Co-reporter:Jonathan M. Goldberg, Karen I. Goldberg, D. Michael Heinekey, Samantha A. Burgess, David B. Lao, and John C. Linehan
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12638-12638
Publication Date(Web):September 1, 2017
DOI:10.1021/jacs.7b06480
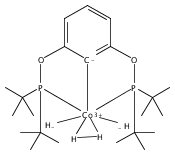
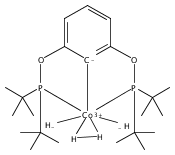
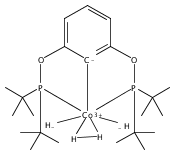
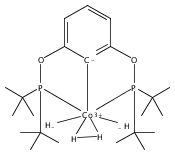
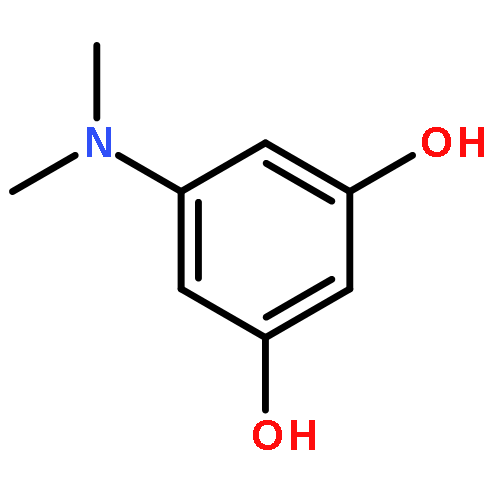
Addition of high pressures of H2 to five-coordinate [(tBu)4(POCOP)Ir(CO)(H)]OTf [(tBu)4(POCOP) = κ3-C6H3-2,6-(OP(tBu)2)2] complexes results in observation of two new iridium–dihydrogen complexes. If the aryl moiety of the POCOP ligand is substituted with an electron withdrawing protonated dimethylamino group at the para position, hydrogen coordination is enhanced. Five-coordinate Ir–H complexes generated by addition of triflic acid to (tBu)4(POCOP)Ir(CO) species show an Ir–H 1H NMR chemical shift dependence on the number of equivalents of acid present. It is proposed that excess triflic acid in solution facilitates triflate dissociation from iridium, resulting in unsaturated five-coordinate Ir–H complexes. The five-coordinate iridium–hydride complexes were found to catalyze H/D exchange between H2 and CD3OD. The existence of the dihydrogen complexes, as well as isotope exchange reactions, provide evidence for proposed ionic hydrogenation intermediates for glycerol deoxygenation.
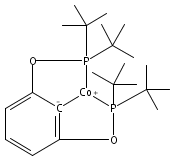
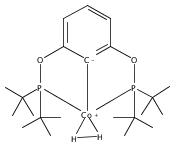
Co-reporter:Louise M. Guard, Travis J. Hebden, Donald E. Linn Jr., and D. Michael Heinekey
Organometallics August 28, 2017 Volume 36(Issue 16) pp:3104-3104
Publication Date(Web):August 18, 2017
DOI:10.1021/acs.organomet.7b00434
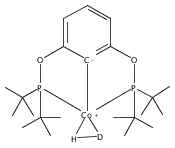
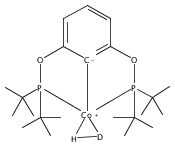
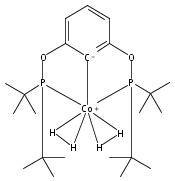
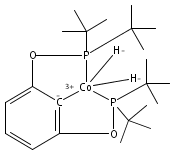
A pair of POCOP-supported mono- and dicarbonyl complexes of Co have been prepared and crystallographically characterized. The reactivity of (tBuPOCOP)Co(CO) with H2, acids, and carbon monoxide has been compared to that of the previously reported Rh and Ir counterparts. Co is found to share reactivity traits with both Rh and Ir.
Co-reporter:Jonathan M. Goldberg, Janna L. Berman, Werner Kaminsky, Karen I. Goldberg, D. Michael Heinekey
Journal of Organometallic Chemistry 2017 Volume 845(Volume 845) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.jorganchem.2017.04.036
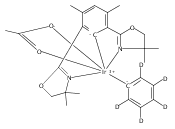
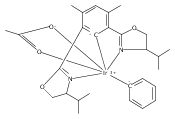
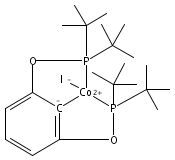
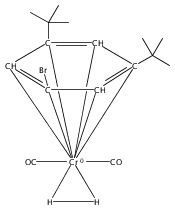
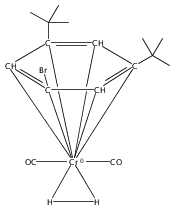
•Formation of stable cis-diiodide iridium carbonyl complexes.•Observation of an oxidative addition intermediate.•I2 addition to square planar Ir(I) complexes likely proceeds via an SN2 mechanism.Reactions of (X)(tBu)4(POCOP)Ir(CO) [(X)(tBu)4(POCOP) = κ3-C6H2-2,6-(OP(tBu)2)2-4-X where X = H, Me, COOMe, OMe, NMe2] complexes with iodine proceed to form Ir(III) iodide carbonyl complexes. When X = H, Me, or COOMe, cis-diiodide carbonyl complexes are observed; however if X = NMe2, a cationic five-coordinate monoiodo carbonyl complex is formed. The stability of the five-coordinate complex is attributed to the unfavorability of iodide coordination to an electron-rich metal center. When X = OMe, iodine addition is proposed to form an equilibrium mixture of a cis-diiodide complex and a five-coordinate monoiodo species. The data are consistent with a mechanism featuring oxidative addition of iodine to Ir(I) to give a cationic monoiodo carbonyl complex, followed by iodide coordination to give a cis-diiodide if the metal center is sufficiently Lewis acidic.Download high-res image (164KB)Download full-size image
Co-reporter:Timothy P. Brewster, Jonathan M. Goldberg, Jeremy C. Tran, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2016 Volume 6(Issue 9) pp:6302
Publication Date(Web):August 23, 2016
DOI:10.1021/acscatal.6b02130
A family of (para-cymene)RuII complexes are shown to be competent precatalysts for the oxidation of aldehydes to carboxylic acids using water as the oxidant. This reaction, known as the “aldehyde–water shift” (AWS), has been previously demonstrated to be in competition with aldehyde disproportionation. For the few reported mononuclear catalysts for this reaction, either high selectivity for AWS and low conversion or low AWS selectivity and high conversion is observed. A homogeneous precatalyst which is both highly selective for the desired AWS and is highly efficient for conversion of the aldehyde to products is reported herein. In addition, catalyst activity is found to be general to a variety of sterically unencumbered aliphatic aldehydes producing the corresponding carboxylic acid and hydrogen gas.Keywords: aldehyde oxidation; aldehyde−water shift; dehydrogenation; homogeneous catalysis; water
Co-reporter:Jonathan M. Goldberg, Sophia D. T. Cherry, Louise M. Guard, Werner Kaminsky, Karen I. Goldberg, and D. Michael Heinekey
Organometallics 2016 Volume 35(Issue 20) pp:3546-3556
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.organomet.6b00598
(POCOP)IrI(CO) [POCOP = κ3-C6H3-2,6-(OPR2)2 for R = tBu, iPr] and (PCP)IrI(CO) [PCP = κ3-C6H3-2,6-(CH2PR2)2 for R = tBu and iPr] complexes can add hydrogen via two distinct pathways. When R = tBu, (POCOP)Ir(CO) and (PCP)Ir(CO) complexes only add hydrogen via a proton-catalyzed pathway due to steric effects, yielding trans-dihydride complexes. For R = iPr, both systems oxidatively add hydrogen to give cis-dihydride complexes which thermally isomerize to more thermodynamically favorable trans-dihydride species, consistent with previous reports. Proton-catalyzed hydrogen addition pathways are also observed for both iPr-substituted (pincer)Ir(CO) complexes. (PCP)Ir(CO) complexes add hydrogen under milder conditions than the analogous POCOP species. Intermediate hydrido-pyridine Ir(III) carbonyl complexes from the proton-catalyzed pathway have been synthesized and characterized. This is the first report of a series of complexes shown to add hydrogen via concerted oxidative addition or a proton-catalyzed pathway to the same iridium center.
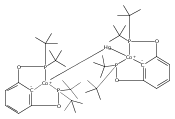
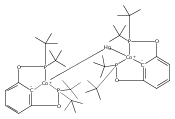
Co-reporter:Sophia D. T. Cherry, Werner Kaminsky, and D. Michael Heinekey
Organometallics 2016 Volume 35(Issue 12) pp:2165-2169
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.organomet.6b00418
Pincer ligand metalation is presumed to proceed via initial coordination to the phosphorus atoms followed by C–H oxidative addition. Few isolated intermediates in this process are known. A rhodium phosphinite complex has been isolated and structurally characterized that exhibits a strong agostic interaction with a C–H bond in the ligand backbone. The [(tBuPOCOP)Rh(CO)H]+ system exhibits greater acidity and reactivity than the analogous iridium species (tBuPOCOP = κ3-C6H3-1,3-[OP(tBu)2]2).
Co-reporter:Samantha J. Connelly;Andrew G. Chanez; Werner Kaminsky ; D. Michael Heinekey
Angewandte Chemie 2015 Volume 127( Issue 20) pp:6013-6016
Publication Date(Web):
DOI:10.1002/ange.201412076
Abstract
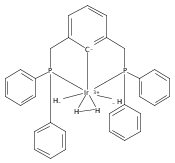
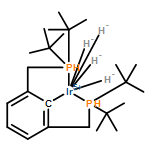
The preparation and isolation of the first palladium dihydrogen complex is described. NMR spectroscopy reveals a very short HH bond length, but the hydrogen molecule is activated toward heterolytic cleavage. An X-ray crystal structure suggests that proton transfer to the tBuPCP (κ3-2,6-(tBu2PCH2)2C6H3) pincer ligand is possible. The basicity of the ipso-carbon atom of the pincer ligand was investigated in a related complex.
Co-reporter:Jonathan M. Goldberg, Gene W. Wong, Kenzie E. Brastow, Werner Kaminsky, Karen I. Goldberg, and D. Michael Heinekey
Organometallics 2015 Volume 34(Issue 4) pp:753-762
Publication Date(Web):February 3, 2015
DOI:10.1021/om501166w
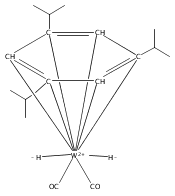
POCOP ligands (POCOP = C6H4-1,3-[OPR2]2) with R= tBu substituents on phosphorus react with Ir(CO)2Cl(p-toluidine) to afford square planar Ir(I) carbonyl complexes. Replacement of tBu groups with iPr groups results in the formation of six-coordinate hydrido-chloride carbonyl complexes of Ir(III). Analogous reactions of POCOP ligands with R groups smaller than iPr require meta-disubstitution in the aromatic ring to prevent precipitation of an insoluble polymer upon metalation with iridium. Structures of five-coordinate cationic hydrido carbonyl species show that a small change in steric demand of the alkyl groups on phosphorus can open the metal coordination sphere, with smaller alkyl groups favoring six-coordinate Ir(III) complexes.
Co-reporter:Samantha J. Connelly;Andrew G. Chanez; Werner Kaminsky ; D. Michael Heinekey
Angewandte Chemie International Edition 2015 Volume 54( Issue 20) pp:5915-5918
Publication Date(Web):
DOI:10.1002/anie.201412076
Abstract
The preparation and isolation of the first palladium dihydrogen complex is described. NMR spectroscopy reveals a very short HH bond length, but the hydrogen molecule is activated toward heterolytic cleavage. An X-ray crystal structure suggests that proton transfer to the tBuPCP (κ3-2,6-(tBu2PCH2)2C6H3) pincer ligand is possible. The basicity of the ipso-carbon atom of the pincer ligand was investigated in a related complex.
Co-reporter:Timothy P. Brewster, William C. Ou, Jeremy C. Tran, Karen I. Goldberg, Susan K. Hanson, Thomas R. Cundari, and D. Michael Heinekey
ACS Catalysis 2014 Volume 4(Issue 9) pp:3034
Publication Date(Web):July 29, 2014
DOI:10.1021/cs500843a
A series of half-sandwich complexes of iridium, rhodium, and ruthenium are shown to be active catalysts for the conversion of aldehydes and water to carboxylic acids. Depending on the catalyst, H2 is either released (the “aldehyde–water shift”) or transferred to a second equivalent of aldehyde (aldehyde disproportionation). Mechanistic studies suggest hydride transfer to be the selectivity-determining step along the reaction pathway. Using [(p-cymene)Ru(bpy)OH2][OTf]2 as precatalyst, we demonstrate a novel example of a highly selective aldehyde–water shift in the absence of a hydrogen acceptor or base.Keywords: aldehyde oxidation; dehydrogenation; disproportionation; homogeneous catalysis; water
Co-reporter:David B. Lao, Alisa C. E. Owens, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2013 Volume 3(Issue 10) pp:2391
Publication Date(Web):September 5, 2013
DOI:10.1021/cs400551g
Iridium pincer complexes (POCOP)Ir(CO) (POCOP = κ3-C6H3-1,3-[OP(tBu)2]2) and substituted POCOP derivatives catalyze deoxygenation of glycerol to n-propanol and 1,3-propanediol in good yield under moderate conditions (acidic aqueous dioxane, 200 °C, 80 bar H2). Catalyst solubility in the polar reaction mixture is improved by incorporation of a polar moiety in the para position of the POCOP phenyl ring, with the best results obtained with a dimethylamino substituent.Keywords: catalysis; deoxygenation; glycerol
Co-reporter:Samantha J. Connelly, Werner Kaminsky, and D. Michael Heinekey
Organometallics 2013 Volume 32(Issue 24) pp:7478-7481
Publication Date(Web):November 11, 2013
DOI:10.1021/om400970j
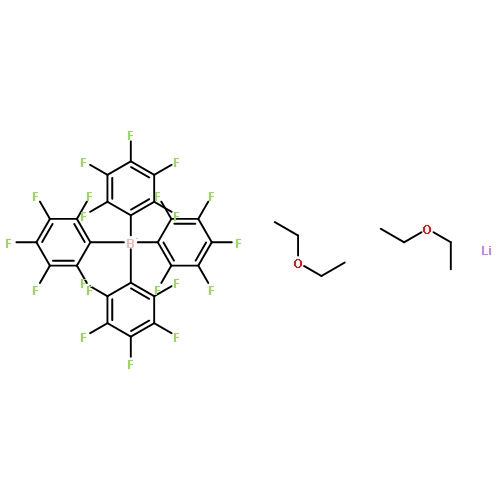
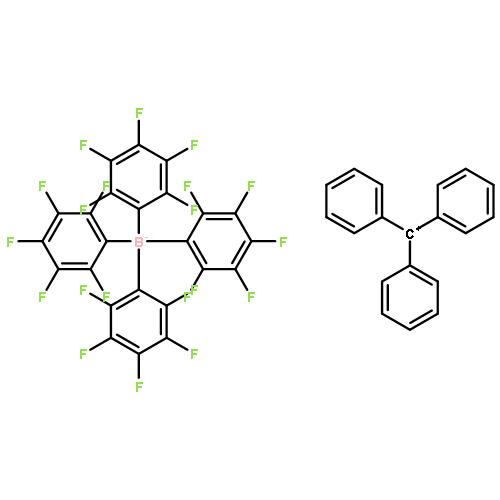
Reaction of triphenylmethylium tetrakis(pentafluorophenyl)borate, [Ph3C][B(C6F5)4], with excess neat triethylsilane affords triethylsilylium(triethylsilane) tetrakis(pentafluorophenyl)borate, [(Et3Si)2(μ-H)][B(C6F5)4] (1), identified by X-ray crystallography. In chlorobenzene and fluorobenzene, 1 is observed in solution. When 1 is dissolved in benzene or toluene, the evolved gas was shown to be hydrogen. Isotope labeling experiments demonstrate that the hydrogen arises from the reaction of Et3SiH with the silylium complex of the arene solvent.
Co-reporter:Takiya J. Ahmed Foskey, D. Michael Heinekey, and Karen I. Goldberg
ACS Catalysis 2012 Volume 2(Issue 6) pp:1285
Publication Date(Web):May 1, 2012
DOI:10.1021/cs300120d
Iridium pincer complex (POCOP)IrH2 (1; POCOP = κ3-C6H3-1,3-[OP(tBu)2]2) catalyzes hydrogenolysis of 1,2-propanediol to n-propanol in good yield under mild conditions (acidic aqueous dioxane, 50–125 °C, 100–600 psi H2). Studies of catalyst speciation revealed that the catalyst reservoir species is (POCOP)Ir(CO) (2), formed by decarbonylation of the substrate. Complex 2 is a superior catalyst precursor, since it is air-stable and readily prepared by treating complex 1 with CO.Keywords: catalysis; diol deoxygenation; hydrogenation; hydrogenolysis;
Co-reporter:Michael Findlater ; Katherine M. Schultz ; Wesley H. Bernskoetter ; Alison Cartwright-Sykes ; D. Michael Heinekey ;Maurice Brookhart
Inorganic Chemistry 2012 Volume 51(Issue 8) pp:4672-4678
Publication Date(Web):February 24, 2012
DOI:10.1021/ic202630x
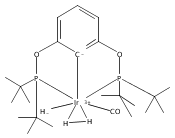
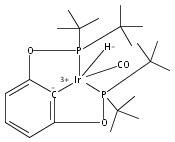
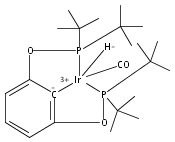
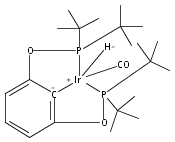
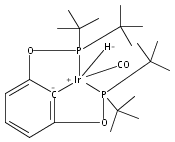
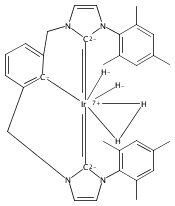
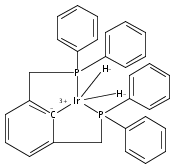
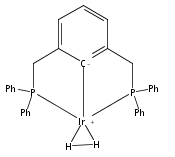
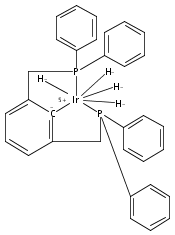
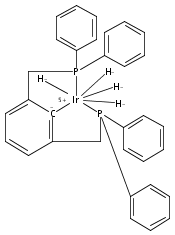
A series of iridium and rhodium pincer complexes have been synthesized and characterized: [(POCOP)Ir(H)(H2)] [BArf4] (1-H3), (POCOP)Rh(H2) (2-H2), [(PONOP)Ir(C2H4)] [BArf4] (3-C2H4), [(PONOP)Ir(H)2)] [BArf4] (3-H2), [(PONOP)Rh(C2H4)] [BArf4] (4-C2H4) and [(PONOP)Rh(H2)] [BArf4] (4-H2) (POCOP = κ3-C6H3-2,6-[OP(tBu)2]2; PONOP = 2,6-(tBu2PO)2C5H3N; BArf4 = tetrakis(3,5-trifluoromethylphenyl)borate). The nature of the dihydrogen–metal interaction was probed using NMR spectroscopic studies. Complexes 1-H3, 2-H2, and 4-H2 retain the H–H bond and are classified as η2-dihydrogen adducts. In contrast, complex 3-H2 is best described as a classical dihydride system. The presence of bound dihydrogen was determined using both T1 and 1JHD coupling values: T1 = 14 ms, 1JHD = 33 Hz for the dihydrogen ligand in 1-H3, T1(min) = 23 ms, 1JHD = 32 Hz for 2-H2, T1(min) = 873 ms for 3-H2, T1(min) = 33 ms, 1JHD = 30.1 Hz for 4-H2.
Co-reporter:Samantha J. Connelly;Ama C. Zimmerman;Dr. Werner Kaminsky ; D. Michael Heinekey
Chemistry - A European Journal 2012 Volume 18( Issue 50) pp:15932-15934
Publication Date(Web):
DOI:10.1002/chem.201203675
Co-reporter:Joseph M. Meredith, Robert Robinson Jr., Karen I. Goldberg, Werner Kaminsky, and D. Michael Heinekey
Organometallics 2012 Volume 31(Issue 5) pp:1879-1887
Publication Date(Web):February 7, 2012
DOI:10.1021/om2012166
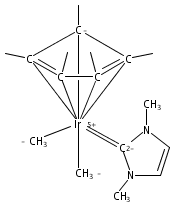
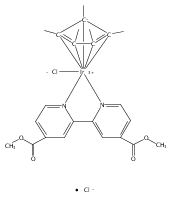
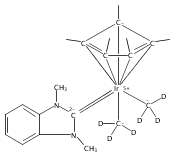
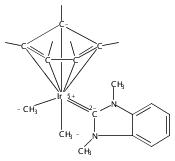
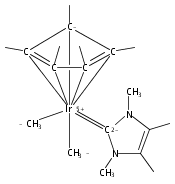
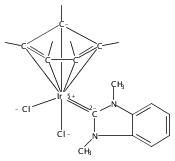
The cationic complexes [Cp*Ir(NHC)Me(solv)]+[MeB(C6F5)3]− were prepared and studied as models for methane oxy-functionalization catalysts (Cp* = η5-C5Me5; NHC = 1,3,4,5-tetramethylimidazol-2-ylidene (MeIMe, 3a), 1,3-dimethylimidazol-2-ylidene (IMe, 3b), 1,3-dimethylbenzimidazol-2-ylidene (BIMe, 3c); solv = solvent or open site). These complexes were targeted on the basis of the C–H bond activation reactions of the previously reported complexes [Cp*Ir(PMe3)R]+ (R = Me, H) and the general robustness of Ir–NHC complexes under oxidizing conditions. The syntheses of the new iridium(III) complexes Cp*Ir(NHC)Me2 are described (NHC = MeIMe (4a), IMe (4b), BIMe (4c)). When 4a–c were allowed to react with B(C6F5)3 in CH2Cl2, the methyl abstraction products [Cp*Ir(NHC)Me(solv)]+[MeB(C6F5)3]— (3a–c) were produced. Complexes 3a–c reacted with arenes to form the aryl complexes [Cp*Ir(NHC)Ar(solv)]+[MeB(C6F5)3]— and methane (Ar = C6H5 (7), C6H4F (8)). Complexes 3a–c reacted very slowly with alkanes; the slow reaction rate is attributed to steric congestion due to the NHC ligand. DFT calculations support this hypothesis: the barriers to C–H activation are in qualitative agreement with the empirical reaction rates, and the C–H activation transition state structures show significant steric crowding. Several of these complexes have been analyzed by X-ray diffraction.
Co-reporter:Joseph M. Meredith, Karen I. Goldberg, Werner Kaminsky, and D. Michael Heinekey
Organometallics 2012 Volume 31(Issue 23) pp:8459-8462
Publication Date(Web):November 26, 2012
DOI:10.1021/om3010492
The reaction of Cp*Ir(NHC)Me2 (Ir-Me2) with [CPh3]+[B(C6F5)4]— in dichloromethane resulted in conversion of the Cp* ligand to an η6-tetramethylfulvene ligand via apparent hydride abstraction (Cp* = η5-C5Me5; NHC = 1,3-N,N-dimethylimidazol-2-ylidene). The products of this reaction are triphenylmethane and the η6-tetramethylfulvene complex [(η6-C5Me4═CH2)Ir(NHC)(Me2)]+[B(C6F5)4]— (fulv-Ir+). When dichloromethane solutions of the related complex [Cp*Ir(NHC)(Ph)(solv)]+[MeB(C6F5)3]− were allowed to stand at room temperature, crystals of the unusual sandwich complex [{Cp*Ir(NHC)}2(μ-η3:η3-C6H6)]2+ (Ir-C6H6-Ir2+) were obtained. Complexes fulv-Ir+ and Ir-C6H6-Ir2+ represent unique architectures in the Cp*Ir(NHC) family. Both compounds were characterized by spectroscopic methods as well as by single-crystal X-ray diffraction.
Co-reporter:Steven L. Matthews and D. Michael Heinekey
Inorganic Chemistry 2011 Volume 50(Issue 17) pp:7925-7927
Publication Date(Web):July 27, 2011
DOI:10.1021/ic2009573
Models for the oxidized form of the FeFe hydrogenase active site have been prepared. These cationic complexes contain two iron atoms, carbonyl ligands, a propanedithiolate bridge, and one other bridging group. Reduction of these complexes with hydrogen gas is demonstrated.
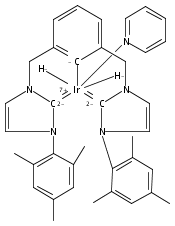
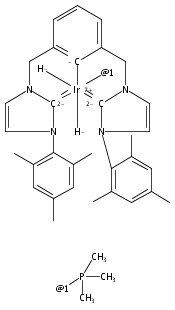
Co-reporter:Dr. Travis J. Hebden;Anthony J. St.John; Dmitry G. Gusev;Dr. Werner Kaminsky; Karen I. Goldberg; D. Michael Heinekey
Angewandte Chemie International Edition 2011 Volume 50( Issue 8) pp:1873-1876
Publication Date(Web):
DOI:10.1002/anie.201005281
Co-reporter:Katherine M. Schultz, Karen I. Goldberg, Dmitry G. Gusev, and D. Michael Heinekey
Organometallics 2011 Volume 30(Issue 6) pp:1429-1437
Publication Date(Web):March 2, 2011
DOI:10.1021/om101024x
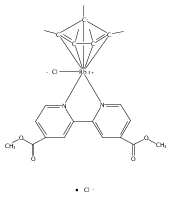
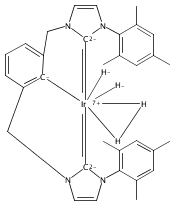
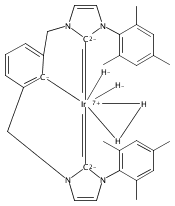
Iridium pincer complexes of the form (C∧C∧CMes)IrCl2 (1), (C∧C∧CMes)IrH4 (2), and (C∧C∧CMes)Ir(H)2L (L = PMe3 (3), pyridine (4)) have been prepared (C∧C∧C = [κ3-1,3-(CH2NHCMes)2C6H3]; NHCMes = N-mesitylimidazol-2-ylidene). Complexes 1−4 are the first examples of iridium complexes with CCC pincer ligands containing methylene bridges. Complex 2 activates C−H bonds of arenes at room temperature, as demonstrated by isotope exchange reactions. Under analogous conditions, no reaction was observed with alkanes.
Co-reporter:Dr. Travis J. Hebden;Anthony J. St.John; Dmitry G. Gusev;Dr. Werner Kaminsky; Karen I. Goldberg; D. Michael Heinekey
Angewandte Chemie 2011 Volume 123( Issue 8) pp:1913-1916
Publication Date(Web):
DOI:10.1002/ange.201005281
Co-reporter:Travis J. Hebden ; Karen I. Goldberg ; D. Michael Heinekey ; Xiawei Zhang ; Thomas J. Emge ; Alan S. Goldman ;Karsten Krogh-Jespersen
Inorganic Chemistry 2010 Volume 49(Issue 4) pp:1733-1742
Publication Date(Web):January 22, 2010
DOI:10.1021/ic902163w
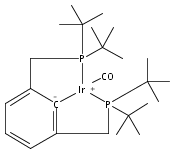
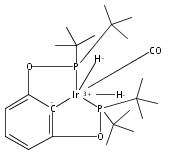
The iridium pincer complexes (PCP)IrH4 (1; PCP = [κ3-1,3-(CH2PtBu2)2C6H3]) and (POCOP)IrH4 (2; POCOP = [κ3-1,3-(OPtBu2)2C6H3]) have proven to be effective catalyst precursors for dehydrogenation of alkanes. The complex (POCOP)IrH2 has also been applied successfully as a catalyst for release of H2 from ammonia borane. Investigation of the “tetrahydride” forms of these complexes by solution NMR methods suggests their formulation as dihydrogen/dihydride species. This is in contrast to the solid state structure of 1, determined by neutron diffraction (at 100 K), which indicates a compressed tetrahydride structure with only weak H−H interactions. Complex 1 (C24H47IrP2) crystallizes in the space group P42, tetragonal, (Z = 2) with a = 11.7006 (19) Å, c = 9.7008(27) Å, and V = 1328.1(5) Å3. Electronic structure calculations on 1 and 2 indicate that the global minima on the potential energy surfaces in the gas phase are tetrahydride structures; however, the dihydrogen/dihydride forms are only slightly higher in energy (1−3 kcal/mol). A dihydrogen/dihydride species is calculated to be the global minimum for 2 when in solution. The barriers to interconversion between the tetrahydride and dihydrogen/dihydride species are almost negligible.
Co-reporter:Steven L. Matthews
Inorganic Chemistry 2010 Volume 49(Issue 21) pp:9746-9748
Publication Date(Web):September 30, 2010
DOI:10.1021/ic1017328
Protonation of Fe2(μ-S2C3H6)(CO)6 with an acid derived from [SiEt3][B(C6F5)4] and HCl affords a hydride-bridged dimer.
Co-reporter:Jonathan D. Egbert and D. Michael Heinekey
Organometallics 2010 Volume 29(Issue 15) pp:3387-3391
Publication Date(Web):July 2, 2010
DOI:10.1021/om100416w
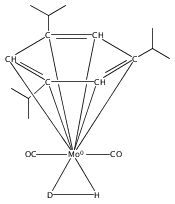
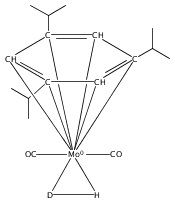
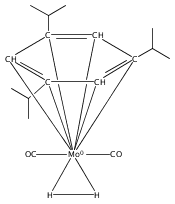
Irradiation of (arene)M(CO)3 complexes (M = Cr, Mo, W; arene = various benzene derivatives) in the presence of H2 results in CO extrusion and subsequent reaction with H2. For chromium and molybdenum, the resulting complexes have moderate thermal stability and are identified as sigma complexes of H2 by NMR spectroscopy. Analogous tungsten reactions require prolonged irradiation in the presence of H2 to afford moderate yields of dihydride complexes. This study allows for comparison of related complexes for Cr, Mo, and W.
Co-reporter:Joseph M. Meredith, Karen I. Goldberg, Werner Kaminsky and D. Michael Heinekey
Organometallics 2009 Volume 28(Issue 12) pp:3546-3551
Publication Date(Web):May 4, 2009
DOI:10.1021/om900186j
The syntheses of the monomeric cationic complexes [Cp*(CO)Ir(R)(CH2Cl2)] were attempted (Cp* = η5-C5Me5, R = Me, H). The target complexes were not obtained; instead, dimeric products were isolated in high yields. The reaction between Cp*(CO)Ir(Me)Cl and [Li(Et2O)2.5][BArF4] afforded the cationic complex [{Cp*(CO)Ir(Me)}2(μ-Cl)][BArF4] (4) in high yield. Complex 4 has been characterized by 1H NMR, 13C{1H} NMR, and IR spectroscopies. Reaction of Cp*(CO)Ir(H)2 with [CPh3][BArF4] produced the dicationic complex [Cp*(CO)Ir(μ-H)]2[BArF4]2 (9) in excellent yield. Complex 9 has been characterized by 1H NMR and IR spectroscopies as well as elemental analysis and single-crystal X-ray diffraction. An X-ray structural determination was also carried out on the related complex [Cp*(Cl)Ir(μ-H)]2 (14). The solid-state structures of 9 and 14 are compared briefly.
Co-reporter:Serena Fantasia Dr.;JonathanD. Egbert;Václav Jur&x10d;ík Dr.;CatherineS.J. Cazin Dr.;Heiko Jacobsen Dr.;Luigi Cavallo Dr.;D.Michael Heinekey Dr.;StevenP. Nolan Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 28) pp:5182-5186
Publication Date(Web):
DOI:10.1002/anie.200900463
Co-reporter:Serena Fantasia Dr.;JonathanD. Egbert;Václav Jur&x10d;ík Dr.;CatherineS.J. Cazin Dr.;Heiko Jacobsen Dr.;Luigi Cavallo Dr.;D.Michael Heinekey Dr.;StevenP. Nolan Dr.
Angewandte Chemie 2009 Volume 121( Issue 28) pp:5284-5288
Publication Date(Web):
DOI:10.1002/ange.200900463
Co-reporter:Inigo Aguirre de Carcer, D. Michael Heinekey
Journal of Organometallic Chemistry 2009 694(6) pp: 840-844
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.06.038
Co-reporter:Brandon L. Dietrich ; Karen I. Goldberg ; D. Michael Heinekey ; Tom Autrey ;John C. Linehan
Inorganic Chemistry 2008 Volume 47(Issue 19) pp:8583-8585
Publication Date(Web):September 12, 2008
DOI:10.1021/ic801161g
Dehydrogenation of amine boranes is catalyzed efficiently by the iridium pincer complex (κ3-1,3-(OPtBu2)2C6H3)Ir(H)2 (1). With CH3NH2BH3 (MeAB) and with AB/MeAB mixtures (AB = NH3BH3), the rapid release of 1 equiv of H2 is observed to yield soluble oligomeric products at rates similar to those previously reported for the dehydrogenation of AB catalyzed by 1. ΔH for the dehydrogenation of AB, MeAB, and AB/MeAB mixtures has been determined by calorimetry. The experimental heats of reaction are compared to results from computational studies.
Co-reporter:Susan Kloek Hanson, D. Michael Heinekey and Karen I. Goldberg
Organometallics 2008 Volume 27(Issue 7) pp:1454-1463
Publication Date(Web):March 4, 2008
DOI:10.1021/om7012259
New rhodium(I) complexes (PNP)Rh(X) (PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) (X = OTf (1), OAc (3), OH (8), OCH2CF3 (9), OC6H5 (10), OC6H4NO2 (11)) have been prepared. Hydroxide complex 8 and trifluoroethoxide complex 9 undergo stoichiometric activation of benzene-d6 to form the phenyl complex (PNP)Rh(C6D5). Acetate and aryloxide complexes 3, 10, and 11 are active catalysts for H−D exchange between arenes and water. Control experiments indicate that the rhodium complexes are the active catalysts and that the observed exchange is not catalyzed by adventitious acid. Mechanistic studies of the H−D exchange reaction support a pathway involving dissociation of aryloxide or acetate ligand. The reaction is accelerated by added alcohol and, for the acetate complex, inhibited by added sodium acetate.
Co-reporter:Daniel F. Brayton
Organometallics 2008 Volume 27(Issue 15) pp:3901-3906
Publication Date(Web):June 28, 2008
DOI:10.1021/om800273t
The synthesis of two bisphosphine ligands (R2P)2C6H4 with a para-disubstituted phenyl moiety is described (R = Cy (3), i-Pr (4)). Solutions of M(CO)3(C7H8) (C7H8 = η6-cycloheptatriene, M = Mo and W) and 3 or 4 were reacted under CO affording dimeric compounds of the general formula [M2(CO)8((R2P)2C6H4)2] (M = W, R = Cy (5); M = Mo, R = Cy (6); and M = W, R = i-Pr (7)). When the same reactions are carried out in the presence of H2, similar W−H2 analogues of the general formula [W2(CO)6((R2P)2C6H4)2(H2)2], R = Cy (8), R = i-Pr (9), can be observed by 1H and 31P NMR spectroscopy. Compounds 8 and 9 are not isolable, due to facile ligand redistribution forming 5 and 7, respectively.
Co-reporter:Susan M. Kloek;D. Michael Heinekey ;Karen I. Goldberg
Angewandte Chemie 2007 Volume 119(Issue 25) pp:
Publication Date(Web):10 MAY 2007
DOI:10.1002/ange.200700270
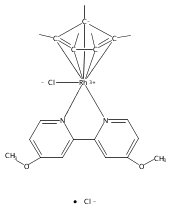
Rhodium(I)-Komplexe der Form [(PNP)Rh(OR)] (R=H, CH2CF3, C6H5; PNP=2,6-Bis[(di-tert-butylphosphanyl)methyl]pyridin) wurden hergestellt. Die Hydroxid- und Trifluorethoxidkomplexe aktivieren Benzol bei der Thermolyse in [D6]Benzol. [(PNP)Rh(OC6H5)] katalysiert den H/D-Austausch zwischen D2O und Benzol sowie zwischen H2O und [D8]Toluol (siehe Schema), bei dem der Austausch selektiv an den meta- und para-Positionen verläuft.
Co-reporter:Susan M. Kloek;D. Michael Heinekey ;Karen I. Goldberg
Angewandte Chemie International Edition 2007 Volume 46(Issue 25) pp:
Publication Date(Web):10 MAY 2007
DOI:10.1002/anie.200700270
Rhodium(I) complexes of the form [(PNP)Rh(OR)] (R=H, CH2CF3, C6H5; PNP=2,6-bis[(di-tert-butylphosphino)methyl]pyridine) have been prepared. Upon thermolysis in [D6]benzene, the hydroxide and trifluoroethoxide complexes undergo benzene activation. [(PNP)Rh(OC6H5)] is an active catalyst for the H/D exchange between D2O and benzene and between H2O and [D8]toluene (see scheme), for which exchange occurs selectively at the meta and para positions.
Co-reporter:D. Michael Heinekey
Journal of Labelled Compounds and Radiopharmaceuticals 2007 Volume 50(Issue 11‐12) pp:1063-1071
Publication Date(Web):26 JUN 2007
DOI:10.1002/jlcr.1385
Complexation of dihydrogen to transition metal centers was discovered by Kubas and coworkers in 1984. The notion that the simplest molecule in chemistry can act as a ligand to form relatively stable transition metal complexes has led to a paradigm shift in coordination chemistry. Crucial to the exploration of this intriguing new chemistry has been the use of isotope substitution. Several aspects of the coordination chemistry of dihydrogen have revealed fascinating isotope effects on reactivity, spectroscopy and in some cases structure. Complexation of HD has been used to diagnose bond distances from measurements of JHD and isotope effects for D2 versus H2 binding have been evaluated. Examples of quantum mechanical exchange coupling in H2 complexes have been described. These effects disappear when one of the H atoms is replaced by D. In molecules with bound hydrogen adjacent to a hydride ligand, non-statistical occupancy of hydrogen versus hydride sites by deuterons has been observed. In some cases, isotope-dependent structures have been established by the study of HD, HT and DT complexes using NMR spectroscopy. Copyright © 2007 John Wiley & Sons, Ltd.
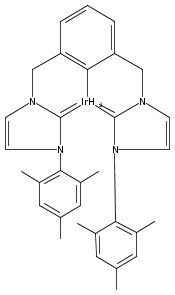
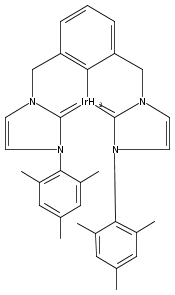
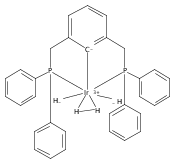
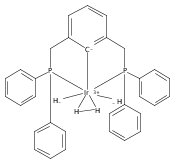
.jpg)





![Phosphine,1,1'-[(1,3-phenylene)bis(methylene)]bis[1,1-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/193100/193084-64-9.png)
![Phosphine,1,1'-[(1,3-phenylene)bis(methylene)]bis[1,1-bis(1-methylethyl)-](http://img.cochemist.com/ccimg/193100/193084-64-9_b.png)











































































































































































































![[2,2'-Bipyridine]-4,4'-diol](/data/chemimg/1008300/90770-88-0.png)
![[2,2'-Bipyridine]-4,4'-diol](/data/chemimg/1008300/90770-88-0_b.png)




![[Ir(η2-ethylene)2Cl]2](http://img.cochemist.com/ccimg/39800/39722-81-1.png)
![[Ir(η2-ethylene)2Cl]2](http://img.cochemist.com/ccimg/39800/39722-81-1_b.png)




![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)






![Molybdenum,tricarbonyl[(1,2,3,4,5,6-h)-1,3,5-trimethylbenzene]- (9CI)](http://img.cochemist.com/ccimg/12100/12089-15-5.png)
![Molybdenum,tricarbonyl[(1,2,3,4,5,6-h)-1,3,5-trimethylbenzene]- (9CI)](http://img.cochemist.com/ccimg/12100/12089-15-5_b.png)
![Chromium,tricarbonyl[(1,2,3,4,5,6-h)-methylbenzene]-](http://img.cochemist.com/ccimg/12100/12083-24-8.png)
![Chromium,tricarbonyl[(1,2,3,4,5,6-h)-methylbenzene]-](http://img.cochemist.com/ccimg/12100/12083-24-8_b.png)