2008 Volume 15(Issue 11) pp:1207-1219
Publication Date(Web):24 November 2008
DOI:10.1016/j.chembiol.2008.10.011
The extensive physiological influence of transmission through the CB2 cannabinoid receptor makes this G protein-coupled receptor (GPCR) a promising therapeutic target for treating neuropathic pain, inflammation, and immune disorders. However, there is little direct structural information pertaining to either GPCR or CB2-receptor ligand recognition and activation. The present work helps characterize experimentally the ligand-binding interactions of the human CB2 (hCB2) receptor. This study illustrates how our overall experimental approach, “ligand-assisted protein structure” (LAPS), affords direct determination of the requirements for ligand binding to the hCB2 receptor and discrimination among the binding motifs for ligands that activate therapeutically relevant GPCRs.
Co-reporter:Nikolai Zvonok, Lakshmipathi Pandarinathan, John Williams, Meghan Johnston, Ioannis Karageorgos, David R. Janero, Srinivasan C. Krishnan, Alexandros Makriyannis
Chemistry & Biology 2008 Volume 15(Issue 8) pp:854-862
Publication Date(Web):25 August 2008
DOI:10.1016/j.chembiol.2008.06.008
The active site of recombinant hexa-histidine-tagged human monoacylglycerol lipase (hMGL) is characterized by mass spectrometry using the inhibitors 5-((biphenyl-4-yl)methyl)-N,N-dimethyl-2H-tetrazole-2-carboxamide (AM6701), and N-arachidonylmaleimide (NAM) as probes. Carbamylation of Ser129 by AM6701 in the putative hMGL catalytic triad demonstrates this residue's essential role in catalysis. Partial NAM alkylation of hMGL cysteine residues 215 and/or 249 was sufficient to achieve ∼80% enzyme inhibition. Although Cys215 and/or Cys249 mutations to alanine(s) did not affect hMGL hydrolytic activity as compared with nonmutated hMGL, the C215A displayed heightened NAM sensitivity, whereas the C249A evidenced reduced NAM sensitivity. These data conclusively demonstrate a sulfhydryl-based mechanism for NAM inhibition of hMGL in which Cys249 is of paramount importance. Identification of amino acids critical to the catalytic activity and pharmacological modulation of hMGL informs the design of selective MGL inhibitors as potential drugs.
Co-reporter:Nikolai Zvonok, John Williams, Meghan Johnston, Lakshmipathi Pandarinathan, David R. Janero, Jing Li, Srinivasan C. Krishnan and Alexandros Makriyannis
Journal of Proteome Research 2008 Volume 7(Issue 5) pp:2158-2164
Publication Date(Web):2017-2-22
DOI:10.1021/pr700839z
The serine hydrolase monoacylglycerol lipase (MGL) modulates endocannabinoid signaling in vivo by inactivating 2-arachidonoylglycerol (2-AG), the main endogenous agonist for central CB1 and peripheral CB2 cannabinoid receptors. To characterize this key endocannabinoid enzyme by mass spectrometry-based proteomics, we first overexpressed recombinant hexa-histidine-tagged human MGL (hMGL) in Escherichia coli and purified it in a single chromatographic step with high yield (≈30 mg/L). With 2-AG as substrate, hMGL displayed an apparent Vmax of 25 µmol/(µg min) and Km of 19.7 µM, an affinity for 2-AG similar to that of native rat-brain MGL (rMGL) (Km = 33.6 µM). hMGL also demonstrated a comparable affinity (Km ≈ 8–9 µM) for the novel fluorogenic substrate, arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE), in a sensitive, high-throughput fluorometric MGL assay. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) unequivocably demonstrated the mass (34 126 Da) and purity of this hMGL preparation. After in-solution tryptic digestion, hMGL full proteomic characterization was carried out, which showed (1) an absence of intramolecular disulfide bridges in the functional, recombinant enzyme and (2) the post-translational removal of the enzyme’s N-terminal methionine. Availability of sufficient quantities of pure, well-characterized hMGL will enable further molecular and structural profiling of this key endocannabinoid-system enzyme.
Co-reporter:Atmaram D. Khanolkar ; Dai Lu ; Mohab Ibrahim ; Richard I. Duclos; Jr. ; Ganesh A. Thakur ; T. Phillip Malan; Jr. ; Frank Porreca ; Vijayabaskar Veerappan ; Xiaoyu Tian ; Clifford George ; Damon A. Parrish ; Demetris P. Papahatjis
Journal of Medicinal Chemistry 2007 Volume 50(Issue 26) pp:6493-6500
Publication Date(Web):November 27, 2007
DOI:10.1021/jm070441u
The identification of the CB2 cannabinoid receptor has provided a novel target for the development of therapeutically useful cannabinergic molecules. We have synthesized benzo[c]chromen-6-one analogs possessing high affinity and selectivity for this receptor. These novel compounds are structurally related to cannabinol (6,6,9-trimethyl-3-pentyl-6H-benzo[c]chromen-1-ol), a natural constituent of cannabis with modest CB2 selectivity. Key pharmacophoric features of the new selective agonists include a 3-(1′,1′-dimethylheptyl) side chain and a 6-oxo group on the cannabinoid tricyclic structure that characterizes this class of compounds as “cannabilactones.” Our results suggest that the six-membered lactone pharmacophore is critical for CB2 receptor selectivity. Optimal receptor subtype selectivity of 490-fold and subnanomolar affinity for the CB2 receptor is exhibited by a 9-hydroxyl analog 5 (AM1714), while the 9-methoxy analog 4b (AM1710) had a 54-fold CB2 selectivity. X-ray crystallography and molecular modeling show the cannabilactones to have a planar ring conformation. In vitro testing revealed that the novel compounds are CB2 agonists, while in vivo testing of cannabilactones 4b and 5 found them to possess potent peripheral analgesic activity.
Co-reporter:Xiaoyu Tian, Spiro Pavlopoulos, De-Ping Yang, Alexandros Makriyannis
Biochimica et Biophysica Acta (BBA) - Biomembranes (September 2011) Volume 1808(Issue 9) pp:
Publication Date(Web):September 2011
DOI:10.1016/j.bbamem.2010.11.026
Two key commonly used cannabinergic agonists, CP55940 and WIN55212-2, are investigated for their effects on the lipid membrane bilayer using 2H solid state NMR, and the results are compared with our earlier work with delta-9-tetrahydrocannabinol (Δ9-THC). To study the effects of these ligands we used hydrated bilayers of dipalmitoylphosphatidylcholine (DPPC) deuterated at the 2′ and 16′ positions of both acyl chains with deuterium atoms serving as probes for the dynamic and phase changes at the membrane interface and at the bilayer center respectively. All three cannabinergic ligands lower the phospholipid membrane phase transition temperature, increase the lipid sn-2 chain order parameter at the membrane interface and decrease the order at the center of the bilayer.Our studies show that the cannabinoid ligands induce lateral phase separation in the lipid membrane at physiological temperatures. During the lipid membrane phase transition, the cooperative dynamic process whereby the C-2H segments at the interface and center of the bilayer spontaneously reach the fast exchange regime (2H NMR timescale) is distinctively modulated by the two cannabinoids. Specifically, CP55940 is slightly more efficient at inducing liquid crystalline-type 2H NMR spectral features at the membrane interface compared to WIN55212-2. In contrast, WIN55212-2 has a far superior ability to induce liquid crystalline-type spectral features at the center of the bilayer, and it increases the order parameter of the sn-1 chain in addition to the sn-2 chain of the lipids. These observations suggest the cannabinoid ligands may influence lipid membrane domain formations and there may be contributions to their cannabinergic activities through lipid membrane microdomain related mechanisms. Our work demonstrates that experimental design strategies utilizing specifically deuterium labeled lipids yield more detailed insights concerning the properties of lipid bilayers.Research Highlights► Lipid membrane effects of two cannabinoid agonists, WIN55212-2 and CP55940, are investigated. ► DPPC lipid membrane with specific deuterium labels provides extra details for solid state 2H NMR studies. ► The cannabinoid agonists induce lipid lateral phase separation and promote microdomain formation. ► The cooperative process of the lipid acyl chain C-2H segment may be modulated by the cannabinoids. ► The agonists lower the lipid phase transition temperature and modulate the chain order distinctively.
Co-reporter:Elvis K. Tiburu, Anna L. Bowman, Jochem O. Struppe, David R. Janero, Hava K. Avraham, Alexandros Makriyannis
Biochimica et Biophysica Acta (BBA) - Biomembranes (May 2009) Volume 1788(Issue 5) pp:
Publication Date(Web):May 2009
DOI:10.1016/j.bbamem.2009.02.002
Little direct information is available regarding the influence of membrane environment on transmembrane (TM) G-protein-coupled receptor (GPCR) conformation and dynamics. The human CB1 cannabinoid receptor (hCB1) is a prominent GPCR pharmacotherapeutic target in which helix 7 appears critical to ligand recognition. We have chemically synthesized a hCB1 peptide corresponding to a segment of TM helix 7 and the entire contiguous helix 8 domain (fourth cytoplasmic loop) and reconstituted it in defined phospholipid-bilayer model membranes. Using an NMR-based strategy combined with molecular dynamics simulations, we provide the first direct experimental description of the orientation of hCB1 helix 7 in phospholipid membranes of varying thickness and the mechanism by which helix-7 conformation adjusts to avoid hydrophobic mismatch. Solid-state 15N NMR data show that hCB1 helices 7 and 8 reconstituted into phospholipid bilayers are oriented in a TM and in-plane (i.e., parallel to the phospholipid membrane surface) fashion, respectively. TM helix orientation is influenced by the thickness of the hydrophobic membrane bilayer as well as the interaction of helix 8 with phospholipid polar headgroups. Molecular dynamics simulations show that a decrease in phospholipid chain-length induces a kink at P394 in TM helix 7 to avoid hydrophobic mismatch. Thus, the NP(X)nY motif found in hCB1 and highly conserved throughout the GPCR superfamily is important for flexing helix 7 to accommodate bilayer thickness. Dynamic modulation of hCB1-receptor TM helix conformation by its membrane environment may have general relevance to GPCR structure and function.
Co-reporter:Jason J. Guo, De-Ping Yang, Xiaoyu Tian, V. Kiran Vemuri, Dali Yin, Chen Li, Richard I. Duclos Jr, Lingling Shen, Xiaoyu Ma, David R. Janero, Alexandros Makriyannis
Biochimica et Biophysica Acta (BBA) - Biomembranes (February 2016) Volume 1858(Issue 2) pp:
Publication Date(Web):February 2016
DOI:10.1016/j.bbamem.2015.11.015
•Estradiol (E2) adopts a “horizontal” orientation within lipid membrane.•The dynamic motions of E2 in membrane is highly asymmetric.•E2 orientation, location, and dynamics are determined by 2H-NMR and 2D 1H-13C HSQC.•All four rings of the E2 molecule are located near the membrane interface.•Both 3- and17β-OH of E2 have H-bonds with polar groups on the phospholipid membrane.Non-genomic membrane effects of estrogens are of great interest because of the diverse biological activities they may elicit. To further our understanding of the molecular features of the interaction between estrogenic hormones and membrane bilayers, we have determined the preferred orientation, location, and dynamic properties of 17β-estradiol (E2) in two different phospholipid membrane environments using 2H-NMR and 2D 1H-13C HSQC in conjunction with molecular dynamics simulations. Unequivocal spectral assignments to specific 2H labels were made possible by synthesizing six selectively deuterated E2 molecules. The data allow us to conclude that the E2 molecule adopts a nearly “horizontal” orientation in the membrane bilayer with its long axis essentially perpendicular to the lipid acyl-chains. All four rings of the E2 molecule are located near the membrane interface, allowing both the E2 3-OH and the 17β-OH groups to engage in hydrogen bonding and electrostatic interactions with polar phospholipid groups. The findings augment our knowledge of the molecular interactions between E2 and membrane bilayer and highlight the asymmetric nature of the dynamic motions of the rigid E2 molecule in a membrane environment.Figure optionsDownload full-size imageDownload high-quality image (366 K)Download as PowerPoint slide

![6H-Dibenzo[b,d]pyran-9-methanol,3-(1,1-dimethylheptyl)-6a,7,8,9,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-,(6aR,9R,10aR)-](http://img.cochemist.com/ccimg/140900/140835-14-9.png)
![6H-Dibenzo[b,d]pyran-9-methanol,3-(1,1-dimethylheptyl)-6a,7,8,9,10,10a-hexahydro-1-hydroxy-6,6-dimethyl-,(6aR,9R,10aR)-](http://img.cochemist.com/ccimg/140900/140835-14-9_b.png)


![1-(4-Chloro-phenyl)-3-[3-(6-pyrrolidin-1-yl-pyridin-2-yl)-phenyl]-urea](http://img.cochemist.com/ccimg/877300/877202-74-9.png)
![1-(4-Chloro-phenyl)-3-[3-(6-pyrrolidin-1-yl-pyridin-2-yl)-phenyl]-urea](http://img.cochemist.com/ccimg/877300/877202-74-9_b.png)
![2-((2'-(5-Ethyl-3,4-diphenyl-1H-pyrazol-1-yl)-[1,1'-biphenyl]-3-yl)oxy)acetic acid](http://img.cochemist.com/ccimg/300700/300657-03-8.png)
![2-((2'-(5-Ethyl-3,4-diphenyl-1H-pyrazol-1-yl)-[1,1'-biphenyl]-3-yl)oxy)acetic acid](http://img.cochemist.com/ccimg/300700/300657-03-8_b.png)

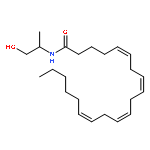
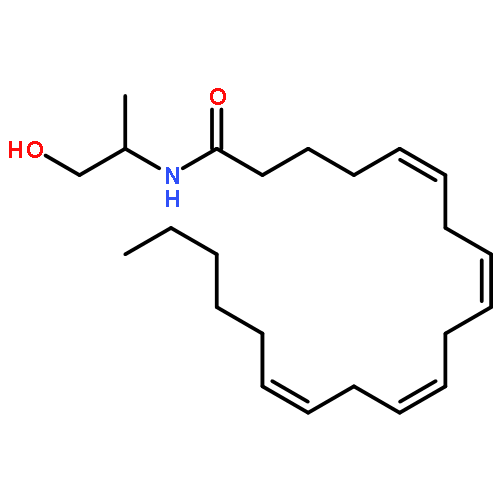
![5,8,11,14-Eicosatetraenamide,N-[(1R)-2-hydroxy-1-methylethyl]-, (5Z,8Z,11Z,14Z)-](http://img.cochemist.com/ccimg/157200/157182-49-5.png)
![5,8,11,14-Eicosatetraenamide,N-[(1R)-2-hydroxy-1-methylethyl]-, (5Z,8Z,11Z,14Z)-](http://img.cochemist.com/ccimg/157200/157182-49-5_b.png)
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2.png)
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2_b.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7.png)
![1-Propanamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/115400/115306-75-7_b.png)
![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7.png)
![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7_b.png)
![CP 55,940;(-)-CIS-3-[2-HYDROXY-4-(1,1-DIMETHYLHEPTYL)PHENYL]-TRANS-4-(3-HYDROXYPROPYL)CYCLOHEXANOL](http://img.cochemist.com/ccimg/83100/83002-04-4.png)
![CP 55,940;(-)-CIS-3-[2-HYDROXY-4-(1,1-DIMETHYLHEPTYL)PHENYL]-TRANS-4-(3-HYDROXYPROPYL)CYCLOHEXANOL](http://img.cochemist.com/ccimg/83100/83002-04-4_b.png)


![2H-Cyclopenta[b]furan-4-carboxaldehyde,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]hexahydro-2-oxo-, [3aR-(3aa,4a,5b,6aa)]- (9CI)](http://img.cochemist.com/ccimg/64100/64091-14-1.png)
![2H-Cyclopenta[b]furan-4-carboxaldehyde,5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]hexahydro-2-oxo-, [3aR-(3aa,4a,5b,6aa)]- (9CI)](http://img.cochemist.com/ccimg/64100/64091-14-1_b.png)
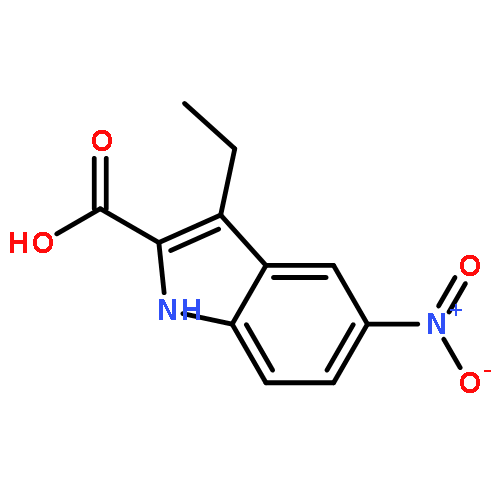
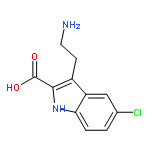
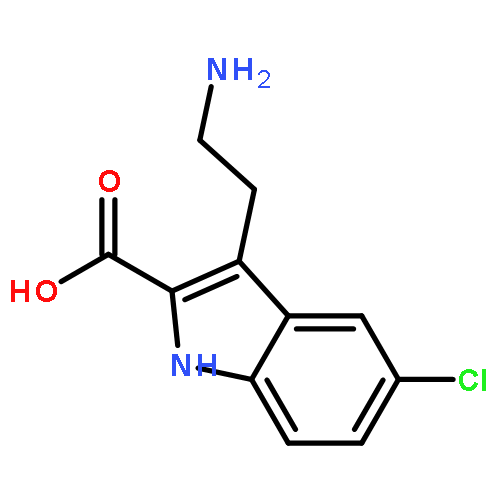
![1H-Indole-2-carboxylic acid,5-chloro-3-[2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/55800/55747-56-3.png)
![1H-Indole-2-carboxylic acid,5-chloro-3-[2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/55800/55747-56-3_b.png)




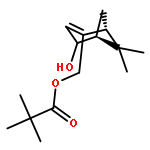
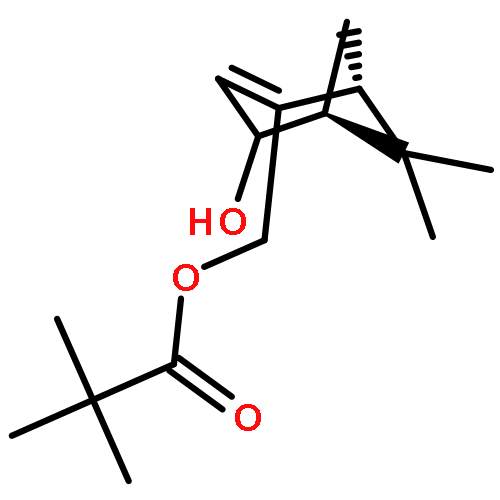
![Bicyclo[3.1.1]hept-3-en-2-one, 6,6-dimethyl-, (1R)-](http://img.cochemist.com/ccimg/35500/35408-03-8.png)
![Bicyclo[3.1.1]hept-3-en-2-one, 6,6-dimethyl-, (1R)-](http://img.cochemist.com/ccimg/35500/35408-03-8_b.png)
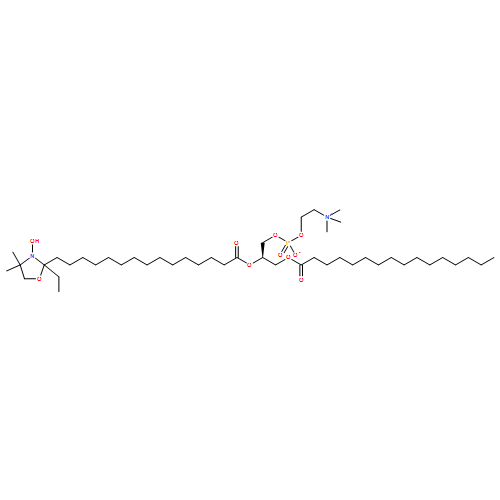
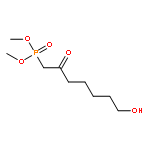
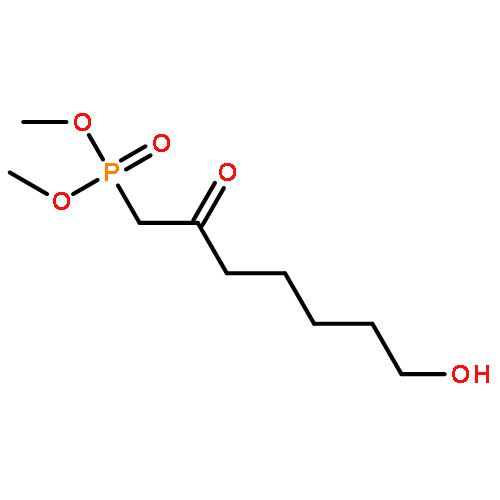
![3,5,9-Trioxa-4-phosphapentadecan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/34600/34506-67-7.png)
![3,5,9-Trioxa-4-phosphapentadecan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxohexyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/34600/34506-67-7_b.png)


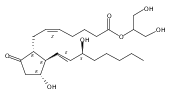
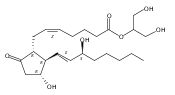
![(3aR,4R,5R,6aS)-5-Hydroxy-4-((S,E)-3-hydroxyoct-1-en-1-yl)hexahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-67-1.png)
![(3aR,4R,5R,6aS)-5-Hydroxy-4-((S,E)-3-hydroxyoct-1-en-1-yl)hexahydro-2H-cyclopenta[b]furan-2-one](http://img.cochemist.com/ccimg/26100/26054-67-1_b.png)

![6-chloro-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/18000/17952-83-9.png)
![6-chloro-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/18000/17952-83-9_b.png)



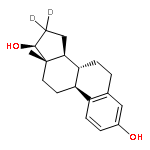
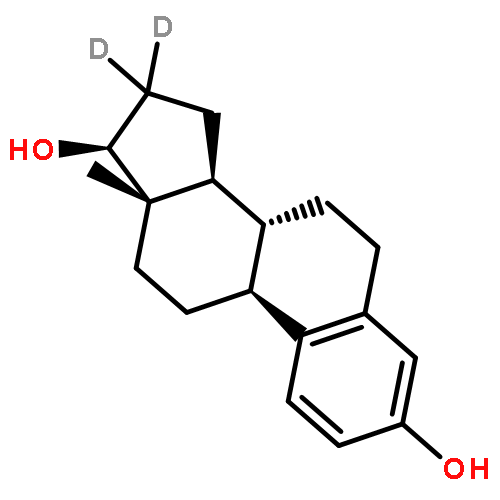


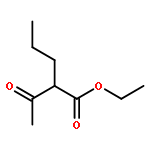
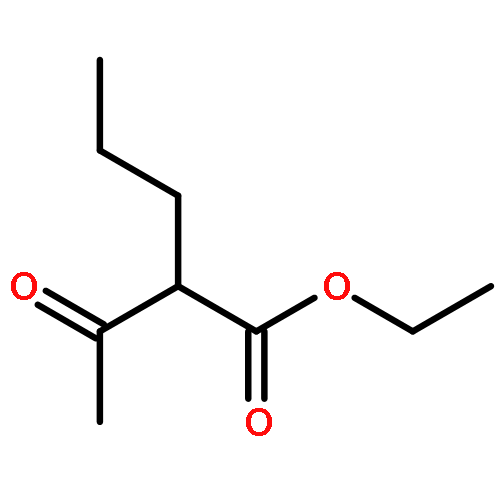



![BICYCLO[3.1.1]HEPT-2-ENE-2,4-DIOL, 6,6-DIMETHYL-, DIACETATE, (1R,5S)-](http://img.cochemist.com/ccimg/487600/487578-94-9.png)
![BICYCLO[3.1.1]HEPT-2-ENE-2,4-DIOL, 6,6-DIMETHYL-, DIACETATE, (1R,5S)-](http://img.cochemist.com/ccimg/487600/487578-94-9_b.png)
![2-Propanamine, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/175800/175717-73-4.png)
![2-Propanamine, 1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/175800/175717-73-4_b.png)


![Bicyclo[3.1.1]hept-3-ene-2,2-diol, 6,6-dimethyl-, diacetate, (1R,5R)-](http://img.cochemist.com/ccimg/82100/82078-77-1.png)
![Bicyclo[3.1.1]hept-3-ene-2,2-diol, 6,6-dimethyl-, diacetate, (1R,5R)-](http://img.cochemist.com/ccimg/82100/82078-77-1_b.png)