Co-reporter:Toshihiro Hirai, Kohki Shibata, Yohei Niwano, Masao Shiozaki, Yoshimitsu Hashimoto, Nobuyoshi Morita, Shintaro Ban, and Osamu Tamura
Organic Letters December 1, 2017 Volume 19(Issue 23) pp:6320-6320
Publication Date(Web):November 20, 2017
DOI:10.1021/acs.orglett.7b03092
The total synthesis of neodysiherbaine A was achieved via 1,3-dipolar cycloaddition of a chiral nitrone template with a sugar-derived allyl alcohol in the presence of MgBr2·OEt2. This cycloaddition constructed the C2 and C4 asymmetric centers in a single step. Then reductive cleavage, intramolecular SN2 reaction of the tertiary alcohol, and oxidation of the primary alcohol afforded neodysiherbaine A.
Co-reporter:Yoshimitsu Hashimoto;Hiromasa Ishiwata;Soko Tachikawa;Shintaro Ban;Nobuyoshi Morita
Chemical Communications 2017 vol. 53(Issue 18) pp:2685-2688
Publication Date(Web):2017/02/28
DOI:10.1039/C7CC00505A
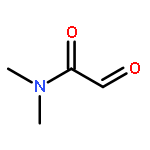
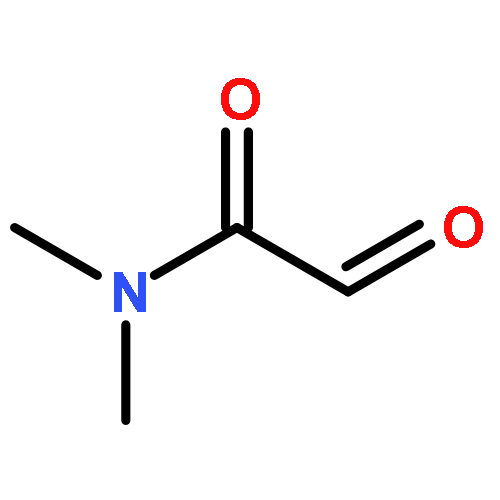
The cycloaddition of nitrones with α,β-unsaturated carbonyl compounds (enones) afforded predominantly 4-acylisoxazolidines, whereas the cycloaddition of the corresponding oximes afforded 5-iminoisoxazolidines. This inverse regioselection is due to HOMO activation by the oxime functionality.
Co-reporter:Nina Shibata;Takahisa Tsuchiya;Yoshimitsu Hashimoto;Nobuyoshi Morita;Shintaro Ban
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 14) pp:3025-3034
Publication Date(Web):2017/04/05
DOI:10.1039/C7OB00279C
ω-Alkynyl O-tert-butyldiphenylsilyloximes, upon treatment with odorless 4-tert-butylbenzenethiol in the presence of azobisisobutyronitrile (AIBN) in refluxing benzene, underwent addition of a thiyl radical to the alkynyl group followed by radical cyclization of the corresponding vinyl radical onto the O-silyloxime moiety to give cyclic O-silylhydroxylamines in good yields. The reactivity of O-silyloximes in radical cyclization was similar to or even higher than that of O-benzyloximes. Facile removal of the silyl group of the cyclization products leading to hydroxylamines and nitrone formation of the hydroxylamines were also demonstrated.
Co-reporter:Nobuyoshi Morita, Masazumi Miyamoto, Akiyoshi Yoda, Mari Yamamoto, Shintaro Ban, Yoshimitsu Hashimoto, Osamu Tamura
Tetrahedron Letters 2016 Volume 57(Issue 40) pp:4460-4463
Publication Date(Web):5 October 2016
DOI:10.1016/j.tetlet.2016.08.045
•Gold-catalyzed synthesis of 1,3-diarylindenes.•Gold-catalyzed dehydrative Friedel-Crafts reaction and Nazarov cyclization.•The reaction proceeds via allene intermediate.An efficient synthesis of 1,3-diarylindenes has been achieved from propargylic alcohols containing aromatic rings via gold-catalyzed dehydrative Friedel–Crafts reaction followed by gold-catalyzed Nazarov cyclization. The reaction proceeds via allene intermediates.
Co-reporter:Nobuyoshi Morita, Arisa Yasuda, Motohiro Shibata, Shintaro Ban, Yoshimitsu Hashimoto, Iwao Okamoto, and Osamu Tamura
Organic Letters 2015 Volume 17(Issue 11) pp:2668-2671
Publication Date(Web):May 21, 2015
DOI:10.1021/acs.orglett.5b01046
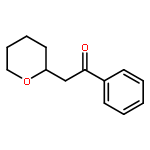
Strategic use of oxophilic (hard) gold(III) and π-philic (soft) gold(I) catalysts provides access to two types of cyclic ethers from propargylic alcohols. Thus, heating propargylic alcohols with an oxophilic gold(III) catalyst (AuBr3) results in cyclization to afford cyclic ethers bearing an acetylenic moiety, due to coordination of gold(III) to the oxygen of the propargylic hydroxyl group. On the other hand, propargylic alcohols with a π-philic gold(I) catalyst (Ph3PAuNTf2) induces Meyer–Schuster rearrangement to afford α,β-unsaturated ketones, which undergo gold(III)-catalyzed intramolecular oxa-Michael addition to afford cyclic ethers bearing a carbonyl group, due to coordination of gold(III) to the oxygen of the carbonyl group.
Co-reporter:Nobuyoshi Morita, Tomonori Tsunokake, Yuji Narikiyo, Mayuka Harada, Tatsuyuki Tachibana, Yuta Saito, Shintaro Ban, Yoshimitsu Hashimoto, Iwao Okamoto, Osamu Tamura
Tetrahedron Letters 2015 Volume 56(Issue 45) pp:6269-6272
Publication Date(Web):4 November 2015
DOI:10.1016/j.tetlet.2015.09.115
Strategic use of hard gold(III) and soft gold(I) catalysts provides facile access to two types of 2-substituted piperidines from propargylic alcohols. Thus, heating propargylic alcohols in the presence of AuBr3 results in cyclization to furnish piperidines having an acetylenic moiety, due to activation of the propargylic hydroxyl group by coordination of gold(III). On the other hand, the catalyst [Ph3PAuNTf2]2PhMe induces Meyer–Schuster rearrangement of propargylic alcohols to give α,β-unsaturated ketones, which undergo intramolecular aza-Michael addition to afford piperidines bearing a carbonyl group, due to activation of the triple bond by coordination of gold(I).
Co-reporter:Nobuyoshi Morita, Rina Kono, Kenji Fukui, Asuka Miyazawa, Hyuma Masu, Isao Azumaya, Shintaro Ban, Yoshimitsu Hashimoto, Iwao Okamoto, and Osamu Tamura
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4797-4802
Publication Date(Web):April 10, 2015
DOI:10.1021/acs.joc.5b00426
A C-amide-substituted O-silylated oxime, (E)-(tert-butyldimethylsiloxyimino)acetic acid N,N-dimethylamide (8b), on treatment with 2.2 equiv of BF3·OEt2, in situ generated boracyclic nitrone-type intermediate BF3·14, which underwent cycloaddition with alkenes to give 3,5-cis-isoxazolidines as the major products. The mechanism was strongly supported by isolation of the reaction intermediate 14 that was characterized by X-ray diffraction and its further reaction. This cycloaddition was successfully applied to the synthesis of syn-HPA-12 known as an inhibitor of CERT that mediates the transport of ceramide.
Co-reporter:Kenichi Kobayashi, Iwao Okamoto, Nobuyoshi Morita, Tamiko Kiyotani and Osamu Tamura
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 16) pp:5825-5832
Publication Date(Web):19 May 2011
DOI:10.1039/C1OB05612C
The first total synthesis of the proposed structure of phaeosphaeride A has been achieved via six-membered-ring formation by means of an intramolecular vinyl-anion aldol reaction as the key step. This synthesis suggests a revised configurational assignment for phaeosphaeride A.
Co-reporter:Osamu Tamura, Kodai Takeda, Naka Mita, Masanori Sakamoto, Iwao Okamoto, Nobuyoshi Morita and Hiroyuki Ishibashi
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 21) pp:7411-7419
Publication Date(Web):05 Aug 2011
DOI:10.1039/C1OB06067H
Stereoselective vinylogous Mannich reaction of 2-trimethylsilyloxyfuran with L-gulose-derived chiral nitrones in the presence of a catalytic amount of trimethylsilyl trifluoromethanesulfonate was investigated. The selectivity was strongly influenced by the bulkiness of the C-substituent of the nitrone: for example, C-benzyloxymethyl nitrone afforded four stereoisomers, whereas bulky C-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]nitrone gave a single stereoisomer. The latter product was elaborated to afford key synthetic intermediates for polyoxin C and dysiherbaine.
Co-reporter:Taku Shibue, Iwao Okamoto, Nobuyoshi Morita, Hiroshi Morita, Yusuke Hirasawa, Takahiro Hosoya, Osamu Tamura
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 1) pp:431-434
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmcl.2010.10.118
The synthesis and biological evaluation of stereoisomers in tubulysin D are described. The stereoselective synthesis of all possible stereoisomers of C-11 and C-13 positions in tubulysin D was achieved by employing 1′-epi-Tuv-Me, 3′-epi-Tuv-Me, and ent-Tuv-Me and their biological properties were evaluated. It is clear that the stereochemistries of the C-11 and C-13 positions in tubulysin D have no practical impact on the inhibition of tubulin polymerization but play a role in the potent antiproliferative activities.
Co-reporter:Haruaki Kurasaki, Iwao Okamoto, Nobuyoshi Morita, Osamu Tamura
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 5) pp:1601-1603
Publication Date(Web):1 March 2010
DOI:10.1016/j.bmcl.2010.01.062
Both enantiomers of elaeocarpenine (1) and its analogs, 21, 22, 25, and 27, were synthesized from bicyclic aldehydes 8–10 via a flexible route previously established for total synthesis of grandisines, and their binding affinities for μ-, κ- and δ-opioid receptor subtypes were evaluated. We found that (9R)-1 exhibited higher affinity than (9S)-1 for all the subtypes, but the enantiomers showed little subtype selectivity. Analogs 21 having a pyrrolizidine skeleton and 27 having a stemona-type skeleton in place of the indolizidine unit of (9S)-1 showed μ-selective and μ-, κ-selective binding, respectively.We report the first total synthesis of elaeocarpenine and its analogs and evaluation of their in vitro binding affinities to μ-, δ- and κ-opioid receptor subtypes.
Co-reporter:Taku Shibue;Toshihiro Hirai;Dr. Iwao Okamoto;Dr. Nobuyoshi Morita;Dr. Hyuma Masu;Dr. Isao Azumaya;Dr. Osamu Tamura
Chemistry - A European Journal 2010 Volume 16( Issue 38) pp:11678-11688
Publication Date(Web):
DOI:10.1002/chem.201000963
Abstract
The total syntheses of tetrapeptides tubulysins D (1 b), U (1 c), and V (1 d), which are potent tubulin polymerization inhibitors, are described. The synthesis of Tuv (2), an unusual amino acid constituent of tubulysins, includes an 1,3-dipolar cycloaddition reaction of chiral nitrone D-6 derived from D-gulose with N-acryloyl camphor sultam (−)-9 employing the double asymmetric induction, whereas the synthesis of Tup (20), another unusual amino acid, involves a stereoselective Evans aldol reaction of (Z)-boron enolate generated from (S)-4-isopropyl-3-propionyl-2-oxazolidinone with N-protected phenylalaninal and a subsequent Barton deoxygenation protocol. We accomplished the total syntheses of tubulysins U (1 c) and V (1 d) by using these methodologies, in which the isoxazolidine ring was used as the effective protective group for γ-amido alcohol functionality. Furthermore, to understand the structure-activity relationship of tubulysins, we synthesized tubulysin D (1 b) and cyclo-tubulysin D (1 e) from 2-Me and 20, and ent-tubulysin D (ent-1 d) from ent-2-Me and ent-20, respectively. The preliminary results regarding their biological activities are also reported.
Co-reporter:Haruaki Kurasaki;Iwao Okamoto Dr.;Nobuyoshi Morita Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 46) pp:12754-12763
Publication Date(Web):
DOI:10.1002/chem.200901843
Abstract
This article describes in detail the first total synthesis of grandisine alkaloids, grandisines B, D, and F, which show affinity for the human δ-opioid receptor. The key steps in this synthesis are construction of the isoquinuclidinone moiety of 2 by intramolecular imine formation and the tetracyclic ring system of 4 by stereoselective ring closure of the enolate of amine 8 generated by 1,4-addition of ammonia to 9. Synthesis of key intermediate 9 featured a highly stereoselective Brønsted acid mediated Morita–Baylis–Hillman (MBH) reaction via the N-acyl iminium ion.
Co-reporter:Taku Shibue, Toshihiro Hirai, Iwao Okamoto, Nobuyoshi Morita, Hyuma Masu, Isao Azumaya, Osamu Tamura
Tetrahedron Letters 2009 50(27) pp: 3845-3848
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.046
Co-reporter:Osamu Tamura, Kodai Takeda, Naka Mita, Masanori Sakamoto, Iwao Okamoto, Nobuyoshi Morita and Hiroyuki Ishibashi
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 21) pp:NaN7419-7419
Publication Date(Web):2011/08/05
DOI:10.1039/C1OB06067H
Stereoselective vinylogous Mannich reaction of 2-trimethylsilyloxyfuran with L-gulose-derived chiral nitrones in the presence of a catalytic amount of trimethylsilyl trifluoromethanesulfonate was investigated. The selectivity was strongly influenced by the bulkiness of the C-substituent of the nitrone: for example, C-benzyloxymethyl nitrone afforded four stereoisomers, whereas bulky C-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]nitrone gave a single stereoisomer. The latter product was elaborated to afford key synthetic intermediates for polyoxin C and dysiherbaine.
Co-reporter:Kenichi Kobayashi, Iwao Okamoto, Nobuyoshi Morita, Tamiko Kiyotani and Osamu Tamura
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 16) pp:NaN5832-5832
Publication Date(Web):2011/05/19
DOI:10.1039/C1OB05612C
The first total synthesis of the proposed structure of phaeosphaeride A has been achieved via six-membered-ring formation by means of an intramolecular vinyl-anion aldol reaction as the key step. This synthesis suggests a revised configurational assignment for phaeosphaeride A.
Co-reporter:Nina Shibata, Takahisa Tsuchiya, Yoshimitsu Hashimoto, Nobuyoshi Morita, Shintaro Ban and Osamu Tamura
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 14) pp:NaN3034-3034
Publication Date(Web):2017/03/06
DOI:10.1039/C7OB00279C
ω-Alkynyl O-tert-butyldiphenylsilyloximes, upon treatment with odorless 4-tert-butylbenzenethiol in the presence of azobisisobutyronitrile (AIBN) in refluxing benzene, underwent addition of a thiyl radical to the alkynyl group followed by radical cyclization of the corresponding vinyl radical onto the O-silyloxime moiety to give cyclic O-silylhydroxylamines in good yields. The reactivity of O-silyloximes in radical cyclization was similar to or even higher than that of O-benzyloximes. Facile removal of the silyl group of the cyclization products leading to hydroxylamines and nitrone formation of the hydroxylamines were also demonstrated.
Co-reporter:Yoshimitsu Hashimoto, Hiromasa Ishiwata, Soko Tachikawa, Shintaro Ban, Nobuyoshi Morita and Osamu Tamura
Chemical Communications 2017 - vol. 53(Issue 18) pp:NaN2688-2688
Publication Date(Web):2017/02/03
DOI:10.1039/C7CC00505A
The cycloaddition of nitrones with α,β-unsaturated carbonyl compounds (enones) afforded predominantly 4-acylisoxazolidines, whereas the cycloaddition of the corresponding oximes afforded 5-iminoisoxazolidines. This inverse regioselection is due to HOMO activation by the oxime functionality.








![Piperidine, 1-[(4-methylphenyl)sulfonyl]-2-(2-oxo-2-phenylethyl)-](http://img.cochemist.com/ccimg/89900/89881-44-7.png)
![Piperidine, 1-[(4-methylphenyl)sulfonyl]-2-(2-oxo-2-phenylethyl)-](http://img.cochemist.com/ccimg/89900/89881-44-7_b.png)