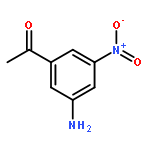
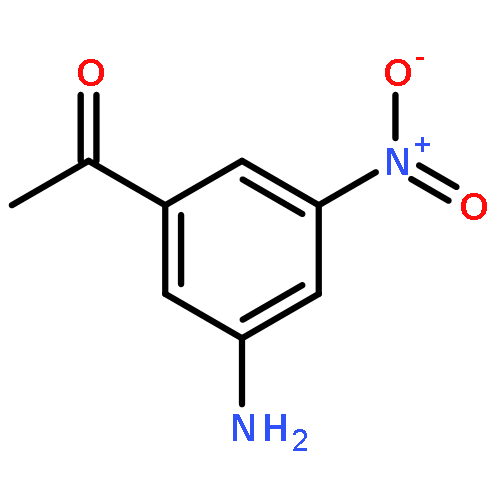
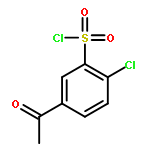
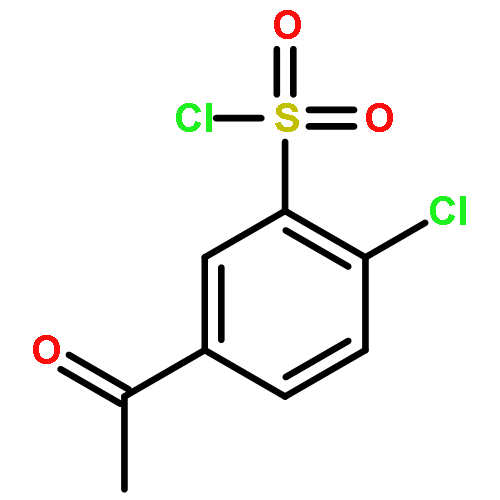
Co-reporter:Na Ye ; Chuan-Huizi Chen ; TianTian Chen ; Zilan Song ; Jin-Xue He ; Xia-Juan Huan ; Shan-Shan Song ; Qiufeng Liu ; Yi Chen ; Jian Ding ; Yechun Xu ; Ze-Hong Miao ;Ao Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 7) pp:2885-2903
Publication Date(Web):March 8, 2013
DOI:10.1021/jm301825t
A series of benzo[de][1,7]naphthyridin-7(8H)-ones possessing a functionalized long-chain appendage have been designed and evaluated as novel PARP1 inhibitors. The initial effort led to the first-generation PARP1 inhibitor 26 bearing a terminal phthalazin-1(2H)-one framework and showing remarkably high PARP1 inhibitory activity (0.31 nM) but only moderate potency in the cell. Further effort generated the second-generation lead 41, showing high potency against both the PARP1 enzyme and BRCA-deficient cells, especially for the BRCA1-deficient MDA-MB-436 cells (CC50 < 0.26 nM). Mechanistic studies revealed that the new PARP1 inhibitors significantly inhibited H2O2-triggered PARylation in SKOV3 cells, induced cellular accumulation of DNA double-strand breaks, and impaired cell-cycle progression in BRCA2-deficient cells. Significant potentiation on the cytotoxicity of Temozolomide was also observed. The unique structural character and exceptionally high potency of 41 made it stand out as a promising drug candidate worthy for further evaluation.
Co-reporter:Yiquan Zou, Lei Xu, Wuyan Chen, Yiping Zhu, Tiantian Chen, Yan Fu, Li Li, Lanping Ma, Bing Xiong, Xin Wang, Jian Li, Jianhua He, Haiyan Zhang, Yechun Xu, Jia Li, Jingkang Shen
European Journal of Medicinal Chemistry 2013 Volume 68() pp:270-283
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.06.027
•We explored the SAR by combining click chemistry and in situ screening assay.•Pro70 and Thr72 were the critical components for binding with these inhibitors.•Based on crystal structures, pyrazole was discovered as a novel C-terminus.•Pyrazole derivatives showed good activities and better selectivity for BACE1.We recently discovered and reported dual inhibitor 5 of AChE and BACE1 with N-benzylpiperidine ethyl as C-terminus. Compound 5 showed potent inhibitory activities for BACE1, and could reduce endogenous Aβ1–40 production in APP transgenic mice. In present work, we rapidly identified substituted triazole as the C-terminus of compound 5 by replacing the benzylpiperidine ethyl group with click chemistry and tested these synthesized compounds by in situ screening assay. As revealed by the crystal structures of BACE1 in complex with our triazole compound 12, we found that Pro70 and Thr72 located in the flap region were the critical components for binding with these inhibitors. With the aid of the crystal structure, a new series of five-membered heterocyclic compounds was prepared in order to explore the structure–activity relationship (SAR) of this class of molecules. From these efforts, pyrazole was discovered as a novel C-terminus of BACE1 inhibitors. After further modification of pyrazole with variable substituents, compound 37 exhibited good potency in enzyme inhibition assay (IC50 = 0.025 μM) and compound 33 showed moderate inhibition effects on Aβ production of APP transfected HEK293 cells. Moreover, these pyrazole derivatives demonstrated good selectivity versus cathepsin D. Our results indicated that the vicinity of Pro70 and Thr72 might be utilized as a subsite, and the discovered pyrazole derivatives might provide useful hints for developing novel BACE1 inhibitors as anti-AD drugs.We combined the click chemistry and in situ screening technologies to fast explore the SAR of C terminus of our previous compound. Further modification identified the pyrazole group as a good substituent for BACE1 inhibitors in the enzymatic, cellular inhibition assays.
Co-reporter:Xudong Gong, Guan Wang, Jing Ren, Zheng Liu, Zhen Wang, Tiantian Chen, Xiaojun Yang, Xiangrui Jiang, Jingshan Shen, Hualiang Jiang, Haji Akber Aisa, Yechun Xu, Jianfeng Li
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 17) pp:4944-4947
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmcl.2013.06.062
The substituents both at the 6-position of the 5-bromopyrimidinone ring and at the 5′-position of the phenyl ring of 5-bromopyrimidin-4(3H)-ones were explored. 5-Bromo-6-isopropyl-2-(2-propoxy-phenyl)pyrimidin-4(3H)-one was identified as a new scaffold for potent PDE5 inhibitors. The crystal structures of PDE5/2e and PDE5/10a complexes provided a structural basis for the inhibition of 5-bromopyrimidinones to PDE5. In addition, it was also found that there is a great tolerance for the substitution at the 5′-position of the phenyl ring of 5-bormopyrimidinones and the resulted compound 13a has the highest inhibition activity to PDE5 (IC50, 1.7 nM).
Co-reporter:Yanxia Liu ; Wei Zhang ; Li Li ; Lilibeth A. Salvador ; Tiantian Chen ; Wuyan Chen ; Kevin M. Felsenstein ; Thomas B. Ladd ; Ashleigh R. Price ; Todd E. Golde ; Jianhua He ; Yechun Xu ; Yingxia Li ;Hendrik Luesch
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10749-10765
Publication Date(Web):November 26, 2012
DOI:10.1021/jm301630s
Inspired by marine cyanobacterial natural products, we synthesized modified peptides with a central statine-core unit, characteristic for aspartic protease inhibition. A series of tasiamide B analogues inhibited BACE1, a therapeutic target in Alzheimer’s disease. We probed the stereospecificity of target engagement and determined additional structure–activity relationships with respect to BACE1 and related aspartic proteases, cathepsins D and E. We cocrystallized selected inhibitors with BACE1 to reveal the structural basis for the activity. Hybrid molecules that combine features of tasiamide B and an isophthalic acid moiety-containing sulfonamide showed nanomolar cellular activity. Compounds were screened in a series of rigorous complementary cell-based assays. We measured secreted APP ectodomain (sAPPβ), membrane bound carboxyl terminal fragment (CTF), levels of β-amyloid (Aβ) peptides and selectivity for β-secretase (BACE1) over γ-secretase. Prioritized compounds showed reasonable stability in vitro and in vivo, and our most potent inhibitor showed efficacy in reducing Aβ levels in the rodent brain.
Co-reporter:Guan Wang ; Zheng Liu ; Tiantian Chen ; Zhen Wang ; Huaiyu Yang ; Mingyue Zheng ; Jing Ren ; Guanghui Tian ; Xiaojun Yang ; Li Li ; Jianfeng Li ; Jin Suo ; Rongxia Zhang ; Xiangrui Jiang ; Nicholas Kenneth Terrett ; Jingshan Shen ; Yechun Xu ;Hualiang Jiang
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10540-10550
Publication Date(Web):November 8, 2012
DOI:10.1021/jm301159y
Cyclic nucleotide phosphodiesterase type 5 (PDE5) is a prime drug target for treating the diseases associated with a lower level of the cyclic guanosine monophosphate (cGMP), which is a specific substrate for PDE5 hydrolysis. Here we report a series of novel PDE5 inhibitors with the new scaffold of the monocyclic pyrimidin-4(3H)-one ring developed using the structure-based discovery strategy. In total, 37 derivatives of the pyrimidin-4(3H)-ones, were designed, synthesized, and evaluated for their inhibitory activities to PDE5, resulting in 25 compounds with IC50 ranging from 1 to 100 nM and 11 compounds with IC50 ranging from 1 to 10 nM. Compound 5, 5,6-diethyl-2-[2-n-propoxy-5-(4-methyl-1-piperazinylsulfonyl)phenyl]pyrimid-4(3H)-one, the most potent compound, has an excellent IC50 (1.6 nM) in vitro and a good efficacy in a rat model of erection. It thus provides a potential candidate for the further development into a new drug targeting PDE5.
Co-reporter:Kui Wu, Jing Ai, Qiufeng Liu, TianTian Chen, Ailing Zhao, Xia Peng, Yuanxiang Wang, Yinchun Ji, Qizheng Yao, Yechun Xu, Meiyu Geng, Ao Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 20) pp:6368-6372
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmcl.2012.08.075
Two series of new analogues were designed by replacing the quinoline scaffold of our earlier lead 2 (zgwatinib) with quinoxaline and pyrido[2,3-d]pyrimidine frameworks. Moderate c-Met inhibitory activity was observed in the quinoxaline series. Among the pyrido[2,3-d]pyrimidine series, compounds 13a–c possessing an O-linkage were inactive, whilst the N-linked analogues 15a–c retained c-Met inhibitory potency. Highest activity was observed in the 3-nitrobenzyl analog 15b that showed an IC50 value of 6.5 nM. Further structural modifications based on this compound were undergoing.
Co-reporter:Zhijian Xu ; Zheng Liu ; Tong Chen ; TianTian Chen ; Zhen Wang ; Guanghui Tian ; Jing Shi ; Xuelan Wang ; Yunxiang Lu ; Xiuhua Yan ; Guan Wang ; Hualiang Jiang ; Kaixian Chen ; Shudong Wang ; Yechun Xu ; Jingshan Shen ;Weiliang Zhu
Journal of Medicinal Chemistry 2011 Volume 54(Issue 15) pp:5607-5611
Publication Date(Web):June 29, 2011
DOI:10.1021/jm200644r
For proof-of-concept of halogen bonding in drug design, a series of halogenated compounds were designed based on a lead structure as new inhibitors of phosphodiesterase type 5. Bioassay results revealed a good correlation between the measured bioactivity and the calculated halogen bond energy. Our X-ray crystal structures verified the existence of the predicted halogen bonds, demonstrating that the halogen bond is an applicable tool in drug design and should be routinely considered in lead optimization.
Co-reporter:Yechun Xu, Jacques-Philippe Colletier, Martin Weik, Guangrong Qin, Hualiang Jiang, Israel Silman, Joel L. Sussman
Biophysical Journal (15 December 2010) Volume 99(Issue 12) pp:
Publication Date(Web):15 December 2010
DOI:10.1016/j.bpj.2010.10.047
The principal role of acetylcholinesterase is termination of nerve impulse transmission at cholinergic synapses, by rapid hydrolysis of the neurotransmitter acetylcholine to acetate and choline. Its active site is buried at the bottom of a deep and narrow gorge, at the rim of which is found a second anionic site, the peripheral anionic site. The fact that the active site is so deeply buried has raised cogent questions as to how rapid traffic of substrate and products occurs in such a confined environment. Various theoretical and experimental approaches have been used to solve this problem. Here, multiple conventional molecular dynamics simulations have been performed to investigate the clearance of the product, thiocholine, from the active-site gorge of acetylcholinesterase. Our results indicate that thiocholine is released from the peripheral anionic site via random pathways, while three exit routes appear to be favored for its release from the active site, namely, along the axis of the active-site gorge, and through putative back- and side-doors. The back-door pathway is that via which thiocholine exits most frequently. Our results are in good agreement with kinetic and kinetic-crystallography studies. We propose the use of multiple molecular dynamics simulations as a fast yet accurate complementary tool in structural studies of enzymatic trafficking.

![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2_b.png)


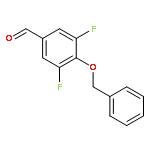
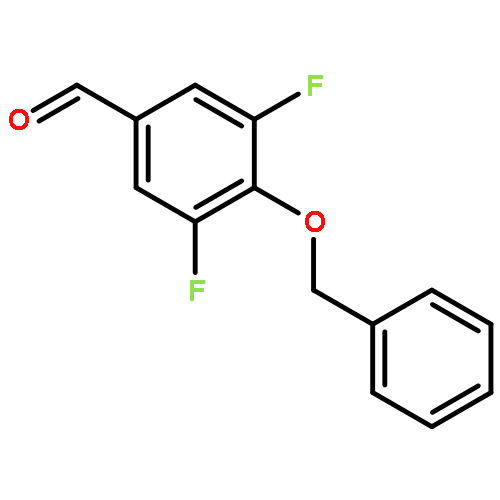
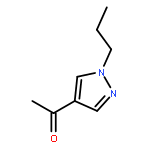
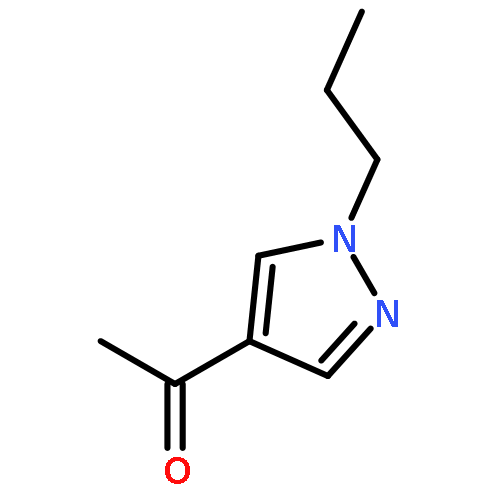
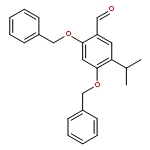
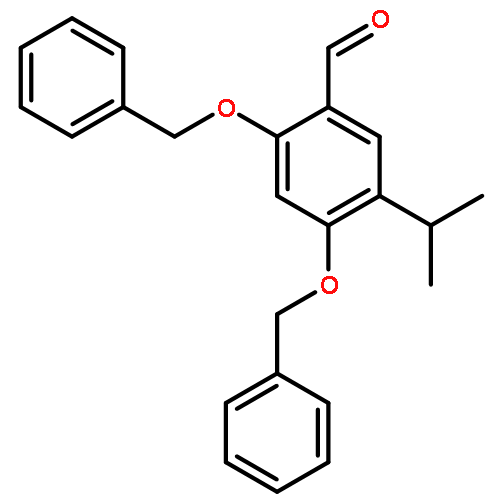
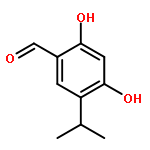
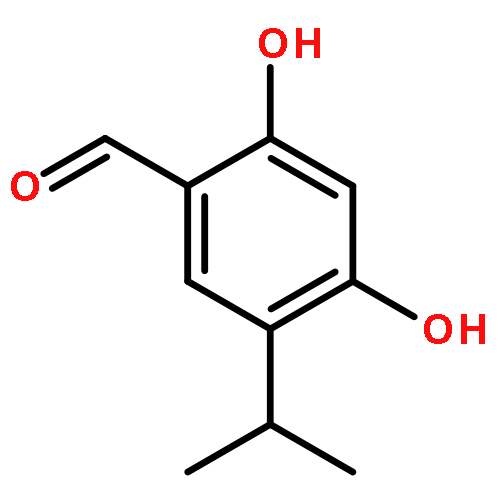
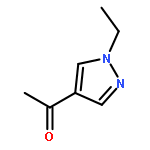
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3.png)
![Benzenamine, 4-[(2-chloro-4-pyridinyl)oxy]-3-fluoro-](http://img.cochemist.com/ccimg/868800/868733-61-3_b.png)

![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0.png)
![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0_b.png)
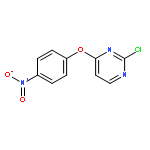
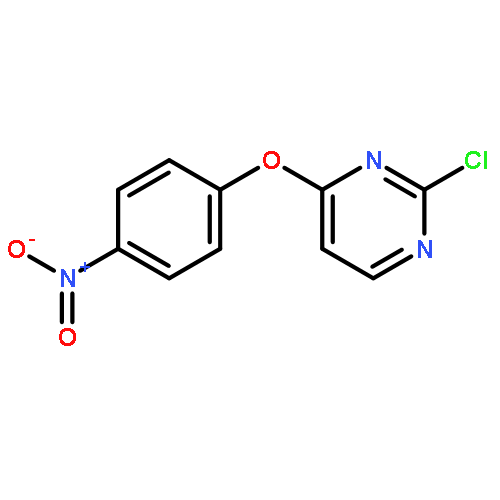


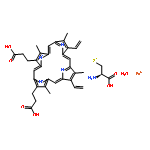
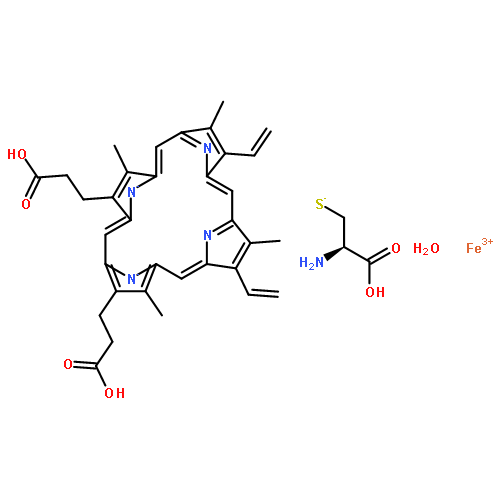






![1H-Benzimidazole, 1-[[2,4,6-tris(1-methylethyl)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/294900/294874-59-2.png)
![1H-Benzimidazole, 1-[[2,4,6-tris(1-methylethyl)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/294900/294874-59-2_b.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4.png)
![Benzenamine, 3-[(methylsulfonyl)methyl]-](http://img.cochemist.com/ccimg/262000/261925-02-4_b.png)


![CYCLOPROPANECARBOXYLIC ACID, 1-[[(3-CHLOROPHENYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/146700/146653-80-7.png)
![CYCLOPROPANECARBOXYLIC ACID, 1-[[(3-CHLOROPHENYL)AMINO]CARBONYL]-](http://img.cochemist.com/ccimg/146700/146653-80-7_b.png)
![Cyclopropanecarboxylic acid, 1-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/145600/145591-80-6.png)
![Cyclopropanecarboxylic acid, 1-[(phenylamino)carbonyl]-](http://img.cochemist.com/ccimg/145600/145591-80-6_b.png)
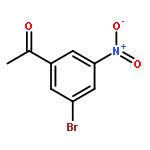
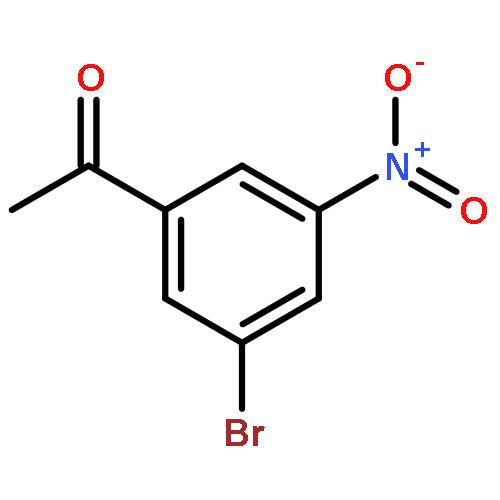
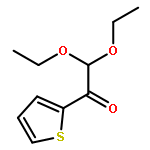
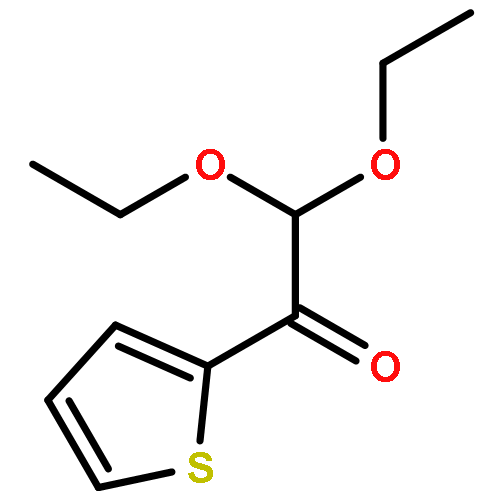
![Cyclopropanecarboxylic acid, 1-[[[4-(trifluoromethyl)phenyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/113200/113136-89-3.png)
![Cyclopropanecarboxylic acid, 1-[[[4-(trifluoromethyl)phenyl]amino]carbonyl]-](http://img.cochemist.com/ccimg/113200/113136-89-3_b.png)
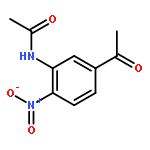
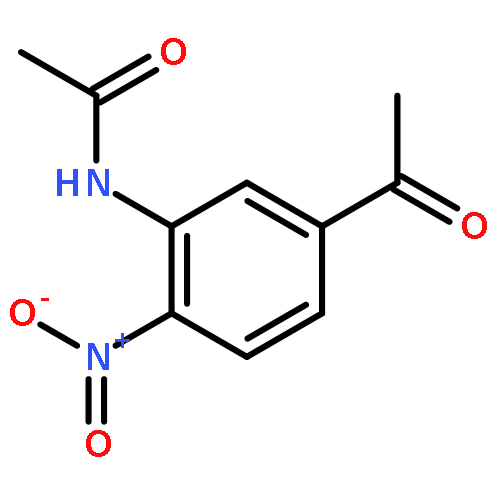
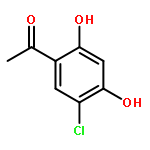
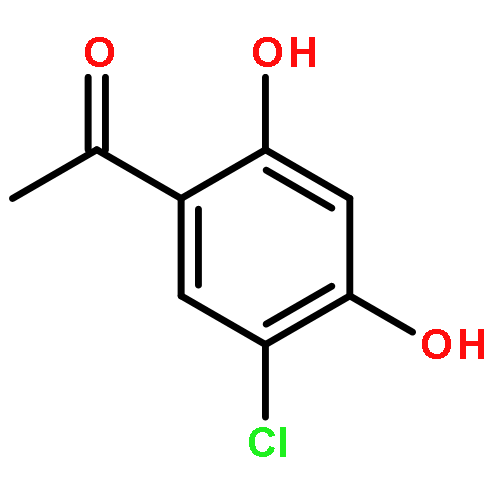


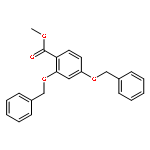
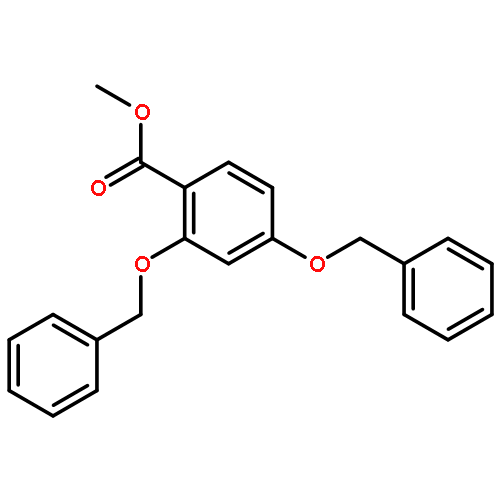
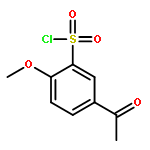
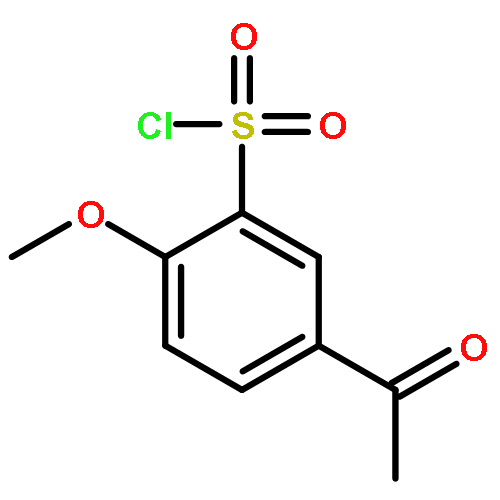
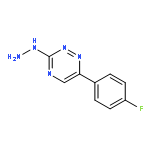
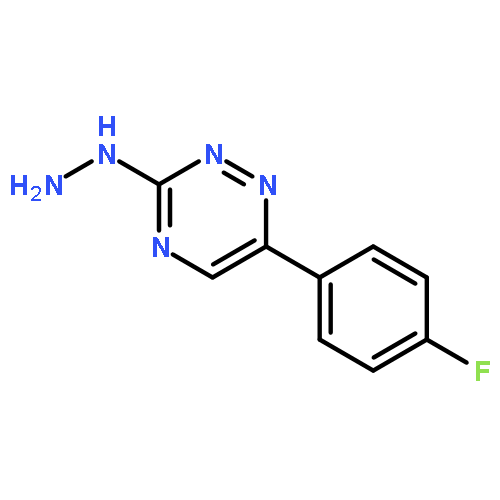


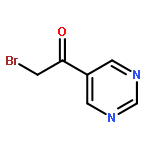
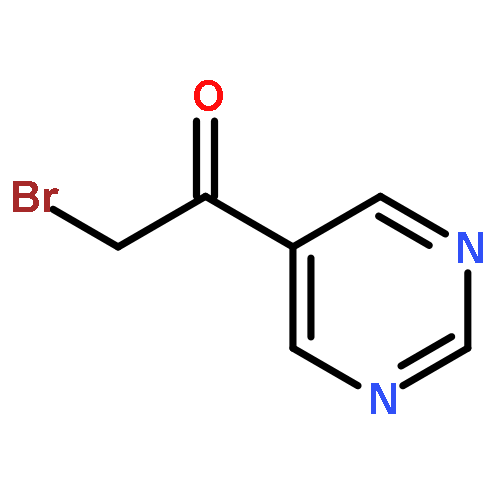

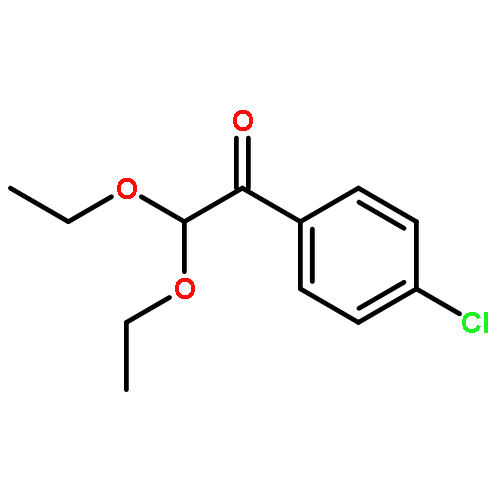
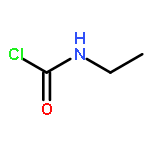
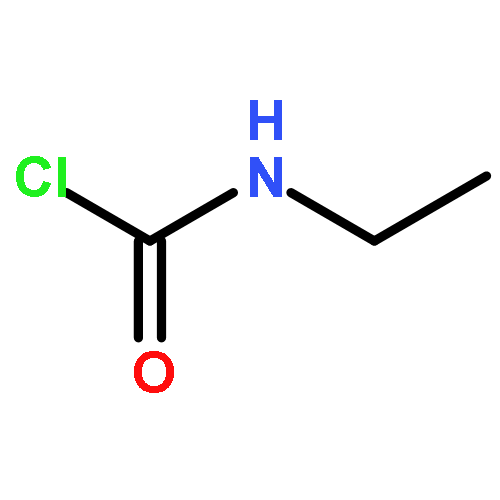
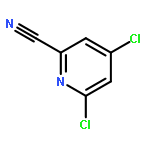


![Benzene, 1-[(methylsulfonyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/25200/25195-60-2.png)
![Benzene, 1-[(methylsulfonyl)methyl]-2-nitro-](http://img.cochemist.com/ccimg/25200/25195-60-2_b.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3.png)
![{4-[(Methylsulfonyl)methyl]phenyl}amine hydrochloride](http://img.cochemist.com/ccimg/24200/24176-70-3_b.png)