Co-reporter:Chao Tian, Meng Wang, Zifei Han, Fang Fang, Zhili Zhang, Xiaowei Wang, Junyi Liu
European Journal of Medicinal Chemistry 2017 Volume 138(Volume 138) pp:
Publication Date(Web):29 September 2017
DOI:10.1016/j.ejmech.2017.07.002
•A series of 6 Substituted pyrrolo[3,2-d]pyrimidine analogues were synthesized as antifolate agents.•The in vitro anti-proliferative effect of these compounds on tumor cells were evaluated.•The anti-proliferative effect of the most potent compound (15a) acted through dual inhibition of TS and DHFR.•The effects of 15a on cell cycle distribution, induction of apoptosis and colony formation inhibition were tested.•Molecular docking of 15a was carried out against TS and DHFR.A series of novel 6-substituted pyrrolo[3,2-d]pyrimidine analogues (10a, 11a-13a, 15a, 17a, 18a, 27a and 28a) have been designed and synthesized as antifolate antitumor agents. The anti-proliferative activities of these compounds against HL60, A549, H1299, Hela, HCT116 and HT29 tumor cells were evaluated. Most of the compounds exhibited micromolar anti-proliferative potencies. Compound 15a, the most potent one, has GI50 value of 0.73, 1.72, and 8.92 μM against A549, H1299 and HL60 cells, respectively. The cell cycle distribution assay displayed that 15a could increase the accumulation of G2/M-phase cells. 15a showed low potency in induction of apoptosis. However, the inhibition of A549 cell colony formation was observed. These indicated that the tumor cell death relied on the irreversible effect of 15a on clonogenicity and cell proliferation. The identification of targeted pathway of 15a implied that the anti-proliferative potencies of 15a probably act through dual inhibition of thymidylate synthase (TS) and dihydrofolate reductase (DHFR).Download high-res image (223KB)Download full-size image
Co-reporter:Shaotong Wu, Qianqian Yin, Liang Zhao, Ningning Fan, Xiaowan Tang, Jianxiong Zhao, Tao Sheng, Ying Guo, Chao Tian, Zhili Zhang, Weisi Xu, Zhenming Liu, Shibo Jiang, Liying Ma, Junyi Liu and Xiaowei Wang
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 4) pp:1413-1420
Publication Date(Web):08 Dec 2015
DOI:10.1039/C5OB02154E
3-Iodo-4-(2′-methylcyclohexyloxy)-6-phenethylpyridin-2(1H)-ones, as effective non-nucleoside reverse transcriptase inhibitors, were synthesized and resolved with different configurations. Biological results revealed that the trans-racemate 2b exhibited more potent activity than the cis-isomers. Noticeably, the trans-(S,S)-enantiomer 2e turned out to be significantly more potent than its counterpart enantiomer 2d against wild-type and double-mutant strains with high selectivity indexes.
Co-reporter:Chao Tian;Zhili Zhang;Shouxin Zhou;Mengmeng Yuan;Xiaowei Wang
Chemical Biology & Drug Design 2016 Volume 87( Issue 3) pp:444-454
Publication Date(Web):
DOI:10.1111/cbdd.12681
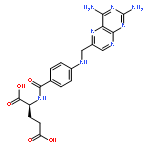
Based on our previous work, seven N5-substituted 8,10-dideazatetrahydrofolate analogues and one 8-deazatetrahydrofolate analogue were designed and synthesized as human dihydrofolate reductase (hDHFR) inhibitors. All compounds were assayed versus DHFR and five different cancer cell lines. The biological assay indicated that replacing N10 with carbon would significantly increase inhibitory activities against DHFR and cytotoxicities against cancer cell lines. Compound 19a with 4-amino and N5-formyl showed great antitumour activities against HL-60, Bel-7402 and BGC823 which were much better than MTX.
Co-reporter:Xianling Ning ; Ying Guo ; Xiaowei Wang ; Xiaoyan Ma ; Chao Tian ; Xueqi Shi ; Renzong Zhu ; Can Cheng ; Yansheng Du ; Zhizhong Ma ; Zhili Zhang
Journal of Medicinal Chemistry 2014 Volume 57(Issue 10) pp:4302-4312
Publication Date(Web):April 3, 2014
DOI:10.1021/jm500258v
Novel (E)-3,4-dihydroxystyryl aralkyl sulfones and sulfoxides were designed and synthesized as new analogues of 1, which showed interesting multifunctional neuroprotective effects, including antioxidative and antineuroinflammatory properties. Specifically, target compounds display excellent potency in scavenging reactive free radicals and demonstrate potent effects against various kinds of toxicities, including H2O2, 6-hydroxydopamine, and lipopolysaccharide in different types of neuronal cells. The antioxidative properties of the target compounds are more potent than that of 1, and the antineuroinflammatory properties are less strong than that of 1. According to the parallel artificial membrane permeation assay for the blood–brain barrier, target compounds possess greater blood–brain barrier (BBB) permeability than 1. In short, due to improvement of the antioxidative effect, stability, and BBB permeability, (E)-3,4-dihydroxystyryl aralkyl sulfones and sulfoxides can thus be considered as potential multifunctional neuroprotective agents and serve as new lead candidates in the treatment of neurodegenerative diseases.
Co-reporter:Amin Li ; Yabo Ouyang ; Ziyun Wang ; Yuanyuan Cao ; Xiangyi Liu ; Li Ran ; Chao Li ; Li Li ; Liang Zhang ; Kang Qiao ; Weisi Xu ; Yang Huang ; Zhili Zhang ; Chao Tian ; Zhenming Liu ; Shibo Jiang ; Yiming Shao ; Yansheng Du ; Liying Ma ; Xiaowei Wang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3593-3608
Publication Date(Web):March 29, 2013
DOI:10.1021/jm400102x
Novel 6-substituted-4-cycloalkyloxy-pyridin-2(1H)-ones were synthesized as non-nucleoside reverse transcriptase inhibitors (NNRTIs), and their biological activity was evaluated. Most of the compounds, especially 26 and 22, bearing a 3-isopropyl and 3-iodine group, respectively, exhibited highly potent activity against wild-type HIV-1 strains and those resistant to reverse transcriptase inhibitors (RTIs). The diastereoisomers of 26-trans and 26-cis were synthesized separately and confirmed with HPLC and NOESY spectra. The 26-trans isomers had an activity about 400-fold more potent than that of 26-cis. The pair of 26-trans enantiomers, one of the most potent inhibitors with EC50 of 4 nM and selectivity index (SI) of 75000, was highly effective against a panel of RTIs-resistant strains with single (Y181C and K103N) or double (A17) mutations in reverse transcriptase. The results suggest that these novel pyridinone derivatives have the potential to be further developed as new antiretroviral drugs with improved antiviral efficacy and drug resistance profile.
Co-reporter:Xianling Ning, Ying Guo, Xiaoyan Ma, Renzong Zhu, Chao Tian, Zhili Zhang, Xiaowei Wang, Zhizhong Ma, Junyi Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:5589-5597
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.05.043
A novel class of (E)-3,4-dihydroxy styryl sulfonamides and their 3,4-diacetylated derivatives as caffeic acid phenethyl ester (CAPE) analogs was designed and prepared for improving stability and solubility of the lead compound. Their neuroprotective properties were assessed by several models. The results showed that target compounds displayed positive free radical quenching abilities, superior to that of CAPE. Compounds 6j–k and 7j–k demonstrated remarkable protection effects against damage induced by hydrogen peroxide which were apparently stronger than that of CAPE. Most of target compounds could inhibit nitric oxide production. Additionally, target compounds showed high blood–brain barrier permeability.A novel class of neuroprotective agent, (E)-3,4-dihydroxy styryl sulfonamides derivatives, was synthesized and evaluated their pharmacological activities by several models in vitro. The results demonstrated that they possess multifunctional neuroprotection effects.
Co-reporter:Xianling Ning, Ying Guo, Xiaoyan Ma, Renzong Zhu, Chao Tian, Xiaowei Wang, Zhizhong Ma, Zhili Zhang, Junyi Liu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3700-3703
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.05.016
E-3,4-Dihydroxy styryl aralkyl ketones as well as their 3,4-diacetylated derivatives as the analogues of neuroprotective agent CAPE were designed and synthesized for improving stability and lipid solubility. The neuroprotective activities of target compounds 10a–g and 11a–g were tested by three models in vitro, including 1,1-diphenyl-2-picrylhydrazyl radical scavenging capacity, neuronal protecting effect against damage induced by H2O2 in PC12 cells and nitric oxide suppression effect in BV2 microglial cells. The results demonstrated that compounds 10f and 11f exhibited the most potent neuroprotective effect against oxidative stress and inflammation, which is higher than that of the lead compound CAPE.A series of neuroprotective agent, E-3,4-dihydroxy styryl aralkyl ketones, was synthesized and their biological activities were evaluated by three models in vitro. The results showed that target compounds had multifunctional neuroprotective effects against oxidative stress and inflammation.
Co-reporter:Li Li;Liying Ma;Xiaowei Wang
Journal of Heterocyclic Chemistry 2013 Volume 50( Issue 1) pp:164-168
Publication Date(Web):
DOI:10.1002/jhet.987
A revised synthetic route to Emivirine (MKC-442) via properly substituted β-keto ester converted from Meldrum's Acid was developed. This method could be applied to the synthesis of a variety of MKC-442 analogues and open the way for their systematic biological evaluation.
Co-reporter:Xiaowei Wang ; Jianfang Zhang ; Yang Huang ; Ruiping Wang ; Liang Zhang ; Kang Qiao ; Li Li ; Chang Liu ; Yabo Ouyang ; Weisi Xu ; Zhili Zhang ; Liangren Zhang ; Yiming Shao ; Shibo Jiang ; Liying Ma
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2242-2250
Publication Date(Web):January 27, 2012
DOI:10.1021/jm201506e
Because the emergence of drug-resistant mutants has limited the efficacy of non-nucleoside reverse transcriptase inhibitors (NNRTIs), it is essential to develop new antivirals with better drug resistance and pharmacokinetic profiles. Here we designed and synthesized a series of 1-[(2-benzyloxyl/alkoxyl)methyl]-5-halo-6-aryluracils, the HEPT analogues, and evaluated their biological activity using nevirapine and 18 (TNK-651) as reference compounds. Most of these compounds, especially 6b, 7b, 9b, 11b, and 7c, exhibited highly potent anti-HIV-1 activity against both wild-type and NNRTI-resistant HIV-1 strains. Compound 7b, which had the highest selectivity index (SI = 38 215), is more potent than nevirapine and 18. These results suggest that the introduction of a halogen at the C-5 position may contribute to the effectiveness of these compounds against RTI-resistant variants. In addition, meta substituents on the C-6 aromatic moiety could significantly enhance activity against NNRTI-resistant HIV-1 strains. These compounds can be further developed as next-generation NNRTIs with an improved antiviral efficacy and drug-resistance profile.
Co-reporter:Gong Li, Xiaowei Wang, Chao Tian, Tongbo Zhang, Zhili Zhang, Junyi Liu
Tetrahedron Letters 2012 Volume 53(Issue 39) pp:5193-5196
Publication Date(Web):26 September 2012
DOI:10.1016/j.tetlet.2012.06.143
A novel pyrimido[4,5-b][1,4]diazepine-2,4,6-trione was synthesized with an efficient strategy. Especially, the key intermediate 2,4-dimethoxypyrimido[4,5-b][1,4]diazepin-6-one was promoted by one pot tandem reduction–cyclization with Na2S2O4. Subsequently, reduction of lactams 6 with LiAlH4 afforded a more flexible scaffold of pyrimidodiazepines. The synthetic strategy was versatile since it facilitated the sequential functionalization on the pyrimidodiazepine at three positions. Thus a convenient and effective method for the rapid preparing of multi-substituted pyrimido[4,5-b][1,4]diazepines was developed.
Co-reporter:Wenjing Chen, Wujie Li, Xiaomei Ling, Xiaowei Wang, Junyi Liu
Journal of Chromatography B 2010 Volume 878(Issue 20) pp:1714-1717
Publication Date(Web):15 June 2010
DOI:10.1016/j.jchromb.2010.04.013
The HIV reverse transcriptase (RT) is an important antiviral target for the chemotherapy of AIDS because of its key role in virus replication. Nevirapine is a first generation of non-nucleoside reverse transcriptase inhibitors (NNRTIs), which is usually used for the therapy of AIDS. In this study, a high-performance analytical method based on capillary electrophoresis (CE) to investigate interactions between HIV RT and nevirapine was developed. Samples containing HIV RT and nevirapine at various ratios were incubated at 37 °C for 45 min and then separated by CE with Tris–acetate buffer at pH 7.3 containing 0.15% SDS. Both qualitative and quantitative characterizations of the binding were determined by CE for the first time. The binding constants of the interactions between HIV RT and nevirapine were calculated as (3.25 ± 0.16) × 104 and (1.25 ± 0.07) × 102 M−1 by Scatchard analysis. HIV RT and nevirapine have two binding sites. The presented methodology should be generally applicable to study the interactions between HIV RT and nevirapine quantitatively and qualitatively.
Co-reporter:Xiao Lu, Yanli Chen, Ying Guo, Zhenming Liu, Yawei Shi, Yang Xu, Xiaowei Wang, Zhili Zhang, Junyi Liu
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 23) pp:7399-7407
Publication Date(Web):1 December 2007
DOI:10.1016/j.bmc.2007.07.058
Novel compounds 1a–u, which can be considered as hybrid analogues of MKC-442 and pyridinon, have been synthesized and evaluated as inhibitors of HIV-1 reverse transcriptase (HIV-1 RT). Starting from 6-methyuracil 2, 1-alkylated-5-bromomethyl-6-methyluracils 8 was prepared in four steps by hydroxylmethylation, etherification, N-1 alkylation, and bromination. Finally, compounds 1a–u were achieved in the displacement of 5-bromomethyl group by nucleophiles with amino compounds. Some of compounds 1a–u showed potent inhibitory activity against HIV-1 RT. The most active compounds showed activity in the low micromolecular range with IC50 values (IC50 0.82–5.09 μM) comparable to that of nevirapine (IC50 10.60 μM). The biological testing results are in accordance with the docking.A synthesis route to get a novel series of hydrid analogues of MKC-442 and pyridinon as inhibitors of HIV-1 reverse transcripts and evaluate results about their potent inhibiting activity against HIV-RT.
Co-reporter:Yixing Sun, Weisi Xu, Ningning Fan, Xuefeng Sun, Xianling Ning, Liying Ma, Junyi Liu, Xiaowei Wang
Bioorganic & Medicinal Chemistry (1 February 2017) Volume 25(Issue 3) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bmc.2016.12.035
Aiming at the limited effectiveness of current clinical therapeutic effect of AIDS, novel series of compounds bearing (E)-3,4-dihydroxystyryl sulfone (or sulfoxide) and anilide fragments were designed and synthesized as dual inhibitors of HIV-1 CCR5/IN. The biological results indicated that several target compounds showed inhibitory activity against HIV-1 Bal (R5) infection in TZM-bl cells. Besides targeting the chemokine receptor on the host cell surface, they also displayed binding affinities with HIV-1 integrase using the surface plasmon resonance (SPR) binding assays. Molecular docking studies have inferred the possible binding mode of target compounds against integrase. These data demonstrate that the structure of (E)-3,4-dihydroxystyryl sulfone and sulfoxide derivatives have the potential to derive potent dual inhibitors of HIV-1 Integrase and CCR5.
Co-reporter:Shaotong Wu, Qianqian Yin, Liang Zhao, Ningning Fan, Xiaowan Tang, Jianxiong Zhao, Tao Sheng, Ying Guo, Chao Tian, Zhili Zhang, Weisi Xu, Zhenming Liu, Shibo Jiang, Liying Ma, Junyi Liu and Xiaowei Wang
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 4) pp:NaN1420-1420
Publication Date(Web):2015/12/08
DOI:10.1039/C5OB02154E
3-Iodo-4-(2′-methylcyclohexyloxy)-6-phenethylpyridin-2(1H)-ones, as effective non-nucleoside reverse transcriptase inhibitors, were synthesized and resolved with different configurations. Biological results revealed that the trans-racemate 2b exhibited more potent activity than the cis-isomers. Noticeably, the trans-(S,S)-enantiomer 2e turned out to be significantly more potent than its counterpart enantiomer 2d against wild-type and double-mutant strains with high selectivity indexes.

![4(1H)-Pyrimidinone, 2-[[(phenylmethoxy)methyl]thio]-6-(phenylmethyl)-](http://img.cochemist.com/ccimg/255900/255897-83-7.png)
![4(1H)-Pyrimidinone, 2-[[(phenylmethoxy)methyl]thio]-6-(phenylmethyl)-](http://img.cochemist.com/ccimg/255900/255897-83-7_b.png)
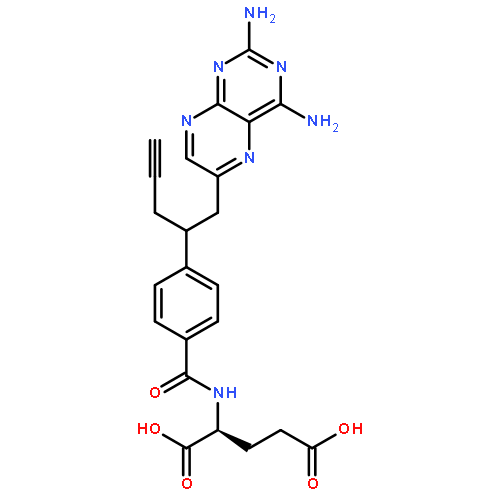




![Acetic acid,2-[(2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/99200/99186-25-1.png)
![Acetic acid,2-[(2-phenylethyl)thio]-](http://img.cochemist.com/ccimg/99200/99186-25-1_b.png)
![Acetic acid,2-[[(4-chlorophenyl)methyl]thio]-](http://img.cochemist.com/ccimg/90700/90649-82-4.png)
![Acetic acid,2-[[(4-chlorophenyl)methyl]thio]-](http://img.cochemist.com/ccimg/90700/90649-82-4_b.png)
![PYRIDO[3,2-D]PYRIMIDIN-4(1H)-ONE, 2-AMINO-6-(BROMOMETHYL)-](http://img.cochemist.com/ccimg/76900/76832-41-2.png)
![PYRIDO[3,2-D]PYRIMIDIN-4(1H)-ONE, 2-AMINO-6-(BROMOMETHYL)-](http://img.cochemist.com/ccimg/76900/76832-41-2_b.png)
![(2,4-DIAMINOPYRIDO[3,2-D]PYRIMIDIN-6-YL)METHANOL](http://img.cochemist.com/ccimg/76900/76822-61-2.png)
![(2,4-DIAMINOPYRIDO[3,2-D]PYRIMIDIN-6-YL)METHANOL](http://img.cochemist.com/ccimg/76900/76822-61-2_b.png)
![6-(BROMOMETHYL)PYRIDO[3,2-D]PYRIMIDINE-2,4-DIAMINE](http://img.cochemist.com/ccimg/76900/76807-56-2.png)
![6-(BROMOMETHYL)PYRIDO[3,2-D]PYRIMIDINE-2,4-DIAMINE](http://img.cochemist.com/ccimg/76900/76807-56-2_b.png)


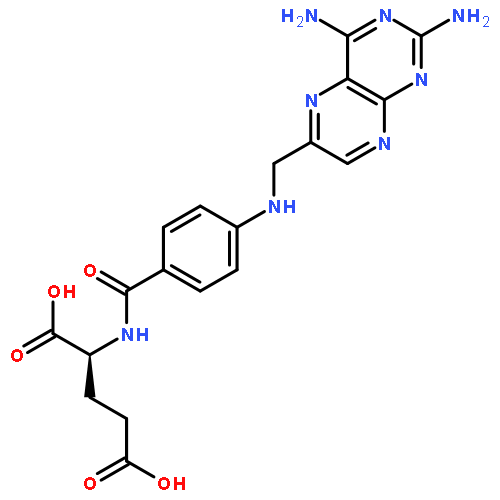
![N-{4-[2-(2,4-diaminopteridin-6-yl)ethyl]benzoyl}glutamic acid](http://img.cochemist.com/ccimg/52500/52454-37-2.png)
![N-{4-[2-(2,4-diaminopteridin-6-yl)ethyl]benzoyl}glutamic acid](http://img.cochemist.com/ccimg/52500/52454-37-2_b.png)

![Acetic acid,2-[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-08-8.png)
![Acetic acid,2-[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-08-8_b.png)
![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3.png)
![Benzene, [(bromomethoxy)methyl]-](http://img.cochemist.com/ccimg/17700/17690-16-3_b.png)


![6-Methylpyrido[3,2-d]pyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/2500/2499-96-9.png)
![6-Methylpyrido[3,2-d]pyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/2500/2499-96-9_b.png)




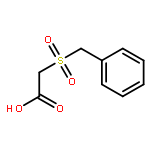
![Acetic acid, [(2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/137600/137524-38-0.png)
![Acetic acid, [(2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/137600/137524-38-0_b.png)
![Acetic acid,2-[[(4-chlorophenyl)methyl]sulfonyl]-](http://img.cochemist.com/ccimg/118700/118672-20-1.png)
![Acetic acid,2-[[(4-chlorophenyl)methyl]sulfonyl]-](http://img.cochemist.com/ccimg/118700/118672-20-1_b.png)
![Acetic acid, [[[4-(1,1-dimethylethyl)phenyl]methyl]sulfonyl]-](http://img.cochemist.com/ccimg/65500/65413-34-5.png)
![Acetic acid, [[[4-(1,1-dimethylethyl)phenyl]methyl]sulfonyl]-](http://img.cochemist.com/ccimg/65500/65413-34-5_b.png)
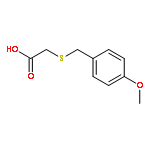
![Acetic acid,2-[[(4-methoxyphenyl)methyl]sulfonyl]-](http://img.cochemist.com/ccimg/28300/28203-60-3.png)
![Acetic acid,2-[[(4-methoxyphenyl)methyl]sulfonyl]-](http://img.cochemist.com/ccimg/28300/28203-60-3_b.png)
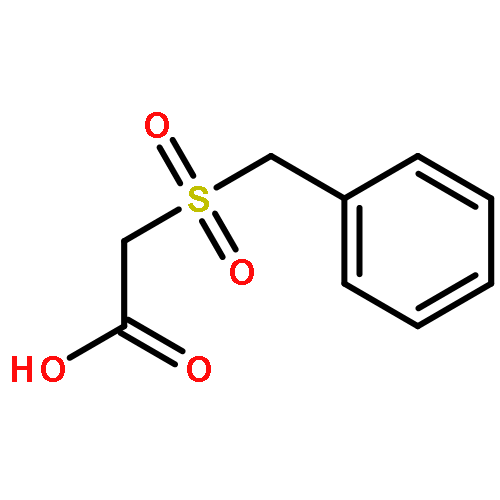
![Acetic acid, [(phenylmethyl)sulfinyl]-](http://img.cochemist.com/ccimg/28300/28203-54-5.png)
![Acetic acid, [(phenylmethyl)sulfinyl]-](http://img.cochemist.com/ccimg/28300/28203-54-5_b.png)





