Co-reporter:Jie Zang, Baowen Shi, Xuewu Liang, Qianwen Gao, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmc.2016.12.001
•A novel series of N-hydroxycinnamamide-based HDAC inhibitors were synthesized.•Most compounds showed superior HDAC inhibition and antiproliferative activity.•These new compounds seem to be pan-HDAC inhibitors as parent compound 1 and SAHA.Histone deacetylase inhibitors (HDACIs) are promising in the treatment of various diseases, among which cancer treatment has achieved the most success. We have previously developed series of HDACIs combining N-hydroxycinnamamide bioactive fragment and indole bioactive fragment, which showed moderate to potent antitumor activities. Herein, further structural derivatization based on our previous structure-activity relationship (SAR) got 25 novel compounds. Most compounds showed much more potent histone deacetylases (HDACs) inhibitory activity than their parent compound 1 and even the positive control SAHA. What’s more, compared with the approved HDACs inhibitor SAHA, compounds 6i, 6k, 6q and 6t displayed better in vitro antiproliferation against multiple tumor cell lines. It is worth noting that though the 4-hydroxycinnamic acid-based compound 2 showed HDAC1/3 dual selectivity, its 4-hydroxy-3-methoxycinnamic acid-based analog 6t turned out to be a pan-HDACs inhibitor as SAHA, indicating that the 3-methoxy group on the N-hydroxycinnamamide fragment could dramatically influence the HDACs isoform selectivity of this series of compounds.Download high-res image (147KB)Download full-size image
Co-reporter:Shuai Gao, Jie Zang, Qianwen Gao, Xuewu Liang, Qinge Ding, Xiaoyang Li, Wenfang Xu, C. James Chou, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmc.2017.03.036
•A novel series of o-phenylenediamine-based HDAC inhibitors were synthesized.•Some compounds showed superior HDAC inhibition and antiproliferative activity.•Compound 9n exhibited moderate HDAC1 selectivity over HDAC2 and HDAC3.As a hot topic of epigenetic studies, histone deacetylases (HDACs) are related to lots of diseases, especially cancer. Further researches indicated that different HDAC isoforms played various roles in a wide range of tumor types. Herein a novel series of HDAC inhibitors with isatin-based caps and o-phenylenediamine-based zinc binding groups have been designed and synthesized through scaffold hopping strategy. Among these compounds, the most potent compound 9n exhibited similar if not better HDAC inhibition and antiproliferative activities against multiple tumor cell lines compared with the positive control entinostat (MS-275). Additionally, compared with MS-275 (IC50 values for HDAC1, 2 and 3 were 0.163, 0.396 and 0.605 µM, respectively), compound 9n with IC50 values of 0.032, 0.256 and 0.311 µM for HDAC1, 2 and 3 respectively, showed a moderate HDAC1 selectivity.Download high-res image (97KB)Download full-size image
Co-reporter:Nan Zhou, Yugang Yan, Chunxi Liu, Jinning Hou, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmc.2017.06.039
Histone deacetylase inhibitors with desirable pharmacokinetic profiles which can be delivered to solid tumor tissues in large amount might be promising to treat solid tumor effectively. Herein, structural modification of a previously reported tetrahydroisoquinoline-based HDAC inhibitor 1 was carried out to improve its plasma stability for more feasible drug delivery. Among three newly synthesized analogs, the in vitro rat plasma stability of compound 2 (t1/2 = 630 min) was over 5-fold improved than its parent 1 (t1/2 = 103 min). In vitro activity evaluation showed that compound 2 and 1 exhibited similar HDACs inhibitory activity, which was validated by western blot analysis and antiproliferative assay. Moreover, compared with 1, compound 2 exhibited comparable, if not higher, in vivo antitumor activity in a human breast carcinoma (MDA-MB-231) xenograft model.Download high-res image (110KB)Download full-size image
Co-reporter:Xiaoyang Li, Yingjie Zhang, Yuqi Jiang, Jingde Wu, Elizabeth S. Inks, C. James Chou, Shuai Gao, Jinning Hou, Qinge Ding, Jingyao Li, Xue Wang, Yongxue Huang, Wenfang Xu
European Journal of Medicinal Chemistry 2017 Volume 134(Volume 134) pp:
Publication Date(Web):7 July 2017
DOI:10.1016/j.ejmech.2017.03.069
•Most of the benzamides exhibited high HDACs inhibitory potency.•11a exhibited selectivity against HDAC1 to some extent.•11a displayed potent oral activity in both hematological and solid tumor xenograft model with no obvious toxicity.•New thienyl and phenyl compounds exhibited dramatic HDAC1/2 dual selectivity.Previously, we reported the discovery of a series of N-hydroxycinnamamide-based HDAC inhibitors, among which compound 11y exhibited high HDAC1/3 selectivity. In this current study, structural derivatization of 11y led to a new series of benzamide based HDAC inhibitors. Most of the compounds exhibited high HDACs inhibitory potency. Compound 11a (with 4-methoxybenzoyl as N-substituent in the cap and 4-(aminomethyl) benzoyl as the linker group) exhibited selectivity against HDAC1 to some extent, and showed potent antiproliferative activity against several tumor cell lines. In vivo studies revealed that compound 11a displayed potent oral antitumor activity in both hematological tumor cell U937 xenograft model and solid tumor cell HCT116 xenograft model with no obvious toxicity. Further modification of benzamide 3, 11a and 19 afforded new thienyl and phenyl compounds (50a, 50b, 63a, 63b and 63c) with dramatic HDAC1 and HDAC2 dual selectivity, and the fluorine containing compound 56, with moderate HDAC3 selectivity.Download high-res image (148KB)Download full-size image
Co-reporter:Yingjie Zhang, Jin Yan, Tso-Pang Yao
European Journal of Medicinal Chemistry 2017 Volume 141(Volume 141) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.ejmech.2017.10.022
•Two novel fluorescent HDAC inhibitors 6a and 6b were designed and synthesized.•6b is a HDAC6 selective inhibitor which can induce hyperacetylation of tubulin but not histone H4.•6b can selectively target and image HDAC6 within the inclusion body.•6b might be useful to visualize other HDAC6 associated protein complex in various physiological and pathological contexts.There is increasing interest in discovering HDAC6 selective inhibitors as chemical probes to elucidate the biological functions of HDAC6 and ultimately as new therapeutic agents. Small-molecular fluorescent probes are widely used to detect target protein location and function, identify protein complex composition in biological processes of interest. In the present study, structural modification of the previously reported compound 4MS leads to two novel fluorescent HDAC inhibitors, 6a and 6b. Determination of IC50 values against the panel of Zn2+ dependent HDACs (HDAC1-11) reveals that 6b is a HDAC6 selective inhibitor, which can induce hyperacetylation of tubulin but not histone H4. Importantly, fluorescent and immunofluorescent analyses of cells treated with the proteasome inhibitor MG132 demonstrates that 6b can selectively target and image HDAC6 within the inclusion body, the aggresome. These results identify 6b not only as a HDAC6 selective inhibitor but also as a fluorescent probe for imaging HDAC6 and investigating the roles of HDAC6 in various physiological and pathological contexts.Download high-res image (212KB)Download full-size image
Co-reporter:Chunhua Ma, Jiangying Cao, Xuewu Liang, Yongxue Huang, Ping Wu, Yingxia Li, Wenfang Xu, Yingjie Zhang
European Journal of Medicinal Chemistry 2016 Volume 108() pp:21-27
Publication Date(Web):27 January 2016
DOI:10.1016/j.ejmech.2015.11.021
•All compounds showed better activities on APN than Bestatin and lead compound 4k.•7a showed better activities than Bestatin in antimetastasis assay.•Introduction of disubstitution on ortho-position could improve APN inhibitory activities.Aminopeptidase N (APN/CD13) over-expressed on tumor cells and tumor microenvironment, plays critical roles in tumor invasion, metastasis and angiogenesis. Here we described the design, synthesis and preliminary activity studies of novel leucine ureido derivatives as aminopeptidase N (APN/CD13) inhibitors. The results showed that compound 7a had the most potent inhibitory activity against APN with the IC50 value of 20 nM, which could be used for further anticancer agent research.7a showed better activity than the positive control Bestatin in inhibiting tumor metastasis.
Co-reporter:Yuqi Jiang, Xiaoyang Li, Jinning Hou, Yongxue Huang, Yuping Jia, Mingming Zou, Jian Zhang, Xuejian Wang, Wenfang Xu, Yingjie Zhang
European Journal of Medicinal Chemistry 2016 Volume 121() pp:649-657
Publication Date(Web):4 October 2016
DOI:10.1016/j.ejmech.2016.05.068
•A novel mutual prodrug (hybrid drug) of ubenimex and fluorouracil was synthesized.•The compound 3 displayed potent inhibitory activity against CD13 enzymatic activity.•Compared with capecitabine, 3 exhibited more potent tumor growth inhibitory and anti-metastasis effects.•Compared with 5-FU, 3 also exhibited a superior antitumor efficiency even in our 5-FU-resistant mice model.We designed and synthesized a novel mutual prodrug, named BC-01 (3), by integrating ubenimex and Fluorouracil (5-FU) into one molecule based on prior research results that showed that a combination of the aminopeptidase N (CD13) inhibitor, ubenimex, and the cytotoxic antitumor agent, 5-FU, exhibited improved in vitro and in vivo antitumor efficiency. 3 showed potent inhibitory activity against CD13 enzymatic activity. Compared with ubenimex, 3 exhibited more potent anti-angiogenesis effects, and compared with the approved 5-FU prodrug, capecitabine, 3 exhibited more potent tumor growth inhibitory and anti-metastasis effects. Additionally, compared with 5-FU or 5-FU plus ubenimex, 3 also exhibited a superior antitumor efficiency even in our 5-FU-resistant mice model. Other antitumor agents could be conjugated with ubenimex using this strategy to obtain novel mutual prodrugs with promising antitumor potency.
Co-reporter:Xuewu Liang, Yongxue Huang, Jie Zang, Qianwen Gao, Binghe Wang, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 12) pp:2660-2672
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmc.2016.04.030
•4-Aminopyrazole derivatives showed inhibitory potency against various JAKs.•The enzymatic activities of 17m were expected to reach nanomolar level.•17m could inhibit the phosphorylation of JAK2 in Hela cells effectively.JAKs inhibitors were widely applied in the treatment of immunodeficiency diseases, inflammation and cancers. We designed and synthesized a novel series of 4-aminopyrazole derivatives, which showed inhibitory potency against various JAKs. The in vitro protein kinase inhibition experiment indicated that compounds 17k, 17l, 17m and 17n could inhibit JAKs effectively. Among them, compound 17m possessed the highest protein kinase inhibitory rates (%) at 10 μM, which were 97, 96 and 100 to JAK1, JAK2 and JAK3, respectively. Further evaluation revealed that the IC50 values of 17m against JAK1, JAK2 and JAK3 were 0.67 μM, 0.098 μM and 0.039 μM, respectively. Moreover, western blotting results showed compound 17m could inhibit the phosphorylation of JAK2 in Hela cells effectively. The data supports the further investigation of these compounds as novel JAKs inhibitors.
Co-reporter:Yuqi Jiang, Jinning Hou, Xiaoyang Li, Yongxue Huang, Xuejian Wang, Jingde Wu, Jian Zhang, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 22) pp:5787-5795
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmc.2016.09.033
•A novel mutual prodrug of ubenimex and gemcitabine was synthesized.•The compound BC-A1 could maintain the anti-CD13 activity of ubenimex in vitro.•BC-A1 was an orally active prodrug of gemcitabine.Herein, a novel mutual prodrug BC-A1 was discovered by integrating ubenimex and gemcitabine into one molecule. Biological characterization revealed that compound BC-A1 could maintain both the anti-CD13 activity of ubenimex and the cytotoxic activity of gemcitabine in vitro. Further characterization also demonstrated that compound BC-A1 exhibited significant anti-invasion and anti-angiogenesis effects in vitro. The preliminary stability test of BC-A1 revealed that it could release gemcitabine in vitro. The in vivo anti-tumor results in liver cancer showed that at the same dosage, oral administration of BC-A1 was as potent as intraperitoneal administration of gemcitabine. This warranted the further research and development of the orally active prodrug BC-A1 because gemcitabine can not be orally administrated in clinic.Download high-res image (64KB)Download full-size image
Co-reporter:Xuewu Liang, Jie Zang, Mengyuan Zhu, Qianwen Gao, Binghe Wang, Wenfang Xu, and Yingjie Zhang
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 10) pp:950
Publication Date(Web):August 23, 2016
DOI:10.1021/acsmedchemlett.6b00247
Abnormalities in the JAK/STAT signaling pathway lead to many diseases such as immunodeficiency, inflammation, and cancer. Herein, we designed and synthesized a series of 4-amino-(1H)-pyrazole derivatives as potent JAKs inhibitors for cancer treatment. Results from in vitro protein kinase inhibition experiments indicated that compounds 3a–f and 11b are potent JAKs inhibitors. For example, the IC50 values of compound 3f against JAK1, JAK2, and JAK3 were 3.4, 2.2, and 3.5 nM, respectively. In cell culture experiments, compound 3f showed potent antiproliferative activity against various cell lines (PC-3, HEL, K562, MCF-7, and MOLT4) at low micromolar levels, while compound 11b showed selective cytotoxicity at submicromolar levels against HEL (IC50: 0.35 μM) and K562 (IC50: 0.37 μM) cell lines. It is worth noting that both 3f and 11b showed more potent antiproliferative activities than the approved JAKs inhibitor Ruxolitinib.Keywords: 4-Amino-(1H)-pyrazole; Anticancer; Inhibitors; JAKs;
Co-reporter:Wenwen Duan; Jin Li; Elizabeth S. Inks; C. James Chou; Yuping Jia; Xiaojing Chu; Xiaoyang Li; Wenfang Xu
Journal of Medicinal Chemistry 2015 Volume 58(Issue 10) pp:4325-4338
Publication Date(Web):April 23, 2015
DOI:10.1021/acs.jmedchem.5b00317
On the basis of the strategy of creating multifunctional drugs, a novel series of phenylsulfonylfuroxan-based hydroxamates with histone deacetylase (HDAC) inhibitory and nitric oxide (NO) donating activities were designed, synthesized, and evaluated. The most potent NO donor–HDAC inhibitor (HDACI) hybrid, 5c, exhibited a much greater in vitro antiproliferative activity against the human erythroleukemia (HEL) cell line than that of the approved drug SAHA (Vorinostat), and its antiproliferative activity was diminished by the NO scavenger hemoglobin in a dose-dependent manner. Further mechanism studies revealed that 5c strongly induced cellular apoptosis and G1 phase arrest in HEL cells. Animal experiment identified 5c as an orally active agent with potent antitumor activity in a HEL cell xenograft model. Interestingly, although compound 5c was remarkably HDAC6-selective at the molecular level, it exhibited pan-HDAC inhibition in a western blot assay, which is likely due to class I HDACs inhibition caused by NO release at the cellular level.
Co-reporter:Xiaoyang Li, Jinning Hou, Xiaoguang Li, Yuqi Jiang, Xueliang Liu, Weiwei Mu, Yiming Jin, Yingjie Zhang, Wenfang Xu
European Journal of Medicinal Chemistry 2015 Volume 89() pp:628-637
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.10.077
•A new series of 3-hydroxycinnamamide-based HDACIs were synthesized.•The series of compounds displayed pan-HDAC inhibitory activity.•Compound 7h showed potent in vitro antiproliferative activity.•7h was a potent, orally active HDACI in in vivo assay.Inhibition of histone deacetylases (HDACs) has diverse effects on cell function, such as causing differentiation, growth arrest and apoptosis in nearly all types of tumor cell lines. In our previous work, we have designed and synthesized a novel series of 4-hydroxycinnamamide-based and 3-hydroxycinnamamide-based HDAC inhibitors (HDACIs), among which, 3-hydroxycinnamamide-based HDACIs 1a–1c exhibited moderate inhibition against HDACs. In this article, we report the development of a more potent class of 3-hydroxycinnamamide-based HDACIs, compound 7o exhibited much higher pan-HDAC inhibitory activity than positive control SAHA. In addition, compound 7h showed excellent in vitro growth inhibitory activity against more than ten cell lines and induced U937 cells apoptosis in micromolar concentration. In vivo assay in U937 xenograft model identified compound 7h as a potent, orally active HDACI.
Co-reporter:Yugang Yan, Xueying Chen, Xinying Yang, Jian Zhang, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 20) pp:6632-6640
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmc.2015.09.013
Compounds 10 (ND-322) and 15 (ND-364) are potent selective inhibitors for gelatinases, matrix metalloproteinase 2 (MMP2) and matrix metalloproteinase 9 (MMP9). However, both of them are racemates. Herein we report facile synthesis of optically active (R)- and (S)-enantiomers of compounds 10 and 15. And the sulfonyl of 15 was transformed to sulfinyl to obtain four epimeric mixtures. All synthesized thiirane-based compounds were evaluated in MMP2 and MMP9 inhibitory assays. Our results indicated that the configuration of thiirane moiety had little effects on gelatinase inhibition, but the substitution of sulfinyl for sulfonyl was detrimental to gelatinase inhibition. Besides, all target compounds exhibited no inhibition against other two Zn2+ dependant metalloproteases, aminopeptidase N (APN) and histone deacetylases (HDACs), which confirmed the unique Zn2+ chelation mechanism of thiirane moiety against gelatinases.Chirality modification of ND-322 and ND-364.
Co-reporter:Wenwen Duan, Jinning Hou, Xiaojing Chu, Xiaoqian Li, Jian Zhang, Jin Li, Wenfang Xu, Yingjie Zhang
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4481-4488
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.015
Co-reporter:Yuanchao Xie ; Dongqing Xu ; Bing Huang ; Xiuli Ma ; Wenbao Qi ; Fangyuan Shi ; Xinyong Liu ; Yingjie Zhang ;Wenfang Xu
Journal of Medicinal Chemistry 2014 Volume 57(Issue 20) pp:8445-8458
Publication Date(Web):September 25, 2014
DOI:10.1021/jm500892k
To discover group-1-specific neuraminidase (NA) inhibitors that are especially involved in combating the H5N1 virus, two series of oseltamivir derivatives were designed and synthesized by targeting the 150-cavity. Among these, compound 20l was the most potent N1-selective inhibitor, with IC50 values of 0.0019, 0.0038, and 0.0067 μM against NAs from three H5N1 viruses. These values are better than those of oseltamivir carboxylate. Compound 32 was another potent N1-selective inhibitor that exhibited a 12-fold increase in activity against the H274Y mutant relative to oseltamivir carboxylate. Molecular docking studies revealed that the 150-cavity was an auxiliary binding site that may contribute to the high selectivity of these compounds. The present work is a significant breakthrough in the discovery of potent group-1-specific neuraminidase inhibitors, which may be further investigated for the treatment of infection by the H5N1 virus.
Co-reporter:Xiaoyang Li ; Elizabeth S. Inks ; Xiaoguang Li ; Jinning Hou ; C. James Chou ; Jian Zhang ; Yuqi Jiang ; Yingjie Zhang ;Wenfang Xu
Journal of Medicinal Chemistry 2014 Volume 57(Issue 8) pp:3324-3341
Publication Date(Web):April 2, 2014
DOI:10.1021/jm401877m
In our previous study, we designed and synthesized a novel series of N-hydroxycinnamamide-based HDAC inhibitors (HDACIs), among which the representative compound 14a exhibited promising HDACs inhibition and antitumor activity. In this current study, we report the development of a more potent class of N-hydroxycinnamamide-based HDACIs, using 14a as lead, among which, compound 11r gave IC50 values of 11.8, 498.1, 3.9, 2000.8, 5700.4, 308.2, and 900.4 nM for the inhibition of HDAC1, HDAC2, HDAC3, HDAC8, HDAC4, HDAC6, and HDAC11, exhibiting dual HDAC1/3 selectivity. Compounds 11e, 11r, 11w, and 11y showed excellent growth inhibition in multiple tumor cell lines. In vivo antitumor assay in U937 xenograft model identified compound 11r as a potent, orally active HDACI. To the best of our knowledge, this work constitutes the first report of oral active N-hydroxycinnamamide-based HDACIs with dual HDAC1/3 selectivity.
Co-reporter:Fuming Xu, Hao Xu, Xuejian Wang, Lei Zhang, Qingli Wen, Yingjie Zhang, Wenfang Xu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 4) pp:1487-1495
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmc.2013.11.052
A novel series of N-(3-((7H-purin-6-yl)thio)-4-hydroxynaphthalen-1-yl)-sulfonamides were designed and synthesized. Biological characterization revealed that several compounds exerted enhanced anti-proliferative activity against human umbilical vein endothelial cells (HUVECs) and several cancer cell lines and high specific protein kinase and angiogenesis inhibitory activities. Compared with our previously synthesized compounds, the substitution of sulfonamide structure for amide fragment played an essential role for the advance of inhibitory activities. In addition, the replacement of 1H-1,2,4-triazole ring by 7H-purine did not result in obvious decrease of inhibition efficacy, indicating that the sulfonamide structure contributes even more to the inhibition efficacy than the 1H-1,2,4-triazole ring. Among these compounds, compound 9n demonstrated comparable in vitro antiangiogenic activities to pazopanib in both HUVEC tube formation assay and the rat thoracic aorta rings (TARs) test. Meanwhile, compound 9n was identified to inhibit Akt1 (IC50 = 1.73 μM) and Abl tyrosine kinase (IC50 = 1.53 μM) effectively.Key compound 9n and its calculated binding mode in the ATP-binding pocket of Akt1 kinase.
Co-reporter:Fuming Xu, Lei Zhang, Yuping Jia, Xuejian Wang, Xiaoguang Li, Qingli Wen, Yingjie Zhang, Wenfang Xu
European Journal of Medicinal Chemistry 2013 Volume 69() pp:191-200
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.07.056
•A series of 4-amino-2-(thio)phenol derivatives were synthesized.•The compounds were evaluated as inhibitors of protein kinase and angiogenesis.•Some of them exhibited remarkably in vitro protein kinase inhibition.•Anti-angiogenic activity of 5i was similar to clinically used drug pazopanib.A novel series of 4-amino-2-(thio)phenol derivatives were well synthesized. The preliminary biological test revealed that several compounds displayed high specific protein kinase and angiogenesis inhibitory activities compared with previous work mainly because of the substitution of sulfonamide structure for amide fragment. Among which, compound 5i was identified to inhibit protein kinase B/AKT (IC50 = 1.26 μM) and ABL tyrosine kinase (IC50 = 1.50 μM) effectively. Meanwhile, compound 5i demonstrated competitive in vitro antiangiogenic activities to Pazopanib in both human umbilical vein endothelial cell (HUVEC) tube formation assay and the rat thoracic aorta rings test.Key compound 5i and its calculated binding mode in the ATP-binding pocket of AKT kinase.





![2'-Deoxy-3',5'-bis-O-[dimethyl(2-methyl-2-propanyl)silyl]-2',2'-d ifluorocytidine](http://img.cochemist.com/ccimg/688100/688009-09-8.png)
![2'-Deoxy-3',5'-bis-O-[dimethyl(2-methyl-2-propanyl)silyl]-2',2'-d ifluorocytidine](http://img.cochemist.com/ccimg/688100/688009-09-8_b.png)


![Benzamide, N-(2-aminophenyl)-4-[(trifluoroacetyl)amino]-](http://img.cochemist.com/ccimg/503100/503043-71-8.png)
![Benzamide, N-(2-aminophenyl)-4-[(trifluoroacetyl)amino]-](http://img.cochemist.com/ccimg/503100/503043-71-8_b.png)







![Benzonitrile, 2-[(2-chloro-4-pyrimidinyl)amino]-](http://img.cochemist.com/ccimg/260100/260045-46-3.png)
![Benzonitrile, 2-[(2-chloro-4-pyrimidinyl)amino]-](http://img.cochemist.com/ccimg/260100/260045-46-3_b.png)
![[2-[[4-(Aminomethyl)benzoyl]amino]phenyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/209800/209784-85-0.png)
![[2-[[4-(Aminomethyl)benzoyl]amino]phenyl]-carbamic Acid tert-Butyl Ester](http://img.cochemist.com/ccimg/209800/209784-85-0_b.png)













![4-Chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/941700/941685-26-3.png)
![4-Chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/941700/941685-26-3_b.png)












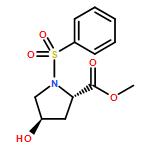
![L-Proline, 4-hydroxy-1-[(4-methoxyphenyl)sulfonyl]-, methyl ester, (4R)-](http://img.cochemist.com/ccimg/204100/204072-16-2.png)
![L-Proline, 4-hydroxy-1-[(4-methoxyphenyl)sulfonyl]-, methyl ester, (4R)-](http://img.cochemist.com/ccimg/204100/204072-16-2_b.png)






















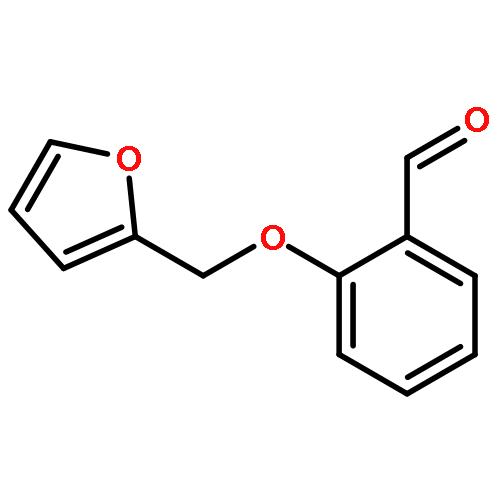
![[1,1':3',1''-Terphenyl]-4'-carboxaldehyde](http://img.cochemist.com/ccimg/136500/136416-26-7.png)
![[1,1':3',1''-Terphenyl]-4'-carboxaldehyde](http://img.cochemist.com/ccimg/136500/136416-26-7_b.png)












![GLYCINE, N-[N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-METHIONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/68200/68164-11-4.png)
![GLYCINE, N-[N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-METHIONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/68200/68164-11-4_b.png)






![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/51900/51803-69-1.png)
![Glycine, N-[(1,1-dimethylethoxy)carbonyl]-L-valyl-, methyl ester](http://img.cochemist.com/ccimg/51900/51803-69-1_b.png)










![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5.png)
![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5_b.png)











![3-ethyl-1-[(4-methoxyphenyl)acetyl]-5-pyridin-4-yl-4,5-dihydro-1H-pyrazol-5-ol](http://img.cochemist.com/ccimg/6100/6009-31-0.png)
![3-ethyl-1-[(4-methoxyphenyl)acetyl]-5-pyridin-4-yl-4,5-dihydro-1H-pyrazol-5-ol](http://img.cochemist.com/ccimg/6100/6009-31-0_b.png)











![[4-[(Hydroxyamino)carbonyl]phenyl]carbamic acid [6-[(diethylamino)methyl]-2-naphthalenyl]methyl ester](http://img.cochemist.com/ccimg/497900/497833-27-9.png)
![[4-[(Hydroxyamino)carbonyl]phenyl]carbamic acid [6-[(diethylamino)methyl]-2-naphthalenyl]methyl ester](http://img.cochemist.com/ccimg/497900/497833-27-9_b.png)
![Benzenemethanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3765100/452095-63-5.png)
![Benzenemethanol, 3-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]-](/data/chemimg/3765100/452095-63-5_b.png)
![Ethanol, 2-[2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]ethoxy]-](/data/chemimg/3765100/452095-54-4.png)
![Ethanol, 2-[2-[[5-oxido-4-(phenylsulfonyl)-1,2,5-oxadiazol-3-yl]oxy]ethoxy]-](/data/chemimg/3765100/452095-54-4_b.png)


![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)












![7-[4-(3-ETHYNYLANILINO)-7-METHOXYQUINAZOLIN-6-YL]OXY-N-HYDROXYHEPTANAMIDE](http://img.cochemist.com/ccimg/1012100/1012054-59-9.png)
![7-[4-(3-ETHYNYLANILINO)-7-METHOXYQUINAZOLIN-6-YL]OXY-N-HYDROXYHEPTANAMIDE](http://img.cochemist.com/ccimg/1012100/1012054-59-9_b.png)


![(E)-3-[2-butyl-1-[2-(diethylamino)ethyl]benzimidazol-5-yl]-N-hydroxyprop-2-enamide](http://img.cochemist.com/ccimg/929100/929016-96-6.png)
![(E)-3-[2-butyl-1-[2-(diethylamino)ethyl]benzimidazol-5-yl]-N-hydroxyprop-2-enamide](http://img.cochemist.com/ccimg/929100/929016-96-6_b.png)
![(E)-3-[1-[4-[(dimethylamino)methyl]phenyl]sulfonylpyrrol-3-yl]-N-hydroxyprop-2-enamide](http://img.cochemist.com/ccimg/864900/864814-88-0.png)
![(E)-3-[1-[4-[(dimethylamino)methyl]phenyl]sulfonylpyrrol-3-yl]-N-hydroxyprop-2-enamide](http://img.cochemist.com/ccimg/864900/864814-88-0_b.png)






![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0.png)
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0_b.png)