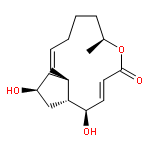
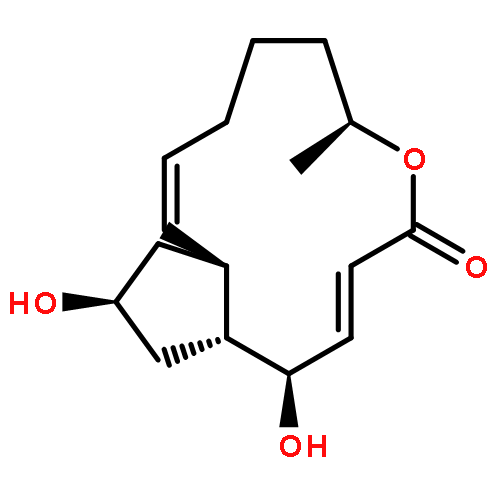
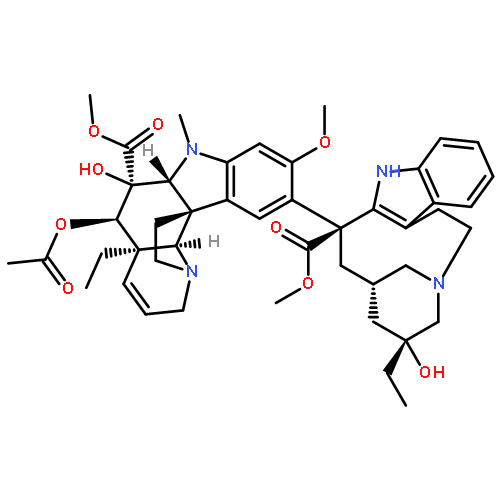
Co-reporter:Masatoshi Takeiri, Kana Horie, Daisuke Takahashi, Mariko Watanabe, Ryoichi Horie, Siro Simizu and Kazuo Umezawa
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 15) pp:3053-3059
Publication Date(Web):03 Feb 2012
DOI:10.1039/C2OB07104E
NF-κB is a transcription factor for the immune activation and tissue stability, but excess activation of NF-κB often causes inflammation and cancer. An NF-κB component RelB is involved in B-cell maturation and autoimmunity. In the present research we studied the role of the RelB DNA binding domain on cellular stability and importin affinity. We prepared a RelB protein mutated at Arg141 to Ala and Tyr142 to Ala (AA mutant) having no DNA binding activity. The stability of this mutant protein was greatly reduced compared with that of the wild-type protein. We also constructed a nuclear localization signal-inactivated mutant of RelB, and found that this mutant was also unstable in the cells. Thus, RelB destabilization was caused by the loss of DNA binding possibly because of the change in cellular localization. The mutation also decreased the affinity to importin-α5 decreasing the nuclear localization. Our newly discovered NF-κB inhibitor (−)-DHMEQ binds to a specific Cys residue in RelB to inhibit DNA binding and also decreased the stability and importin affinity. These findings would indicate that the DNA binding activity of this transcription factor is a crucial for its stability and intracellular localization.
Co-reporter:Yukihiro Niitsu, Masatoshi Hakamata, Yuko Goto, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai and Kazuo Umezawa
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 12) pp:4635-4641
Publication Date(Web):29 Mar 2011
DOI:10.1039/C1OB05205E
Dehydroxymethylepoxyquinomicin (DHMEQ, 1a) is a specific and potent inhibitor of NF-κB, and it is now being developed as an anti-inflammatory and anticancer agent. While previously only the (2S,3S,4S)-form had been available from the racemate by using lipase-catalyzed enantioselective resolution, in the present study a new route for production of the (2R,3R,4R)-form was established by use of a chemoenzymatic approach. (1R*,2R*,3R*)-2,3-Epoxy-5-N-[(2-hydroxybenzoyl)amino]-4,4-dimethoxycyclohex-5-en-1-ol (2a) was hexanoylated on both secondary and phenolic hydroxy groups, and subjected to Burkholderia cepacialipase-catalyzed hydrolysis. The reaction proceeded in a highly enantioselective manner (E >500) to give (1S,2S,3S)-2a in an enantiomerically pure state. Several chemical steps of transformation from the enzyme reaction product gave (2R,3R,4R)-DHMEQ (1a) without any loss of stereochemical purity. Moreover, we newly found that (2R,3R,4R)-DHMEQ activated Nrf2, which is a transcription factor that induces the expression of multiple antioxidant enzymes. It activated Nrf2 in a promoter reporter assay. It also increased the expression of target antioxidant proteins and cancelled ROS-induced cell death in a neuronal cell line. Thus, (2R,3R,4R)-DHMEQ was efficiently prepared by a newly designed route using lipase, and it may be useful as a new anti-inflammatory agent.
Co-reporter:Masatoshi Takeiri;Miyuki Tachibana;Ayumi Kaneda;Ayumi Ito
Inflammation Research 2011 Volume 60( Issue 9) pp:
Publication Date(Web):2011 September
DOI:10.1007/s00011-011-0348-z
We have previously synthesized a novel piperidine compound, 3-[(dodecylthiocarbonyl)methyl]glutarimide (DTCM-glutarimide), that inhibits LPS-induced NO production, and in the present research we studied further the anti-inflammatory activity of DTCM-glutarimide in a macrophage cell line and in mice bearing transplanted hearts.Mouse macrophage-like RAW264.7 cells were employed for the evaluation of cellular inflammatory activity. DTCM-glutarimide was synthesized in our laboratory. The AP-1 activity was measured by nuclear translocation and phosphorylation. For the heart transplantation experiment, male C57BL/6 (H-2b) and BALB/c (H-2d) mice were used as donor and recipient, respectively. DTCM-glutarimide was administered intraperitoneally.DTCM-glutarimide inhibited the LPS-induced expression of iNOS and COX-2 in macrophages; but, unexpectedly, it did not inhibit LPS-induced NF-κB activation. Instead, it inhibited the nuclear translocation of both c-Jun and c-Fos. It also inhibited LPS-induced c-Jun phosphorylation. Moreover, it inhibited the mixed lymphocyte reaction in primary cultures of mouse spleen cells; and furthermore, in mice it prolonged the graft survival in heart transplantation experiments.The novel piperidine compound, DTCM-glutarimide, was found to be a new inhibitor of macrophage activation, inhibiting AP-1 activity. It also inhibited graft rejection in mice, and thus may be a candidate for an anti-inflammatory agent.
Co-reporter:Masahiko Morioka, Akihito Kamizono, Hirosato Takikawa, Akihisa Mori, Hiroaki Ueno, Shu-ichiro Kadowaki, Yoshihide Nakao, Kuniki Kato, Kazuo Umezawa
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1143-1148
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.041
Bone deficiency causes osteoporosis and often decreases quality of life in patients with rheumatoid arthritis. Estrogens are known to protect elderly women from bone loss. Synthesis of new estradiol–bisphosphonate conjugates (E2–BPs) was accomplished and their in vivo activity as bone-specific estrogens were examined. Among them, MCC-565 showed selective estrogenic activity in bones; but it showed little estrogenic activity in the uterus. We also found that the linker moiety in E2–BPs was essential for the absorption and specificity of the conjugates.Synthesis of new estradiol–bisphosphonate conjugates (E2–BPs) was accomplished and their in vivo activity as bone-specific estrogens examined. Among them, MCC-565 showed selective estrogenic activity in bones; but it showed little estrogenic activity in the uterus.
Co-reporter:Eriko Suzuki, Hideki Ogura, Kuniki Kato, Izumi Takei, Yasuaki Kabe, Hiroshi Handa, Kazuo Umezawa
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 17) pp:6188-6195
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmc.2009.07.062
Conophylline, a vinca alkaloid extracted from the tropical plant Ervatamia microphylla, has been shown to induce the differentiation of insulin-producing β-cells in cultured cells and in animals. However, its mechanism of action and the molecular target have remained unclear. Therefore, we prepared a fishing probe with conophylline to identify the target protein by using latex nano-beads, which are newly innovated tools for affinity-purification. With these conophylline-linked nano-beads, we found that conophylline directly interacted with ARL6IP. ARL6IP may thus be involved in the mechanism of cellular differentiation of β-cells, and this probe should be useful to find other target proteins.We prepared conophylline-linked nano-beads and conophylline was found to directly interact with ARL6IP.
Co-reporter:Ikuko Kozawa, Kuniki Kato, Toshiaki Teruya, Kiyotake Suenaga, Kazuo Umezawa
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 18) pp:5380-5382
Publication Date(Web):15 September 2009
DOI:10.1016/j.bmcl.2009.07.123
Previously we found that (−)-DHMEQ, a specific NF-κB inhibitor, covalently bound to a specific cysteine of NF-κB component proteins. In the course of formation of the (−)-DHMEQ and protected cysteine conjugate, we observed an unusual intramolecular N→O acyl group migration.We observed an unusual intramolecular N→O acyl group migration in the course or formation of the (−)-DHMEQ and protected cysteine conjugate.
Co-reporter:Mizuki Yamamoto ; Ryouichi Horie ; Masatoshi Takeiri ; Ikuko Kozawa
Journal of Medicinal Chemistry 2008 Volume 51(Issue 18) pp:5780-5788
Publication Date(Web):August 26, 2008
DOI:10.1021/jm8006245
Previously, we designed and synthesized a potent NF-κB inhibitor, DHMEQ. Although DHMEQ showed potent anti-inflammatory and anticancer activities in animals, its molecular target has not been elucidated. In the present study, its target protein was found to be p65 and other Rel homology proteins. We found that (−)-DHMEQ bound to p65 covalently with a 1:1 stoichiometry by conducting SPR and MALDI-TOF MS analyses. MS analysis of the chymotrypsin-digested peptide suggested the binding of (−)-DHMEQ to a Cys residue. Formation of Cys/(−)-DHMEQ adduct in the protein was supported by chemical synthesis of the adduct. Substitution of specific Cys in p65 and other Rel homology proteins resulted in the loss of (−)-DHMEQ binding. (−)-DHMEQ is the first NF-κB inhibitor that was proven to bind to the specific Cys by chemical methodology. These findings may explain the highly selective inhibition of NF-κB and the low toxic effect of (−)-DHMEQ in cells and animals.
Co-reporter:Taichi Takahashi, Osamu Ohno, Yoko Ikeda, Ryuichi Sawa, Yoshiko Homma, Masayuki Igarashi and Kazuo Umezawa
The Journal of Antibiotics 2006 59(1) pp:35-43
Publication Date(Web):
DOI:10.1038/ja.2006.6
Compounds that inactivate lipopolysaccharide (LPS) activity have the potential of being new anti-inflammatory agents. Therefore, we searched among microbial secondary metabolites for compounds that inhibited LPS-stimulated adhesion between human umbilical vein endothelial cells (HUVEC) and HL-60 cells. By this screening, we found a cyclic lipopeptide surfactin from the culture broth of Bacillus sp. BML752-121F2 to be inhibitory. The addition of the surfactin prior to the LPS stimulation decreased HL-60 cell-HUVEC adhesion without showing any cytotoxicity. We confirmed that surfactin inhibited LPS-induced expression of ICAM-1 and VCAM-1 in HUVEC. It also inhibited the cellular adhesion induced by lipid A, the active component of LPS; but it did not inhibit TNF- or IL-1-induced cell adhesion. Then, surfactin was shown to suppress the interaction of lipid A with LPS-binding protein (LBP) that mediates the transport of LPS to its receptors. Finally, surface plasmon resonance (SPR) analysis revealed the surfactin to interact reversibly with lipid A. Thus, this Bacillus surfactin was shown to be an inhibitor of LPS-induced signal transduction, directly interacting with LPS.
Co-reporter:Kazuo Umezawa, Yoko Ikeda, Osamu Kawase, Hiroshi Naganawa and Shinichi Kondo
Organic & Biomolecular Chemistry 2001 (Issue 13) pp:1550-1553
Publication Date(Web):15 Jun 2001
DOI:10.1039/B101942M
Polyoxypeptin A is a hexadepsipeptide, isolated from Streptomyces, containing the novel amino acid (2S,3R)-3-hydroxy-3-methylproline. Its biosynthetic pathway is studied by incorporation of stable isotope-labelled amino acids and carboxylic acids. (2S,3R)-3-Hydroxy-3-methylproline is shown to be derived from isoleucine but not from proline. Isoleucine is also incorporated into the alkyl moiety of the C15 acyl side chain, possibly through α-methylbutyryl-CoA. The piperazic acid moieties are synthesized from glutamate. Other hydroxylated amino acids are shown to be derived from each corresponding amino acid.
Co-reporter:Naoki Matsumoto, Akiko Ariga, Sakino To-e, Hikaru Nakamura, Naoki Agata, Shin-ichi Hirano, Jun-ichiro Inoue, Kazuo Umezawa
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 9) pp:865-869
Publication Date(Web):1 May 2000
DOI:10.1016/S0960-894X(00)00114-1
In order to develop new inhibitors of NF-κB activation, we designed and synthesized dehydroxymethyl derivatives of epoxyquinomicin C, namely, DHM2EQ and its regioisomer DHM3EQ. These derivatives were synthesized from 2,5-dimethoxyaniline in 5 steps. Since DHM2EQ was more active and less toxic than DHM3EQ, its stereochemical configuration was determined by X-ray crystallographic analysis. Each enantiomer of the protected DHM2EQ was separated by a chiral column and deprotected. DHM2EQ inhibited TNF-α-induced activation of NF-κB in human T cell leukemia cells, and also inhibited collagen-induced arthritis in a rheumatoid model in mice.
Co-reporter:Masaki Itabashi, Kaoru Segawa, Yoko Ikeda, Shinichi Kondo, Hiroshi Naganawa, Takashi Koyano, Kazuo Umezawa
Carbohydrate Research 1999 Volume 323(1–4) pp:57-62
Publication Date(Web):12 January 1999
DOI:10.1016/S0008-6215(99)00255-4
Microbial and plant secondary metabolites were screened for compounds that are selectively cytotoxic to mutant p53-expressing mouse fibroblasts. As a result, furcreastatin, a novel steroidal saponin, was isolated from an EtOH extract of the leaves of Furcraeafoetida. Furcreastatin consisted of hecogenin as the aglycone and a hexasaccharide containing d-galactose, l-rhamnose and four d-glucose residues. The structure was determined to be (3β,5α,25R)-3-hydroxyspirostan-12-one 3-O-[α-l-Rhap-(1→4)-β-d-Glcp-(1→3)-{β-d-Glcp-(1→3)-β-d-Glcp-(1→2)}-β-d-Glcp-(1→4)-β-d-Galp] by extensive NMR spectroscopic studies. Furcreastatin decreased the viability of mutant p53-overexpressing cells with an ED50 of 4.0 μg/mL, and decreased that of the parental cell-line with an ED50 of 9.6 μg/mL.
Co-reporter:Naoyuki Komura, Yoko Ikeda, Natsuko Masuda, Yoji Umezawa, Keisuke Ito, Masahiro Kizaki, Kazuo Umezawa
Leukemia Research (March 2007) Volume 31(Issue 3) pp:301-313
Publication Date(Web):1 March 2007
DOI:10.1016/j.leukres.2006.07.015
All-trans retinoic acid (ATRA) induces the differentiation of acute promyelocytic leukemia (APL) cells into neutrophils. UF-1 cells were established from an ATRA-resistant APL patient, and were previously shown to possess a single amino acid (or nucleotide) substitution, Arg276Trp, in their ATRA receptor. In the present research, we designed several ATRA derivatives having a hydrophobic alkyl ketone moiety instead of the negatively charged carboxylic acid moiety. Among them the ethyl ketone derivative, Et-ketone ATRA, was shown to induce the differentiation of UF-1 cells when assessed in terms of intracellular ROS production. It also induced the formation of PML NBs and expression of CD11b antigen marker and p21, transcriptional targets of RARα. Et-ketone ATRA did not induce these phenotypic changes in wild-type APL NB4 cells. Furthermore, we found that Et-ketone ATRA induced apoptosis selectively in UF-1 cells, i.e., not in other leukemic cells. The induction of apoptosis was shown to be partly due to the up-regulation of Bax protein. Thus, Et-ketone ATRA selectively induced differentiation and apoptosis in ATRA-resistant APL UF-1 cells, and is likely to be useful for the clinical treatment of the Arg276Trp-type of ATRA-resistant APL.
Co-reporter:Tamami Yasukagawa, Yuki Niwa, Siro Simizu, Kazuo Umezawa
FEBS Letters (21 May 2012) Volume 586(Issue 10) pp:1504-1509
Publication Date(Web):21 May 2012
DOI:10.1016/j.febslet.2012.04.007
It has been demonstrated that potassium channels (K+ channels) play significant roles in some malignant phenotypes. Here, we provide the first evidence that treatment with glybenclamide, an ATP-sensitive K+ channel blocker, inhibited cell migration in an ovarian clear cell carcinoma cell line, ES-2. Treatment with glybenclamide or knockdown by siRNA targeted against K+ channel subunits demonstrated the suppression of ovarian cancer cell invasion, which occurred via inhibition of PDGF-AA secretion. Therefore, our findings suggest that K+ channel blockers may be useful chemotherapeutic drugs for blocking the invasiveness of ovarian cancers.Highlights► Glybenclamide is an orally active K-channel blocker widely used for type II diabetes. ► It was found to inhibit PDGF-mediated cellular invasion in ovarian carcinoma cells. ► Thus, K-channel activity is likely to be a new target of anti-metastasis agent.
Co-reporter:Masatoshi Takeiri, Kana Horie, Daisuke Takahashi, Mariko Watanabe, Ryoichi Horie, Siro Simizu and Kazuo Umezawa
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 15) pp:NaN3059-3059
Publication Date(Web):2012/02/03
DOI:10.1039/C2OB07104E
NF-κB is a transcription factor for the immune activation and tissue stability, but excess activation of NF-κB often causes inflammation and cancer. An NF-κB component RelB is involved in B-cell maturation and autoimmunity. In the present research we studied the role of the RelB DNA binding domain on cellular stability and importin affinity. We prepared a RelB protein mutated at Arg141 to Ala and Tyr142 to Ala (AA mutant) having no DNA binding activity. The stability of this mutant protein was greatly reduced compared with that of the wild-type protein. We also constructed a nuclear localization signal-inactivated mutant of RelB, and found that this mutant was also unstable in the cells. Thus, RelB destabilization was caused by the loss of DNA binding possibly because of the change in cellular localization. The mutation also decreased the affinity to importin-α5 decreasing the nuclear localization. Our newly discovered NF-κB inhibitor (−)-DHMEQ binds to a specific Cys residue in RelB to inhibit DNA binding and also decreased the stability and importin affinity. These findings would indicate that the DNA binding activity of this transcription factor is a crucial for its stability and intracellular localization.
Co-reporter:Yukihiro Niitsu, Masatoshi Hakamata, Yuko Goto, Toshinori Higashi, Mitsuru Shoji, Takeshi Sugai and Kazuo Umezawa
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 12) pp:NaN4641-4641
Publication Date(Web):2011/03/29
DOI:10.1039/C1OB05205E
Dehydroxymethylepoxyquinomicin (DHMEQ, 1a) is a specific and potent inhibitor of NF-κB, and it is now being developed as an anti-inflammatory and anticancer agent. While previously only the (2S,3S,4S)-form had been available from the racemate by using lipase-catalyzed enantioselective resolution, in the present study a new route for production of the (2R,3R,4R)-form was established by use of a chemoenzymatic approach. (1R*,2R*,3R*)-2,3-Epoxy-5-N-[(2-hydroxybenzoyl)amino]-4,4-dimethoxycyclohex-5-en-1-ol (2a) was hexanoylated on both secondary and phenolic hydroxy groups, and subjected to Burkholderia cepacialipase-catalyzed hydrolysis. The reaction proceeded in a highly enantioselective manner (E >500) to give (1S,2S,3S)-2a in an enantiomerically pure state. Several chemical steps of transformation from the enzyme reaction product gave (2R,3R,4R)-DHMEQ (1a) without any loss of stereochemical purity. Moreover, we newly found that (2R,3R,4R)-DHMEQ activated Nrf2, which is a transcription factor that induces the expression of multiple antioxidant enzymes. It activated Nrf2 in a promoter reporter assay. It also increased the expression of target antioxidant proteins and cancelled ROS-induced cell death in a neuronal cell line. Thus, (2R,3R,4R)-DHMEQ was efficiently prepared by a newly designed route using lipase, and it may be useful as a new anti-inflammatory agent.

![2-Hydroxy-N-((1S,2S,6S)-2-hydroxy-5-oxo-7-oxabicyclo[4.1.0]hept-3-en-3-yl)benzamide](http://img.cochemist.com/ccimg/287200/287194-40-5.png)
![2-Hydroxy-N-((1S,2S,6S)-2-hydroxy-5-oxo-7-oxabicyclo[4.1.0]hept-3-en-3-yl)benzamide](http://img.cochemist.com/ccimg/287200/287194-40-5_b.png)
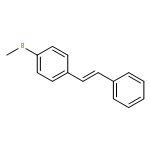






















![Benzenemethanamine, N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/58300/58259-35-1.png)
![Benzenemethanamine, N-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/58300/58259-35-1_b.png)










![2H-Pyrano[2'',3'':5',6']benz[1',2':6,7]indeno[1,2-b]indol-3(4bH)-one,5,6,6a,7,12,12b,12c,13,14,14a-decahydro-4b-hydroxy-2-(1-hydroxy-1-methylethyl)-12b,12c-dimethyl-,(2R,4bS,6aS,12bS,12cR,14aS)-](http://img.cochemist.com/ccimg/57200/57186-25-1.png)
![2H-Pyrano[2'',3'':5',6']benz[1',2':6,7]indeno[1,2-b]indol-3(4bH)-one,5,6,6a,7,12,12b,12c,13,14,14a-decahydro-4b-hydroxy-2-(1-hydroxy-1-methylethyl)-12b,12c-dimethyl-,(2R,4bS,6aS,12bS,12cR,14aS)-](http://img.cochemist.com/ccimg/57200/57186-25-1_b.png)