Co-reporter:Carmine D'Agostino, Sarayute Chansai, Isabelle Bush, Chensong Gao, Mick D. Mantle, Christopher Hardacre, Stuart L. James and Lynn F. Gladden
Catalysis Science & Technology 2016 vol. 6(Issue 6) pp:1661-1666
Publication Date(Web):30 Oct 2015
DOI:10.1039/C5CY01508A
The selective catalytic reduction (SCR) of NOx in the presence of different reducing agents over Ag/Al2O3 prepared by wet impregnation was investigated by probing catalyst activity and using NMR relaxation time analysis to probe the strength of surface interaction of the various reducing agent species and water. The results reveal that the strength of surface interaction of the reducing agent relative to water, the latter present in engine exhausts as a fuel combustion product and, in addition, produced during the SCR reaction, plays an important role in determining catalyst performance. Reducing agents with weak strength of interaction with the catalyst surface, such as hydrocarbons, show poorer catalytic performance than reducing agents with a higher strength of interaction, such as alcohols. This is attributed to the greater ability of oxygenated species to compete with water in terms of surface interaction with the catalyst surface, hence reducing the inhibiting effect of water molecules blocking catalyst sites. The results support the observations of earlier work in that the light off-temperature and maximum NOx conversion and temperature at which that occurs are sensitive to the reducing agent present during reaction, and the proposal that improved catalyst performance is caused by increased adsorption strength of the reducing agent, relative to water, at the catalyst surface. Importantly, the NMR relaxation time analysis approach to characterising the strength of adsorption more readily describes the trends in catalytic behaviour than does a straightforward consideration of the polarity (i.e., relative permittivity) of the reducing agents studied here. In summary, this paper describes a simple approach to characterising the interaction energy of water and reducing agent so as to aid the selection of reducing agent and catalyst to be used in SCR conversions.
Co-reporter:Marta Falkowska, Sarayute Chansai, Haresh G. Manyar, Lynn F. Gladden, Daniel T. Bowron, Tristan G. A. Youngs and Christopher Hardacre
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 26) pp:17237-17243
Publication Date(Web):29 Mar 2016
DOI:10.1039/C6CP01494A
Total neutron scattering has been used to follow the hydrogenation of toluene-d8 to methylcyclohexane-d14 over 3 wt% platinum supported on highly ordered mesoporous silica (MCM-41) at 298 K and under 150 mbar D2 pressure. The detailed kinetic information so revealed indicates that liquid reorganisation inside pores is the slowest step of the whole process. Additionally, the results were compared with the reaction performed under 250 mbar D2 pressure as well as with toluene-h8 hydrogenation using D2 at 150 mbar.
Co-reporter:Alex R. Neale, Peilin Li, Johan Jacquemin, Peter Goodrich, Sarah C. Ball, Richard G. Compton and Christopher Hardacre
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 16) pp:11251-11262
Publication Date(Web):17 Mar 2016
DOI:10.1039/C5CP07160G
This paper reports on the solubility and diffusivity of dissolved oxygen in a series of ionic liquids (ILs) based on the bis{(trifluoromethyl)sulfonyl}imide anion with a range of related alkyl and ether functionalised cyclic alkylammonium cations. Cyclic voltammetry has been used to observe the reduction of oxygen in ILs at a microdisk electrode and chronoamperometric measurements have then been applied to simultaneously determine both the concentration and the diffusion coefficient of oxygen in different ILs. The viscosity of the ILs and the calculated molar volume and free volume are also reported. It is found that, within this class of ILs, the oxygen diffusivity generally increases with decreasing viscosity of the neat IL. An inverse relationship between oxygen solubility and IL free volume is reported for the two IL families implying that oxygen is not simply occupying the available empty space. In addition, it is reported that the introduction of an ether-group into the IL cation structure promotes the diffusivity of dissolved oxygen but reduces the solubility of the gas.
Co-reporter:Bo Yang, Robbie Burch, Christopher Hardacre, P. Hu, Philip Hughes
Surface Science 2016 Volume 646() pp:45-49
Publication Date(Web):April 2016
DOI:10.1016/j.susc.2015.07.015
•Surface carbide is more readily formed over Pd(100) than Pd(111).•Pd(100) carbide shows better performance for acetylene hydrogenation.•Hydrogen atoms are less stable over Pd(100) carbide than over Pd(111) carbide.A recent experimental investigation (Kim et al. J. Catal. 306 (2013) 146–154) on the selective hydrogenation of acetylene over Pd nanoparticles with different shapes concluded that Pd(100) showed higher activity and selectivity than Pd(111) for acetylene hydrogenation. However, our recent density functional calculations (Yang et al. J. Catal. 305 (2013) 264–276) observed that the clean Pd(111) surface should result in higher activity and ethylene selectivity compared with the clean Pd(100) surface for acetylene hydrogenation. In the current work, using density functional theory calculations, we find that Pd(100) in the carbide form gives rise to higher activity and selectivity than Pd(111) carbide. These results indicate that the catalyst surface is most likely in the carbide form under the experimental reaction conditions. Furthermore, the adsorption energies of hydrogen atoms as a function of the hydrogen coverage at the surface and subsurface sites over Pd(100) are compared with those over Pd(111), and it is found that the adsorption of hydrogen atoms is always less favoured on Pd(100) over the whole coverage range. This suggests that the Pd(100) hydride surface will be less stable than the Pd(111) hydride surface, which is also in accordance with the experimental results reported.
Co-reporter:Corina McCrellis; S. F. Rebecca Taylor; Johan Jacquemin
Journal of Chemical & Engineering Data 2016 Volume 61(Issue 3) pp:1092-1100
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.jced.5b00710
The miscibility of monoethanolamine (MEA) in five superbase ionic liquids (ILs), namely the trihexyl-tetradecylphosphonium benzotriazolide ([P66614][Bentriz]), trihexyl-tetradecylphosphonium benzimidazolide ([P66614][Benzim]), trihexyl-tetradecylphosphonium 1,2,3-triazolide ([P66614][123Triz]), trihexyl-tetradecylphosphonium 1,2,4-triazolide ([P66614][124Triz]), and trihexyl-tetradecylphosphonium imidazolide ([P66614][Im]) was determined at 295.15 K using 1H NMR spectroscopy. The solubility of carbon dioxide (CO2) in equimolar (IL + MEA) mixtures was then studied experimentally using a gravimetric technique at 295.15 K and 0.1 MPa. The effect of MEA on the CO2 capture ability of these ILs was investigated together with the viscosity of these systems in the presence or absence of CO2 to evaluate their practical application in CO2 capture processes. The effect of the presence of MEA on the rate of CO2 uptake was also studied. The study showed that the MEA can enhance CO2 absorption over the ideal values in the case of [P66614][123Triz] and [P66614][Bentriz] while in the other systems the mixtures behave ideally. A comparison of the effect of MEA addition with the addition of water to these superbase ILs showed that similar trends were observed in each case for the individual ILs studied.
Co-reporter:Marta Falkowska;Dr. Daniel T. Bowron;Dr. Haresh G. Manyar; Christopher Hardacre;Dr. Tristan G. A. Youngs
ChemPhysChem 2016 Volume 17( Issue 13) pp:2043-2055
Publication Date(Web):
DOI:10.1002/cphc.201600149
Abstract
Organic solvents, such as cyclohexane, cyclohexene, methylcyclohexane, benzene and toluene, are widely used as both reagents and solvents in industrial processes. Despite the ubiquity of these liquids, the local structures that govern the chemical properties have not been studied extensively. Herein, we report neutron diffraction measurements on liquid cyclohexane, cyclohexene, methylcyclohexane, benzene and toluene at 298 K to obtain a detailed description of the local structure in these compounds. The radial distribution functions of the centres of the molecules, as well as the partial distribution functions for the double bond for cyclohexene and methyl group for methylcyclohexane and toluene have been calculated. Additionally, probability density functions and angular radial distribution functions were extracted to provide a full description of the local structure within the chosen liquids. Structural motifs are discussed and compared for all liquids, referring specifically to the functional group and aromaticity present in the different liquids.
Co-reporter:C. E. Stere, W. Adress, R. Burch, S. Chansai, A. Goguet, W. G. Graham, and C. Hardacre
ACS Catalysis 2015 Volume 5(Issue 2) pp:956
Publication Date(Web):December 23, 2014
DOI:10.1021/cs5019265
The current paper reports on a newly developed DRIFTS-MS system for the investigation of non-thermal plasma (NTP) assisted heterogeneously catalyzed reactions. Specifically, this methodology has been utilized to investigate the surface changes during the NTP-activated hydrocarbon selective catalytic reduction (HC-SCR) deNOx reaction over a silver-based catalyst at ambient temperature using simulated diesel fuels (toluene and n-octane). The experimental setup and the methods used to investigate the plasma activation operating with helium as the carrier gas in order to examine low-temperature reactions are described. The technique has identified the importance, even at low temperatures, of isocyanate species in the HC-SCR deNOx reaction as well as the critical role of water in the formation of N2.Keywords: Ag/Al2O3; DRIFTS-MS; low-temperature hydrocarbon-selective catalytic reduction; non-thermal plasma; NOx reduction; octane; toluene
Co-reporter:Kevin Morgan, Alexandre Goguet, and Christopher Hardacre
ACS Catalysis 2015 Volume 5(Issue 6) pp:3430
Publication Date(Web):April 23, 2015
DOI:10.1021/acscatal.5b00535
Catalyst deactivation is ultimately inevitable, and one of the processes known to cause deactivation is sintering of metal particles. Consequently, numerous methods to reverse the sintering process by redispersing metal nanoparticles have been developed. These methods are discussed in this perspective, and the reported mechanisms of redispersion are summarized. Additionally, the longer-term practical use of such treatments and the benefits this can bring are briefly disclosed.Keywords: catalyst deactivation; catalyst recycling; catalyst regeneration; metal redispersion; supported metal catalysts
Co-reporter:V. K. Puthiyapura, D. J. L. Brett, A. E. Russell, W. F. Lin and C. Hardacre
Chemical Communications 2015 vol. 51(Issue 69) pp:13412-13415
Publication Date(Web):20 Jul 2015
DOI:10.1039/C5CC04188K
Pt and PtSn catalysts were studied for n-butanol electro-oxidation at various temperatures. PtSn showed a higher activity towards butanol electro-oxidation compared to Pt in acidic media. The onset potential for n-butanol oxidation on PtSn is ∼520 mV lower than that found on Pt, and significantly lower activation energy was found for PtSn compared with that for Pt.
Co-reporter:Octavian Dumitru Pavel, Peter Goodrich, Liliana Cristian, Simona M. Coman, Vasile I. Pârvulescu and Christopher Hardacre
Catalysis Science & Technology 2015 vol. 5(Issue 5) pp:2696-2704
Publication Date(Web):23 Feb 2015
DOI:10.1039/C5CY00011D
The immobilization of a ruthenium complex (Ru2Cl4(az-tpy)2) within a range of supported ionic liquids ([C4C1im]Cl, [C4C1im][NTf2], [C6C1im]Cl, [C4C1pyrr]Br, [C4C1im]Br, [C4C1pyrr]Cl) dispersed silica (SILP) operates as an efficient heterogeneous catalyst in oxidation of long chain linear primary amines to corresponding nitriles. This reaction follows a “green” route using a cheap and easy to handles oxidant (oxygen or air). The conversion was found to be strongly influenced by the alkyl chain length of the amine substrate and the choice of oxidant. No condensation reaction was observed between the starting amines and the selectivity to nitrile is 100%. Moving from a composition of 20 atm N2/5 atm O2 to 5 atm N2/20 atm O2 led to enhancements in the conversion (n-alkylamines) and selectivity (benzonitrile) which have been correlated with an increase of the solubilized oxygen. This was further supported by using different inert gas (nitrogen, helium, argon)/oxygen mixtures indicating that the O2 solubility in the SILP system, has an important effect on conversions and TON in this reaction using SILP catalysts. Experiments performed in the presence of CO2 led to a different behaviour due to the formation of amine-CO2 adducts. The application of the Weisz–Prater criterion confirmed the absence of any diffusional constraints.
Co-reporter:K. Anderson, M.P. Atkins, P. Goodrich, C. Hardacre, A.S. Hussain, R. Pilus, D.W. Rooney
Fuel 2015 Volume 146() pp:60-68
Publication Date(Web):15 April 2015
DOI:10.1016/j.fuel.2015.01.015
•Carbonate-based ionic liquids can be used to remove naphthenic acids from crude oil.•The reaction mechanism involves the formation of an ionic liquid-naphthenic acid complex.•The complex can be reacted with carbonic acid to reform the ionic liquid.•Recycling of the ionic liquid has been demonstrated.A number of tetraalkylammonium methylcarbonate and hydrogencarbonate based ionic liquids are shown to be capable of reacting with the naphthenic acids contained in Doba crude oil via a neutralisation reaction. Spectral studies show that the ionic liquids neutralisation mechanism involves the formation of an ionic liquid-naphthenate complex, liberating methanol and carbon dioxide. Extraction of the neutralised complex into a separate methanol phase and subsequent regeneration using aqueous carbonic acid results in ∼70% of the ionic liquid being recovered for recycle. Isolation of the naphthenic acids shows that these make up to 0.85 wt% of the crude oil. Speciation of the naphthenic acids shows a mixture of monocyclic, through to tetracyclic structures with carbon numbers in the range C12–C40.
Co-reporter:S. F. Rebecca Taylor;Corina McCrellis;Claire McStay
Journal of Solution Chemistry 2015 Volume 44( Issue 3-4) pp:511-527
Publication Date(Web):2015 April
DOI:10.1007/s10953-015-0319-z
The solubility of carbon dioxide in five tetraalkylphosphonium superbase ionic liquids, namely the trihexyltetradecylphoshonium phenoxide, trihexyltetradecylphoshonium benzotriazolide, trihexyltetradecylphoshonium benzimidazolide, trihexyltetradecylphoshonium 1,2,3-triazolide, and trihexyltetradecylphoshonium 1,2,4-triazolide was studied experimentally under dry and wet conditions at 22 °C and at atmospheric pressure, using a gravimetric saturation technique. The effects of anion structure and of the presence or absence of water in the solution on the carbon dioxide solubility were then deduced from the data. 1H and 13C-NMR spectroscopy and ab initio calculations were also conducted to probe the interactions in these solutions, as carbon dioxide and water can compete in the ionic liquid structure during the absorption process. Additionally, the viscosity of selected superbase ionic liquids was measured under dry and wet conditions, in the presence or absence of CO2, to evaluate their practical application in carbon dioxide capture processes. Finally, the recyclability of the trihexyltetradecylphoshonium 1,2,4-triazolide under dry and wet conditions was determined to probe the ability of selected solvents to solubilize chemically a high concentration of carbon dioxide and then release it in a low energy demand process.
Co-reporter:Dr. Nathan Hollingsworth;Dr. S. F. Rebecca Taylor;Miguel T. Galante;Dr. Johan Jacquemin;Dr. Claudia Longo;Dr. Katherine B. Holt; Nora H. deLeeuw; Christopher Hardacre
Angewandte Chemie 2015 Volume 127( Issue 47) pp:14370-14374
Publication Date(Web):
DOI:10.1002/ange.201507629
Abstract
A new low-energy pathway is reported for the electrochemical reduction of CO2 to formate and syngas at low overpotentials, utilizing a reactive ionic liquid as the solvent. The superbasic tetraalkyl phosphonium ionic liquid [P66614][124Triz] is able to chemisorb CO2 through equimolar binding of CO2 with the 1,2,4-triazole anion. This chemisorbed CO2 can be reduced at silver electrodes at overpotentials as low as 0.17 V, forming formate. In contrast, physically absorbed CO2 within the same ionic liquid or in ionic liquids where chemisorption is impossible (such as [P66614][NTf2]) undergoes reduction at significantly increased overpotentials, producing only CO as the product.
Co-reporter:Dr. Nathan Hollingsworth;Dr. S. F. Rebecca Taylor;Miguel T. Galante;Dr. Johan Jacquemin;Dr. Claudia Longo;Dr. Katherine B. Holt; Nora H. deLeeuw; Christopher Hardacre
Angewandte Chemie International Edition 2015 Volume 54( Issue 47) pp:14164-14168
Publication Date(Web):
DOI:10.1002/anie.201507629
Abstract
A new low-energy pathway is reported for the electrochemical reduction of CO2 to formate and syngas at low overpotentials, utilizing a reactive ionic liquid as the solvent. The superbasic tetraalkyl phosphonium ionic liquid [P66614][124Triz] is able to chemisorb CO2 through equimolar binding of CO2 with the 1,2,4-triazole anion. This chemisorbed CO2 can be reduced at silver electrodes at overpotentials as low as 0.17 V, forming formate. In contrast, physically absorbed CO2 within the same ionic liquid or in ionic liquids where chemisorption is impossible (such as [P66614][NTf2]) undergoes reduction at significantly increased overpotentials, producing only CO as the product.
Co-reporter:Bo Yang, Robbie Burch, Christopher Hardacre, Gareth Headdock, and P. Hu
ACS Catalysis 2014 Volume 4(Issue 1) pp:182
Publication Date(Web):December 9, 2013
DOI:10.1021/cs400727f
The fundamental understanding of the activity in heterogeneous catalysis has long been the major subject in chemistry. This paper shows the development of a two-step model to understand this activity. Using the theory of chemical potential kinetics with Brønsted–Evans–Polanyi relations, the general adsorption energy window is determined from volcano curves, using which the best catalysts can be searched. Significant insights into the reasons for catalytic activity are obtained.Keywords: adsorption energy window; Brønsted−Evans−Polanyi relation; catalyst design; chemical potential; heterogeneous catalysis; two-step model; volcano curve
Co-reporter:Cristina E. Stere, Wameedh Adress, Robbie Burch, Sarayute Chansai, Alexandre Goguet, William G. Graham, Fabio De Rosa, Vincenzo Palma, and Christopher Hardacre
ACS Catalysis 2014 Volume 4(Issue 2) pp:666
Publication Date(Web):January 10, 2014
DOI:10.1021/cs4009286
Atmospheric pressure nonthermal-plasma-activated catalysis for the removal of NOx using hydrocarbon selective catalytic reduction has been studied utilizing toluene and n-octane as the hydrocarbon reductant. When the plasma was combined with a Ag/Al2O3 catalyst, a strong enhancement in activity was observed when compared with conventional thermal activation with high conversions of both NOx and hydrocarbons obtained at temperature ≤250 °C, where the silver catalyst is normally inactive. Importantly, even in the absence of an external heat source, significant activity was obtained. This low temperature activity provides the basis for applying nonthermal plasmas to activate emission control catalysts during cold start conditions, which remains an important issue for mobile and stationary applications.Keywords: Ag/Al2O3; low temperature hydrocarbon selective catalytic reduction; n-octane; nonthermal plasma; NOx reduction; toluene
Co-reporter:H. Daly, H. G. Manyar, R. Morgan, J. M. Thompson, J.-J. Delgado, R. Burch, and C. Hardacre
ACS Catalysis 2014 Volume 4(Issue 8) pp:2470
Publication Date(Web):June 16, 2014
DOI:10.1021/cs500185n
A new experimental procedure based on attenuated total reflection infrared spectroscopy has been developed to investigate surface species under liquid phase reaction conditions. The technique has been tested by investigating the enhanced selectivity in the hydrogenation of α,β-unsaturated aldehyde citral over a 5% Pt/SiO2 catalyst toward unsaturated alcohols geraniol/nerol, which occurs when citronellal is added to the reaction. The change in selectivity is proposed to be the result of a change in the citral adsorption mode in the presence of citronellal. Short time on stream attenuated total internal reflection infrared spectroscopy has allowed identification of the adsorption modes of citral. With no citronellal, citral adsorbs through both the C═C and C═O groups; however, in the presence of citronellal, citral adsorption occurs through the C═O group only, which is proposed to be the cause of the altered reaction selectivity.Keywords: ATR; citral; hydrogenation; infrared; Pt
Co-reporter:Simon Doherty, Julian G. Knight, Jack R. Ellison, Peter Goodrich, Leanne Hall, Christopher Hardacre, Mark J. Muldoon, Soomin Park, Ana Ribeiro, Carlos Alberto Nieto de Castro, Maria José Lourenço and Paul Davey
Green Chemistry 2014 vol. 16(Issue 3) pp:1470-1479
Publication Date(Web):09 Dec 2013
DOI:10.1039/C3GC41378K
The asymmetric Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene and the Mukaiyama-aldol reaction between methylpyruvate and 1-phenyl-1-trimethylsilyloxyethene have been catalysed by heterogeneous copper(II)-bis(oxazoline)-based polymer immobilised ionic liquid phase (PIILP) systems generated from a range of linear and cross linked ionic polymers. In both reactions selectivity and ee were strongly influenced by the choice of polymer. A comparison of the performance of a range of Cu(II)-bis(oxazoline)-PIILP catalyst systems against analogous supported ionic liquid phase (SILP) heterogeneous catalysts as well as their homogeneous counterparts has been undertaken and their relative merits evaluated.
Co-reporter:Kevin Morgan, Robbie Burch, Muhammad Daous, Juan José Delgado, Alexandre Goguet, Christopher Hardacre, Lachezar A. Petrov and David W. Rooney
Catalysis Science & Technology 2014 vol. 4(Issue 3) pp:729-737
Publication Date(Web):02 Jan 2014
DOI:10.1039/C3CY00915G
Methods to control the dispersion of gold in supported heterogeneous catalysts are very valuable due to the strong nanoparticle size dependence on their activity and selectivity towards many reactions. Additionally, the ability to disperse large, inactive gold nanoparticles to smaller nanoparticles provides an opportunity to reactivate, stabilise and increase the lifetime of gold catalysts making them more practical for industrial applications. Previously it has been demonstrated that the use of gas phase iodomethane (J. Am. Chem. Soc., 2009, 131, 6973; Angew. Chem., Int. Ed., 2011, 50, 8912) was able to re-disperse gold from >20 nm particles to dimers and trimers. In the current work, we show that this technique can be applied using less hazardous halohydrocarbons treatments, both in the gas phase and the liquid phase. The ability of these individual halohydrocarbons to re-disperse gold as well as the extent to which leaching occurs is assessed.
Co-reporter:Kathryn Ralphs, Carmine D'Agostino, Robbie Burch, Sarayute Chansai, Lynn F. Gladden, Christopher Hardacre, Stuart L. James, Jonathan Mitchell and Sarah F. R. Taylor
Catalysis Science & Technology 2014 vol. 4(Issue 2) pp:531-539
Publication Date(Web):03 Dec 2013
DOI:10.1039/C3CY00945A
The surface modification of a mechanochemically prepared Ag/Al2O3 catalyst compared with catalysts prepared by standard wet impregnated methods has been probed using two-dimensional T1–T2 NMR correlations, H2O temperature programmed desorption (TPD) and DRIFTS. The catalysts were examined for the selective catalytic reduction of NOx using n-octane in the presence and absence of H2. Higher activities were observed for the ball milled catalysts irrespective of whether H2 was added. This higher activity is thought to be related to the increased affinity of the catalyst surface towards the hydrocarbon relative to water, following mechanochemical preparation, resulting in higher concentrations of the hydrocarbon and lower concentrations of water at the surface. DRIFTS experiments demonstrated that surface isocyanate was formed significantly quicker and had a higher surface concentration in the case of the ball milled catalyst which has been correlated with the stronger interaction of the n-octane with the surface. This increased interaction may also be the cause of the reduced activation barrier measured for this catalyst compared with the wet impregnated system. The decreased interaction of water with the surface on ball milling is thought to reduce the effect of site blocking whilst still providing a sufficiently high surface concentration of water to enable effective hydrolysis of the isocyanate to form ammonia and, thereafter, N2.
Co-reporter:Jia-Mei Jin, Tian Sheng, Xiao Lin, Richard Kavanagh, Philip Hamer, Peijun Hu, Christopher Hardacre, Alex Martinez-Bonastre, Jonathan Sharman, David Thompsett and Wen-Feng Lin
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 20) pp:9432-9440
Publication Date(Web):01 Apr 2014
DOI:10.1039/C4CP00859F
The most active binary PtSn catalyst for direct ethanol fuel cell applications has been studied at 20 °C and 60 °C, using variable temperature electrochemical in situ FTIR. In comparison with Pt, binary PtSn inhibits ethanol dissociation to CO(a), but promotes partial oxidation to acetaldehyde and acetic acid. Increasing the temperature from 20 °C to 60 °C facilitates both ethanol dissociation to CO(a) and then further oxidation to CO2, leading to an increased selectivity towards CO2; however, acetaldehyde and acetic acid are still the main products. Potential-dependent phase diagrams for surface oxidants of OH(a) formation on Pt(111), Pt(211) and Sn modified Pt(111) and Pt(211) surfaces have been determined using density functional theory (DFT) calculations. It is shown that Sn promotes the formation of OH(a) with a lower onset potential on the Pt(111) surface, whereas an increase in the onset potential is found upon modification of the (211) surface. In addition, Sn inhibits the Pt(211) step edge with respect to ethanol C–C bond breaking compared with that found on the pure Pt, which reduces the formation of CO(a). Sn was also found to facilitate ethanol dehydrogenation and partial oxidation to acetaldehyde and acetic acid which, combined with the more facile OH(a) formation on the Pt(111) surface, gives us a clear understanding of the experimentally determined results. This combined electrochemical in situ FTIR and DFT study provides, for the first time, an insight into the long-term puzzling features of the high activity but low CO2 production found on binary PtSn ethanol fuel cell catalysts.
Co-reporter:Weiqing Zhang, Shuguang Jiang, Christopher Hardacre, Peter Goodrich, Kai Wang, Zhengyan Wu, and Hao Shao
Energy & Fuels 2014 Volume 28(Issue 7) pp:4333-4341
Publication Date(Web):June 19, 2014
DOI:10.1021/ef402229z
To develop a chemical inhibitor that can efficiently suppress coal oxidation, nine tetraalkylphosphonium-based ionic liquids (ILs) and one imidazolium-based IL [1-allyl-3-methylimidazolium chloride ([AMIm]Cl)] were examined as additives. These ILs were used to treat and investigate the inhibitory effect on the oxidation activity and the structure of lignite coal. Characterization using thermogravimetric analysis showed that phosphonium-based ILs are able to inhibit coal oxidation up to 400 °C with the tributylethylphosphonium diethylphosphate ([P4,4,4,2][DEP]) found to be the most effective. In contrast to the tetraalkylphosphonium-based ILs, inhibition using [AMIm]Cl was only found to be effective at temperatures below 250 °C, indicating that the tetraalkylphosphonium-based ILs may be more suitable for the future application of suppressing coal spontaneous combustion over a wide range of temperatures. Fourier transform infrared spectroscopic data showed that the various functional groups change in the coal following IL treatment, which are a decrease in the minerals and hydrogen bonds in all treated coals, while decreased aliphatic hydrocarbon and increased carbonyl bonds only appeared in some samples. During the oxidation of coal, the decomposition of aliphatic hydrocarbon groups is inhibited and the formation of carbonyl groups is delayed, so that the evolved gas concentration decreased, as shown by the temperature-programmed oxidation–mass spectrometry results. The deployment of the [P4,4,4,2][DEP] and tributylmethylphosphonium methylsulfate ILs as additives also show good inhibitory effect on coal oxidation over the temperature range studied, and a relatively stronger interaction between [P4,4,4,2][DEP] and coal is demonstrated by the additive model.
Co-reporter:Bo Yang ; Robbie Burch ; Christopher Hardacre ; P. Hu ;Philip Hughes
The Journal of Physical Chemistry C 2014 Volume 118(Issue 7) pp:3664-3671
Publication Date(Web):January 21, 2014
DOI:10.1021/jp412255a
Boron-modified Pd catalysts have shown excellent performance for the selective hydrogenation of alkynes experimentally. In the current work, we investigated the hydrogenation of acetylene on boron-modified Pd(111) and Pd(211) surfaces, utilizing density functional theory calculations. The activity of acetylene hydrogenation has been studied by estimating the effective barrier of the whole process. The selectivity of ethylene formation is investigated from a comparison between the desorption and the hydrogenation of ethylene as well as comparison between the ethylene and the 1,3-butadiene formation. Formation of subsurface carbon and hydrogen on both boron-modified Pd(111) and Pd(211) surfaces has also been evaluated, since these have been reported to affect both the activity and the selectivity of acetylene hydrogenation to produce ethylene on Pd surfaces. Our results provide some important insights into the Pd–B catalysts for selective hydrogenation of acetylene and also for more complex hydrogenation systems, such as stereoselective hydrogenation of longer chain alkynes and selective hydrogenation of vegetable oil.
Co-reporter:Bo Yang, Robbie Burch, Christopher Hardacre, P. Hu, and Philip Hughes
The Journal of Physical Chemistry C 2014 Volume 118(Issue 3) pp:1560-1567
Publication Date(Web):December 31, 2013
DOI:10.1021/jp408807c
Green oil, which leads to the deactivation of the catalysts used for the selective hydrogenation of acetylene, has long been observed but its formation mechanism is not fully understood. In this work, the formation of 1,3-butadiene, known to be the precursor of green oil, on both Pd(111) and Pd(211) surfaces is examined using density functional theory calculations. The pathways containing C2 + C2 coupling reactions as well as the corresponding hydrogenation reactions are studied in detail. Three pathways for 1,3-butadiene production, namely coupling plus hydrogenation and further hydrogenation, hydrogenation plus coupling plus hydrogenation, and a two step hydrogenation followed by coupling, are determined. By comparing the effective barriers, we identify the favored pathway on both surfaces. A general understanding toward the deactivation process of the industrial catalysts is also provided. In addition, the effects of the formation of subsurface carbon atoms as well as the Ag alloying on the 1,3-butadiene formation on Pd-based catalysts are also investigated and compared with experimental results.
Co-reporter:Kathryn Ralphs, Christopher Hardacre and Stuart L. James
Chemical Society Reviews 2013 vol. 42(Issue 18) pp:7701-7718
Publication Date(Web):11 Jun 2013
DOI:10.1039/C3CS60066A
Mechanochemical synthesis has the potential to provide more sustainable preparative routes to catalysts than the current multistep solvent-based routes. In this review, the mechanochemical synthesis of catalysts is discussed, with emphasis placed on catalysts for environmental, energy and chemical synthesis applications. This includes the formation of mixed-metal oxides as well as the process of dispersing metals onto solid supports. In most cases the process involves no solvent. Encouragingly, there are several examples where the process is advantageous compared with the more normal solvent-based methods. This can be because of process cost or simplicity, or, notably, where it provides more active/selective catalysts than those made by conventional wet chemical methods. The need for greater, and more systematic, exploration of this currently unconventional approach to catalyst synthesis is highlighted.
Co-reporter:Sofiane Saouane, Sarah E. Norman, Christopher Hardacre and Francesca P. A. Fabbiani
Chemical Science 2013 vol. 4(Issue 3) pp:1270-1280
Publication Date(Web):02 Jan 2013
DOI:10.1039/C2SC21959J
The solid-state polymorphism of the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate, [bmim][PF6], has been investigated via low-temperature and high-pressure crystallisation experiments. The samples have been characterised by single-crystal X-ray diffraction, optical microscopy and Raman spectroscopy. The solid-state phase behaviour of the compound is confirmed and clarified with respect to previous phase diagrams. The structures of the previously reported γ-form, which essentially exhibits a G′T cation conformation, as well as those of the elusive β- and α-forms, are reported. Crystals of the β-phase are twinned and the structure is heavily disordered; the cation conformation in this form is predominantly TT, though significant contributions from other less frequently encountered conformers are also observed at low temperature and high pressure. The cation conformation in the α-form is GT; the presence of the G′T conformer at 193 K in this phase can be eliminated on cooling to 100 K. Whilst X-ray structural data are overall in good agreement with previous interpretations based on Raman and NMR studies, they also reveal a more subtle interplay of intermolecular interactions, which give rise to a wider range of conformers than previously considered.
Co-reporter:Tristan G. A. Youngs, Haresh Manyar, Daniel T. Bowron, Lynn F. Gladden and Christopher Hardacre
Chemical Science 2013 vol. 4(Issue 9) pp:3484-3489
Publication Date(Web):17 Jun 2013
DOI:10.1039/C3SC51477C
Using benzene hydrogenation over Pt/SiO2 as an industrially-relevant example, we show that state-of-the-art neutron total scattering methods spanning a wide Q-range now permit relevant time-resolved catalytic chemistry to be probed directly in situ within the pore of the catalyst. The method gives access to the reaction rates on both nanometric and atomic length scales, whilst simultaneously providing an atomistic structural viewpoint on the reaction mechanism itself.
Co-reporter:Haresh G. Manyar, Richard Morgan, Kevin Morgan, Bo Yang, P. Hu, Jakub Szlachetko, Jacinto Sá and Christopher Hardacre
Catalysis Science & Technology 2013 vol. 3(Issue 6) pp:1497-1500
Publication Date(Web):09 Apr 2013
DOI:10.1039/C3CY00031A
The change in the Pt electronic structure following the adsorption of an α,β-unsaturated aldehyde and ketone was followed by in situ HERFD-XANES in the liquid phase. The resulting shift in the Pt Fermi energy is in good agreement with the molecule adsorption energy trends calculated by DFT and provides insight into the reaction selectivity.
Co-reporter:Jamal Touitou, Robbie Burch, Christopher Hardacre, Colin McManus, Kevin Morgan, Jacinto Sá and Alexandre Goguet
Analyst 2013 vol. 138(Issue 10) pp:2858-2862
Publication Date(Web):15 Mar 2013
DOI:10.1039/C3AN00250K
This paper reports the detailed description and validation of a fully automated, computer controlled analytical method to spatially probe the gas composition and thermal characteristics in packed bed systems. As an exemplar, we have examined a heterogeneously catalysed gas phase reaction within the bed of a powdered oxide supported metal catalyst. The design of the gas sampling and the temperature recording systems are disclosed. A stationary capillary with holes drilled in its wall and a moveable reactor coupled with a mass spectrometer are used to enable sampling and analysis. This method has been designed to limit the invasiveness of the probe on the reactor by using the smallest combination of thermocouple and capillary which can be employed practically. An 80 μm (O.D.) thermocouple has been inserted in a 250 μm (O.D.) capillary. The thermocouple is aligned with the sampling holes to enable both the gas composition and temperature profiles to be simultaneously measured at equivalent spatially resolved positions. This analysis technique has been validated by studying CO oxidation over a 1% Pt/Al2O3 catalyst and the spatial resolution profiles of chemical species concentrations and temperature as a function of the axial position within the catalyst bed are reported.
Co-reporter:Mark D. Garrett, Stephen C. Bennett, Christopher Hardacre, Robin Patrick and Gary N. Sheldrake
RSC Advances 2013 vol. 3(Issue 44) pp:21552-21557
Publication Date(Web):06 Sep 2013
DOI:10.1039/C3RA44382E
Hydrogenolysis of bark from three different species of tree using heterogeneous platinum group metal catalysts produces two major product streams. Aromatic substituted guaiacols are produced from lignin and the lignin-like regions of suberin and a range of saturated fatty acids and alcohols, including α,ω-functionalised species, are produced from the polyester regions of suberin. Control experiments demonstrate clear advantages of catalytic hydrogenolysis over base hydrolysis, both in terms of conversion and product selectivity.
Co-reporter:A. Abdelkader, H. Daly, Y. Saih, K. Morgan, M.A. Mohamed, S.A. Halawy, C. Hardacre
International Journal of Hydrogen Energy 2013 Volume 38(Issue 20) pp:8263-8275
Publication Date(Web):9 July 2013
DOI:10.1016/j.ijhydene.2013.04.009
•Co–Fe oxide catalyst produced with intimate contact between the oxide phases.•DRIFTS-MS showed that adsorption properties of the individual oxides are maintained in the mixed oxide catalyst.•Increased OH at the surface in the Co–Fe catalyst.•Promotes reforming rather than decomposition reactions enhancing H2 yield and reducing by-product formation (CH4 and coke).Co3O4, Fe2O3 and a mixture of the two oxides Co–Fe (molar ratio of Co3O4/Fe2O3 = 0.67 and atomic ratio of Co/Fe = 1) were prepared by the calcination of cobalt oxalate and/or iron oxalate salts at 500 °C for 2 h in static air using water as a solvent/dispersing agent. The catalysts were studied in the steam reforming of ethanol to investigate the effect of the partial substitution of Co3O4 with Fe2O3 on the catalytic behaviour. The reforming activity over Fe2O3, while initially high, underwent fast deactivation. In comparison, over the Co–Fe catalyst both the H2 yield and stability were higher than that found over the pure Co3O4 or Fe2O3 catalysts. DRIFTS-MS studies under the reaction feed highlighted that the Co–Fe catalyst had increased amounts of adsorbed OH/water; similar to Fe2O3. Increasing the amount of reactive species (water/OH species) adsorbed on the Co–Fe catalyst surface is proposed to facilitate the steam reforming reaction rather than decomposition reactions reducing by-product formation and providing a higher H2 yield.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dr. Haresh G. Manyar;Bo Yang;Dr. Helen Daly;Dr. Helen Moor;Stephen McMonagle;Yu Tao; Ganapati D. Yadav;Dr. Alexre Goguet; P. Hu; Christopher Hardacre
ChemCatChem 2013 Volume 5( Issue 2) pp:506-512
Publication Date(Web):
DOI:10.1002/cctc.201200447
Abstract
The selective hydrogenation of α,β-unsaturated aldehydes and ketones has been studied using ketoisophorone and cinnamaldehyde as model substrates using manganese oxide octahedral molecular sieve (OMS-2) based catalysts. For the first time, OMS-2 has been shown to be an efficient and selective hydrogenation catalyst. High selectivities for either the CC or CO double bond at ≈100 % conversion were achieved by using OMS-2 and platinum supported on OMS-2 catalysts. Density functional theory (DFT) calculations showed that the dissociation of H2 on OMS-2 was water assisted and occurred on the surface Mn of OMS-2(0 0 1) that had been modified by an adsorbed H2O molecule. The theoretically calculated activation barrier was in good agreement with the experimentally determined value for the hydrogenation reactions, indicating that H2 dissociation on OMS-2 is likely to be the rate-determining step. A significant increase in the rate of reaction was observed in the presence of Pt as a result of the enhancement of H2 dissociative adsorption and subsequent reaction on the Pt or spillover of the hydrogen to the OMS-2 support. The relative adsorption strengths of ketoisophorone and cinnamaldehyde on the OMS-2 support compared with the Pt were found to determine the product selectivity.
Co-reporter:Jacinto Sá, Sarah Frances Rebecca Taylor, Helen Daly, Alexandre Goguet, Ramchandra Tiruvalam, Qian He, Christopher J. Kiely, Graham J. Hutchings, and Christopher Hardacre
ACS Catalysis 2012 Volume 2(Issue 4) pp:552
Publication Date(Web):March 2, 2012
DOI:10.1021/cs300074g
Although many gold heterogeneous catalysts have been shown to exhibit significant activity and high selectivity for a wide range of reactions in both the liquid and gas phases, they are prone to irreversible deactivation. This is often associated with sintering or loss of the interaction of the gold with the support. Herein, we report on the use of methyl iodide as a method of dispersing gold nanoparticles supported on silica, titania, and alumina supports. In the case of titania- and alumina-based catalysts, the gold was transformed from nanometer particles into small clusters and some atomically dispersed gold. In contrast, although there was a drop in the gold particle size on the silica support following CH3I treatment, the size remained in the submicrometer range. The structural changes were correlated with changes in the selectivity and activity for ethanol dehydration and benzyl alcohol oxidation. From these observations, it is clear that this treatment provides a method by which deactivated gold catalysts can be reactivated via redispersion of the gold.Keywords: dehydration; gold; methyl iodide; oxidation; oxides; redispersion;
Co-reporter:Bo Yang, Robbie Burch, Christopher Hardacre, Gareth Headdock, and P. Hu
ACS Catalysis 2012 Volume 2(Issue 6) pp:1027
Publication Date(Web):April 18, 2012
DOI:10.1021/cs2006789
Activity and selectivity are both important issues in heterogeneous catalysis and recent experimental results have shown that Ni catalysts doped by gold exhibit high activity for the hydrogenation of acetylene with good selectivity of ethylene formation. To unravel the underlying mechanism for this observation, the general trend of activity and selectivity of Ni surfaces doped by Au, Ag, and Cu has been investigated using density functional theory calculations. Complete energy profiles from C2H2 to C2H4 on Ni(111), Au/Ni(111), Ag/Ni(111) and Cu/Ni(111) are obtained and their turnover frequencies (TOFs) are computed. The results show that acetylene adsorption on Ni catalyst is strong which leads to the low activity while the doping of Au, Ag, and Cu on the Ni catalyst weakens the acetylene adsorption, giving rise to the increase of activity. The selectivity of ethylene formation is also quantified by using the energy difference between the hydrogenation barriers and the absolute value of the adsorption energies of ethylene. It is found that the selectivity of ethylene formation increases by doping Au and Ag, while those of Cu/Ni and Ni are similar.Keywords: acetylene; activity; Ag; Au; Cu; DFT; ethylene; Ni; selective hydrogenation; selectivity;
Co-reporter:Simon Doherty, Julian G. Knight, Jack R. Ellison, David Weekes, Ross W. Harrington, Christopher Hardacre and Haresh Manyar
Green Chemistry 2012 vol. 14(Issue 4) pp:925-929
Publication Date(Web):06 Mar 2012
DOI:10.1039/C2GC16679H
A linear cation-decorated polymeric support with tuneable surface properties and microstructure has been prepared by ring-opening metathesis polymerisation (ROMP) of a pyrrolidinium-functionalised norbornene-based monomer with cyclooctene. The derived peroxophosphotungstate-based polymer-immobilised ionic liquid phase (PIILP) catalyst is an efficient and recyclable system for the epoxidation of allylic alcohols and alkenes, with only a minor reduction in performance on successive cycles.
Co-reporter:Kerri Crossey, Christopher Hardacre and Marie E. Migaud
Chemical Communications 2012 vol. 48(Issue 98) pp:11969-11971
Publication Date(Web):24 Oct 2012
DOI:10.1039/C2CC36367D
A range of nucleoside phosphoramidites incorporating small amino substituents have been readily synthesised using ionic liquid stabilised phosphorodiamidites coupled with mechanochemistry.
Co-reporter:Kevin Morgan, Burapat Inceesungvorn, Alexandre Goguet, Christopher Hardacre, Frederic C. Meunier and Sergiy O. Shekhtman
Catalysis Science & Technology 2012 vol. 2(Issue 10) pp:2128-2133
Publication Date(Web):25 Jun 2012
DOI:10.1039/C2CY20295F
Catalysts currently employed for the polymerization of ethylene have previously been found to deactivate in the presence of oxygen. It is, therefore, important that oxygen is removed from the ethylene feedstock prior to the polymerization. The Ag/γ–Al2O3 catalyst exhibits excellent activity and selectivity toward oxygen reduction with hydrogen in the presence of ethylene. TAP vacuum pulse experiments have been utilised to understand the catalytic behaviour of the Ag/γ–Al2O3 catalyst. TAP multi-pulse experiments have determined the types of active sites that are found on the Ag/γ–Al2O3 catalyst, and the intrinsic activity of these sites. The lifetime of the reactive adsorbed oxygen intermediate has also been determined through TAP consecutive pulse experiments. Multi-pulse and consecutive pulse data have been combined with ethylene adsorption/desorption rate constants to provide an overview of the Ag/γ–Al2O3 catalyst system.
Co-reporter:Ayten Ates, Christopher Hardacre
Journal of Colloid and Interface Science 2012 Volume 372(Issue 1) pp:130-140
Publication Date(Web):15 April 2012
DOI:10.1016/j.jcis.2012.01.017
Two different natural zeolites having different phase compositions were obtained from different regions of Turkey and modified by ion-exchange (0.5 M NH4NO3) and acid leaching using 1 M HCl. The natural and modified samples were treated at low temperature (LT), high temperature (HT) and steam (ST) conditions and characterised by XRF, XRD, BET, FTIR, DR–UV–Vis, NH3-TPD and TGA. Ion-exchange with NH4+ of natural zeolites results in the exchange of the Na+ and Ca2+ cations and the partial exchange of the Fe3+ and Mg2+ cations. However, steam and acidic treatments cause significant dealumination and decationisation, as well as loss of crystalline, sintering of phases and the formation of amorphous material. The presence of mordenite and quartz phases in the natural zeolites increases the stability towards acid treatment, whereas the structure of clinoptilolite-rich zeolites is mostly maintained after high temperature and steam treatments. The natural and modified zeolites treated at high temperature and in steam were found to be less stable compared with synthetic zeolites, resulting in a loss of crystallinity, a decrease in the surface area and pore volume, a decrease in the surface acidity as well as dealumination, and decationisation.Graphical abstractHighlights► The extraction of the embedded iron in natural zeolites into the extra-framework. ► The homogenisation of zeolites via ion exchange, acid and steam treatments. ► Determination of a relationship between the composition and stability of the zeolites. ► Improvement of characteristics of natural zeolites by a range of chemical and temperature treatments.
Co-reporter:Kerri Crossey, Christopher Hardacre, Marie E. Migaud and Sarah E. Norman
RSC Advances 2012 vol. 2(Issue 7) pp:2988-2993
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2RA20131C
A series of phosphorodiamidite reagents have been readily prepared using bis{(trifluoromethyl)sulfonyl}imide based ionic liquids and compared with their syntheses in conventional organic solvents. This method demonstrates a versatile procedure that allows access to both known and novel phosphorodiamidite reagents, whilst addressing issues such as moisture sensitivity and product selectivity present in current molecular based protocols. This method negates the need for reagent purification, whilst allowing for the reactions to be conducted at high concentrations.
Co-reporter:Dr. Johan Jacquemin;Dr. Magdalena Bendová;Dr. Zuzana Sedláková;Dr. Marijana Blesic;Dr. John D. Holbrey;Dr. Claire L. Mullan;Dr. Tristan G. A. Youngs;Laure Pison;Dr. Zden&x11b;k Wagner;Dr. Karel Aim; Margarida F. Costa Gomes; Christopher Hardacre
ChemPhysChem 2012 Volume 13( Issue 7) pp:1825-1835
Publication Date(Web):
DOI:10.1002/cphc.201100952
Abstract
We present a study on the phase equilibrium behaviour of binary mixtures containing two 1-alkyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}imide-based ionic liquids, [Cnmim] [NTf2] (n=2 and 4), mixed with diethylamine or triethylamine as a function of temperature and composition using different experimental techniques. Based on this work, two systems showing an LCST and one system with a possible hourglass shape are measured. Their phase behaviours are then correlated and predicted by using Flory–Huggins equations and the UNIQUAC method implemented in Aspen. The potential of the COSMO-RS methodology to predict the phase equilibria was also tested for the binary systems studied. However, this methodology is unable to predict the trends obtained experimentally, limiting its use for systems involving amines in ionic liquids. The liquid-state structure of the binary mixture ([C2mim] [NTf2]+diethylamine) is also investigated by molecular dynamics simulation and neutron diffraction. Finally, the absorption of gaseous ethane by the ([C2mim][NTf2]+diethylamine) binary mixture is determined and compared with that observed in the pure solvents.
Co-reporter:Tristan G. A. Youngs, John D. Holbrey, Claire L. Mullan, Sarah E. Norman, M. Cristina Lagunas, Carmine D'Agostino, Mick D. Mantle, Lynn F. Gladden, Daniel T. Bowron and Christopher Hardacre
Chemical Science 2011 vol. 2(Issue 8) pp:1594-1605
Publication Date(Web):09 Jun 2011
DOI:10.1039/C1SC00241D
β-D-glucose dissolved in the ionic liquid 1-ethyl-3-methylimidazolium acetate in a 6:1 molar ratio (ionic liquid:glucose) has been studied by neutron scattering, NMR and molecular dynamics simulations. Good agreement was found between simulated neutron scattering profiles generated for isotopically substituted liquid systems and those experimentally determined as well as between simulated and experimental diffusion coefficients obtained by Pulsed Field Gradient NMR spectroscopy. The overriding glucose–ionic liquid interactions in the liquid are hydrogen-bonding between acetate oxygens and sugarhydroxyl groups. The ionic liquid cation was found to play only a minor role in the solvation of the sugar and does not participate in hydrogen-bonding with the sugar to any significant degree. NOESY experiments lend further evidence that there is no direct interaction between sugarhydroxyl groups and acidic hydrogens on the ionic liquid cation.
Co-reporter:Uraiwan Kamolphop, Sarah. F. R. Taylor, John P. Breen, Robbie Burch, Juan J. Delgado, Sarayute Chansai, Christopher Hardacre, Sunantha Hengrasmee, and Stuart L. James
ACS Catalysis 2011 Volume 1(Issue 10) pp:1257
Publication Date(Web):August 25, 2011
DOI:10.1021/cs200326m
Low-temperature (<200 °C) hydrocarbon selective catalytic reduction of NOx has been achieved for the first time in the absence of hydrogen using a solvent-free mechanochemically prepared Ag/Al2O3 catalyst. Catalysts prepared by this ball-milling method show a remarkable increase in activity for the reduction of nitrogen oxides with octane by lowering the light-off temperature by up to 150 °C compared with a state-of-the-art 2 wt % Ag/Al2O3 catalyst prepared by wet impregnation. The best catalyst prepared from silver oxide showed 50% NOx conversion at 240 °C and 99% at 302 °C. The increased activity is not due to an increased surface area of the support, but may be associated with a change in the defect structure of the alumina surface, leading to the formation of the small silver clusters necessary for the activation of the octane without leading to total combustion. On the other hand, since one possible role of hydrogen is to remove inhibiting species from the silver, we cannot exclude some change in the chemical properties of the silver as a result of the ball-milling treatment.Keywords: Ag/Al2O3; ball-milling; hydrocarbon selective catalytic reduction; mechanochemistry; NOx; octane;
Co-reporter:Christopher Hardacre, Haifeng Huang, Stuart L. James, Marie E. Migaud, Sarah E. Norman and William R. Pitner
Chemical Communications 2011 vol. 47(Issue 20) pp:5846-5848
Publication Date(Web):18 Apr 2011
DOI:10.1039/C1CC11025J
Ionic liquids have been used in combination with ball milling on a range of chlorophosphoramidite reagents to phosphitylate nucleosides and 2-deoxynucleosides. The enhanced stability offered by the ionic liquid mediated processes combined with efficient mass transfer induced by ball milling has enabled excellent yields to be obtained even when using small dialkyl amino groups as well as the more commonly used diisopropylamino protection.
Co-reporter:P. Goodrich;C. Hardacre;C. Paun;A. Ribeiro;S. Kennedy;M. J. V. Lourenço;H. Manyar;C. A. Nieto de Castro;M. Besnea;V. I. Pârvulescu
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 6) pp:995-1004
Publication Date(Web):
DOI:10.1002/adsc.201000953
Abstract
A series of bis(oxazoline) metal(II) complexes has been supported on silica and carbon supports by non-covalent immobilisation using an ionic liquid. The catalytic performance of these solids was compared for the enantioselective Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene and the Mukaiyama-aldol reaction between methyl pyruvate and 1-methoxy-1-trimethylsilyloxypropene. In both reactions the enantioselectivity was strongly influenced by the choice of support displaying enantioselectivies (ee values) up to 40% higher than those conducted under homogeneous reaction conditions.
Co-reporter:Alexandre Goguet, Christopher Hardacre, Burapat Inceesungvorn, Kevin Morgan and Sergiy O. Shekhtman
Catalysis Science & Technology 2011 vol. 1(Issue 5) pp:760-767
Publication Date(Web):14 Feb 2011
DOI:10.1039/C0CY00075B
All TAP micro-reactor configurations contain inert particles which are used so that the catalyst zone can be maintained under isothermal conditions. Even on “inert” particles adsorption will occur to some degree; however, the extent to which this occurs has a critical influence on the analysis of the TAP data. In many cases the assumption that there is no interaction between probe molecules and inert particles is required as reversible adsorption over inert material is problematic when the TAP model has to be solved. Moreover, as the TAP pulse response experiments are designed to be conducted within the Knudsen diffusion regime, central to TAP data analysis is the characterization of the diffusional transport of reagent molecules through the micro-reactor, which is achieved via “diffusion only” experiments over an inert one zone packing. Therefore, if there are any processes occurring in addition to Knudsen diffusion over the inert material, such as reversible adsorption, it is important to factor these into the analysis. If these additional processes are not included, the entire data analysis would be questionable. The current work discloses the development of a function which accounts for the adsorption over the inert material, so that the TAP data analysis can be accurately determined. This newly developed analysis method has been exemplified using the selective reduction of oxygen in a hydrogen rich ethylene feed over silver catalysts as a case study.
Co-reporter:Ninie S. A. Manan, Leigh Aldous, Yatimah Alias, Paul Murray, Lesley J. Yellowlees, M. Cristina Lagunas, and Christopher Hardacre
The Journal of Physical Chemistry B 2011 Volume 115(Issue 47) pp:13873-13879
Publication Date(Web):October 12, 2011
DOI:10.1021/jp208159v
The electrochemistry of elemental sulfur (S8) and the polysulfides Na2S4 and Na2S6 has been studied for the first time in nonchloroaluminate ionic liquids. The cyclic voltammetry of S8 in the ionic liquids is different to the behavior reported in some organic solvents, with two reductions and one oxidation peak observed. Supported by in situ UV–vis spectro-electrochemical experiments, the main reduction products of S8 in [C4mim][DCA] ([C4mim] = 1-butyl-3-methylimidazolium; DCA = dicyanamide) have been identified as S62– and S42–, and plausible pathways for the formation of these species are proposed. Dissociation and/or disproportionation of the polyanions S62– and S42– appears to be slow in the ionic liquid, with only small amounts of the blue radical species S3•– formed in the solutions at r.t., in contrast with that observed in most molecular solvents.
Co-reporter:Sarah F. R. Taylor;Jacinto Sá ; Christopher Hardacre
ChemCatChem 2011 Volume 3( Issue 1) pp:119-121
Publication Date(Web):
DOI:10.1002/cctc.201000337
Co-reporter:Dr. Jacinto Sá;Matthew Ace;Dr. Juan José Delgado;Dr. Alexre Goguet; Christopher Hardacre;Kevin Morgan
ChemCatChem 2011 Volume 3( Issue 2) pp:394-398
Publication Date(Web):
DOI:10.1002/cctc.201000285
Abstract
Functionalization of alkanes is much sought after for the production of fine and bulk chemicals. In particular, the oxidative activation of alkanes and their conversion to ethene and propene has been studied extensively, owing to the use of these alkenes in polymerization reactions. The greater reactivity of the products in comparison with the reactants has proven a major issue in this reaction as this can result in overoxidation, producing CO and CO2 and, therefore, reducing the alkene yield. Herein, the first application of supported gold catalysts for the direct activation of C2+ aliphatic alkanes with oxygen to form alkenes is demonstrated. This catalyst is particularly notable as it is highly active, selective to propene and ethene, and stable on stream over a 48 h period. Maintaining cationic gold is thought to be critical for the stability and this catalyst design provides the possibility of applying gold-based catalysts over a much wider temperature range than has been reported.
Co-reporter:Ninie S. A. Manan, Leigh Aldous, Yatimah Alias, Richard G. Compton, M. Cristina Lagunas, and Christopher Hardacre
The Journal of Physical Chemistry B 2011 Volume 115(Issue 11) pp:2574-2581
Publication Date(Web):February 28, 2011
DOI:10.1021/jp1120096
The electrochemistry of HgCl2 and [Hg(NTf2)2] ([NTf2]− = bis{(trifluoromethyl)sulfonyl}imide) has been studied in room temperature ionic liquids. It has been found that the cyclic voltammetry of Hg(II) is strongly dependent on a number of factors (e.g., concentration, anions present in the mixture, and nature of the working electrode) and differs from that found in other media. Depending on conditions, the cyclic voltammetry of Hg(II) can give rise to one, two, or four reduction peaks, whereas the reverse oxidative scans show two to four peaks. Diffuse reflectance UV−vis spectroscopy and X-ray powder diffraction have been used to aid the assignment of the voltammetric waves.
Co-reporter:Dr. Jacinto Sá;Dr. Alexre Goguet;S. F. Rebecca Taylor;Ramchra Tiruvalam; Christopher J. Kiely;Dr. Maarten Nachtegaal; Graham J. Hutchings; Christopher Hardacre
Angewandte Chemie International Edition 2011 Volume 50( Issue 38) pp:8912-8916
Publication Date(Web):
DOI:10.1002/anie.201102066
Co-reporter:Dr. Jacinto Sá;Dr. Alexre Goguet;S. F. Rebecca Taylor;Ramchra Tiruvalam; Christopher J. Kiely;Dr. Maarten Nachtegaal; Graham J. Hutchings; Christopher Hardacre
Angewandte Chemie 2011 Volume 123( Issue 38) pp:9074-9078
Publication Date(Web):
DOI:10.1002/ange.201102066
Co-reporter:Haresh G. Manyar, Cristina Paun, Rashidah Pilus, David W. Rooney, Jillian M. Thompson and Christopher Hardacre
Chemical Communications 2010 vol. 46(Issue 34) pp:6279-6281
Publication Date(Web):27 Jul 2010
DOI:10.1039/C0CC01365J
Selective hydrogenation of carboxylic acids to alcohols and alkanes has been achieved under remarkably mild reaction temperatures and H2 pressures (333 K, 0.5 MPa) using Pt/TiO2 catalyst.
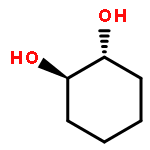
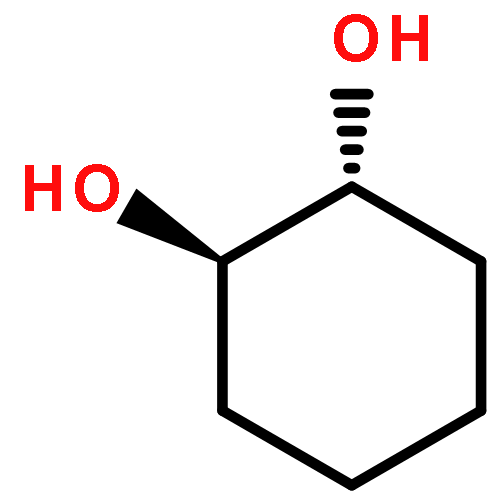
Co-reporter:D.R. Boyd;N.D. Sharma;M.V. Berberian;K.S. Dunne;C. Hardacre;M. Kaik;B. Kelly;J.F. Malone;S.T. McGregor;P.J. Stevenson
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 5) pp:855-868
Publication Date(Web):
DOI:10.1002/adsc.200900818
Abstract
Enantiopure cis-dihydro-1,2-diol metabolites, obtained from toluene dioxygenase-catalysed cis-dihydroxylation of six monosubstituted benzene substrates, have been converted to their corresponding cis-hexahydro-1,2-diol derivatives by catalytic hydrogenation via their cis-tetrahydro-1,2-diol intermediates. Optimal reaction conditions for total catalytic hydrogenation of the cis-dihydro-1,2-diols have been established using six heterogeneous catalysts. The relative and absolute configurations of the resulting benzene cis-hexahydro-1,2-diol products have been unequivocally established by X-ray crystallography and NMR spectroscopy. Methods have been developed to obtain enantiopure cis-hexahydro-1,2-diol diastereoisomers, to desymmetrise a meso-cis-hexahydro-1,2-diol and to synthesise 2-substituted cyclohexanols. The potential of these enantiopure cyclohexanols as chiral reagents was briefly evaluated through their application in the synthesis of two enantiomerically enriched phosphine oxides from the corresponding racemic phosphine precursors.
Co-reporter:Maria Valnice Boldrin Zanoni, Emma I. Rogers, Christopher Hardacre, Richard G. Compton
Analytica Chimica Acta 2010 Volume 659(1–2) pp:115-121
Publication Date(Web):5 February 2010
DOI:10.1016/j.aca.2009.11.026
The reduction of guanine was studied by microelectrode voltammetry in the room temperature ionic liquids (RTILs) N-hexyltriethylammonium bis (trifluoromethanesulfonyl) imide [N6,2,2,2][N(Tf)2], 1-butyl-3-methylimidazolium hexafluorosphosphate [C4mim][PF6], N-butyl-N-methyl-pyrrolidinium bis(trifluoromethanesulfonyl)imide [C4mpyrr][N(Tf)2], 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide [C4mim][N(Tf)2], N-butyl-N-methyl-pyrrolidinium dicyanamide [C4mpyrr][N(NC)2] and tris(P-hexyl)-tetradecylphosphonium trifluorotris(pentafluoroethyl)phosphate [P14,6,6,6][FAP] on a platinum microelectrode. In [N6,2,2,2][NTf2] and [P14,6,6,6][FAP], but not in the other ionic liquids studied, guanine reduction involves a one-electron, diffusion-controlled process at very negative potential to produce an unstable radical anion, which is thought to undergo a dimerization reaction, probably after proton abstraction from the cation of the ionic liquid. The rate of this subsequent reaction depends on the nature of the ionic liquid, and it is faster in the ionic liquid [P14,6,6,6][FAP], in which the formation of the resulting dimer can be voltammetrically monitored at less negative potentials than required for the reduction of the parent molecule. Adenine showed similar behaviour to guanine but the pyrimidines thymine and cytosine did not; thymine was not reduced at potentials less negative than required for solvent (RTIL) decomposition while only a poorly defined wave was seen for cytosine. The possibility for proton abstraction from the cation in [N6,2,2,2][NTf2] and [P14,6,6,6][FAP] is noted and this is thought to aid the electrochemical dimerization process. The resulting rapid reaction is thought to shift the reduction potentials for guanine and adenine to lower values than observed in RTILs where the scope for proton abstraction is not present. Such shifts are characteristic of so-called EC processes where reversible electron transfer is followed by a chemical reaction.
Co-reporter:Ramesh L. Gardas, Rile Ge, Peter Goodrich, Christopher Hardacre, Azlan Hussain and David W. Rooney
Journal of Chemical & Engineering Data 2010 Volume 55(Issue 4) pp:1505-1515
Publication Date(Web):December 22, 2009
DOI:10.1021/je900660x
Ionic liquids (ILs) having either cations or anions derived from naturally occurring amino acids have been synthesized and characterized as amino acid-based ionic liquids (AAILs). In this work, the experimental measurements of the temperature dependence of density, viscosity, heat capacity, and thermal conductivity of several AAILs, namely, tributylmethylammonium serinate ([N4441][Ser]), tributylmethylammonium taurinate ([N4441][Tau]), tributylmethylammonium lysinate ([N4441][Lys]), tributylmethylammonium threonate ([N4441][Thr]), tetrabutylphosphonium serinate ([P4444][Ser]), tetrabutylphosphonium taurinate ([P4444][Tau]), tetrabutylphosphonium lysinate ([P4444][Lys]), tetrabutylphosphonium threonate ([P4444][Thr]), tetrabutylphosphonium prolinate ([P4444][Pro]), tetrabutylphosphonium valinate ([P4444][Val]), and tetrabutylphosphonium cysteinate ([P4444][Cys]), are presented. The influence of cations and anions on studied properties is discussed. On the basis of experimental data, the QSPR (quantitative structure−property relationship) correlations and group contribution methods for thermophysical properties of AAILs have been developed, which form the basis for the development of the computer-aided molecular design (CAMD) of AAILs. It has also been demonstrated that that the predictive data obtained by correlation methods are in good agreement with the experimental data. The correlations developed, herein, can thus be used to evaluate the studied thermophysical properties of AAILs for use in process design or in the CAMD of new AAILs.
Co-reporter:D. T. Bowron, C. D’Agostino, L. F. Gladden, C. Hardacre, J. D. Holbrey, M. C. Lagunas, J. McGregor, M. D. Mantle, C. L. Mullan and T. G. A. Youngs
The Journal of Physical Chemistry B 2010 Volume 114(Issue 23) pp:7760-7768
Publication Date(Web):May 19, 2010
DOI:10.1021/jp102180q
The liquid state structure of the ionic liquid, 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]), an excellent nonderivitizing solvent for cellulosic biomass, has been investigated at 323 K by molecular dynamics (MD) simulation and by neutron diffraction using the SANDALS diffractometer at ISIS to provide experimental differential neutron scattering cross sections from H/D isotopically substituted materials. Ion−ion radial distribution functions both calculated from MD and derived from the empirical potential structure refinement (EPSR) model to the experimental data show the alternating shell structure of anions around the cation, as anticipated. Spatial probability distributions reveal the main anion-to-cation features as in-plane interactions of anions with the three imidazolium ring hydrogens and cation−cation planar stacking above/below the imidazolium rings. Interestingly, the presence of the polarized hydrogen-bond acceptor (HBA) anion (acetate) leads to an increase in anion−anion tail−tail structuring within each anion shell, an indicator of the onset of hydrophobic regions within the anion regions of the liquid. MD simulations show the importance of scaling of the effective ionic charges in the basic simulation approach to accurately reproduce both the observed experimental neutron scattering cross sections and ion self-diffusion coefficients.
Co-reporter:Alexandre Goguet ; Christopher Hardacre ; Ian Harvey ; Katabathini Narasimharao ; Youssef Saih ;Jacinto Sa
Journal of the American Chemical Society 2009 Volume 131(Issue 20) pp:6973-6975
Publication Date(Web):April 30, 2009
DOI:10.1021/ja9021705
The active site in supported gold catalysts for the carbonylation of methanol has been identified as dimers/trimers of gold which are formed from large gold particles >10 nm in diameter. Methyl iodide was found to be critical for this dispersion process and to maintain the catalyst in the active form. This study also shows that it may be possible to redisperse gold catalysts, in general, after reaction.
Co-reporter:Eric J. Amigues, Christopher Hardacre, Gillian Keane, Marie E. Migaud, Sarah E. Norman and William R. Pitner
Green Chemistry 2009 vol. 11(Issue 9) pp:1391-1396
Publication Date(Web):16 Jun 2009
DOI:10.1039/B905000K
A range of chlorophosphoramidites have been prepared in ionic liquids and compared with material synthesised in molecular solvents. Through the use of ionic liquids as reaction media the moisture sensitivity and impurity issues hampering existing traditional synthetic routes have been eased. Not only can stock chemicals be used without purification, but the reactions may be conducted at room temperature and at high concentrations. Furthermore, reaction times are reduced and rapid addition of reagents is possible whilst retaining tight control over product selectivity. Beyond their role as reaction media, ionic liquids also present a unique storage medium for these highly moisture sensitive chlorophosphoramidites.
Co-reporter:R. Ge, R. W. K. Allen, L. Aldous, M. R. Bown, Nicola Doy, C. Hardacre, J. M. MacInnes, G. McHale and M. I. Newton
Analytical Chemistry 2009 Volume 81(Issue 4) pp:1628
Publication Date(Web):January 22, 2009
DOI:10.1021/ac802406k
A microfluidic device designed for electrochemical studies on a microliter scale has been utilized for the examination of impurity levels in ionic liquids (ILs). Halide impurities are common following IL synthesis, and this study demonstrates the ability to quantify low concentrations of halide in a range of ILs to levels of ∼5 ppm, even in ILs not currently measurable using other methods such as ion chromatography. To validate the mixer device, the electrochemistry of ferrocene was also examined and compared with spectroscopic and bulk electrochemistry measurements. An automated “sample preparation, delivery, and calibration” method was developed, and the chip successfully used for linear sweep, cyclic voltammetry (under both quiescent and steady-state flowing conditions), square wave voltammetry, and differential pulse voltammetry. An effective method of electrochemically cleaning the electrodes is also presented.
Co-reporter:Nicolas Legagneux, Jean-Marie Basset, Amélie Thomas, Frédéric Lefebvre, Alexandre Goguet, Jacinto Sá and Christopher Hardacre
Dalton Transactions 2009 (Issue 12) pp:2235-2240
Publication Date(Web):04 Feb 2009
DOI:10.1039/B816806G
Dodecatungsto-silicic H4SiW12O40 and -phosphoric acids H3PW12O40 were deposited on silica by a classical impregnation technique. The resulting materials were studied by in situ Raman and infrared spectroscopy, XPS and by solid-state 1H MAS NMR as a function of their dehydroxylation temperature. The data show that in the case of H3PW12O40 three silanol groups are protonated while in the case of H4SiW12O40 at least one acidic proton remains. Upon heating this proton reacts leading to a disordered structure and a broadening of the W–O Raman bands.
Co-reporter:Christopher Hardacre, Ivano Messina, Marie E. Migaud, Kerry A. Ness, Sarah E. Norman
Tetrahedron 2009 65(32) pp: 6341-6347
Publication Date(Web):
DOI:10.1016/j.tet.2009.06.013
Co-reporter:Norfaizah Ab Manan, Christopher Hardacre, Johan Jacquemin, David W. Rooney and Tristan G. A. Youngs
Journal of Chemical & Engineering Data 2009 Volume 54(Issue 7) pp:2005-2022
Publication Date(Web):April 30, 2009
DOI:10.1021/je800857x
As the range of available ionic liquids increases, methods by which important engineering parameters such as gas solubilities can be estimated from simple structural information become ever more desirable. COSMO-based thermodynamic models, such as that used by COSMOthermX, allow the determination of such data for pure and mixed component systems. Herein, we evaluate the predictive capability of COSMOthermX through a comparison with literature data obtained from the IUPAC database which contains data for 15 gases in 27 ionic liquids. To determine any effect inherent to ionic liquids, gas solubility predictions were first performed for selected molecular solvents at constant temperature and pressure. Further estimations of gas solubility at temperatures ranging from (278 to 368) K at 0.1 MPa in water were performed for 14 gases. The study has demonstrated that COSMOthermX is capable of predicting, qualitatively, gas solubilities in ionic liquids and, hence, reducing the amount of unnecessary experimental measurements prior to specific applications using ionic liquids.
Co-reporter:Eric Jean Amigues, Christopher Hardacre, Gillian Keane and Marie Eugenie Migaud
Green Chemistry 2008 vol. 10(Issue 6) pp:660-669
Publication Date(Web):20 Mar 2008
DOI:10.1039/B718849H
The reactivity of phosphorus trichloride towards amines in ionic liquids has been investigated and compared with that in conventional organic solvents. In addition to easing the synthetic procedures thus far required for the preparation of aminochlorophosphines in molecular solvents, for example the ability to operate at 20 °C, under non-anhydrous and high concentration conditions with short reaction times and rapid addition of reagents, bis{(trifluoromethyl)sulfonyl}imide-based ionic liquids offer a tight control over the selectivity during the reaction and offer access to monoamination and bisamination products in high yields and chemoselectively. Furthermore, these ionic liquids offer a unique storage medium for these highly moisture sensitive aminochlorophosphines.
Co-reporter:Burapat Inceesungvorn, Frederic C. Meunier, Chris Hardacre, Robbie Burch and Ken Griffin
Chemical Communications 2008 (Issue 46) pp:6212-6214
Publication Date(Web):21 Oct 2008
DOI:10.1039/B813470G
The selective reduction of molecular oxygen with excess H2 in the presence of alkenes was achieved successfully for the first time: silver supported on alumina catalysts exhibited full conversion of O2 at temperature as low as 50 °C, while the conversion of ethene or propene remained essentially zero up to 250 °C.
Co-reporter:P. Goodrich;C. Hardacre;C. Paun;V.I. Pârvulescu;I. Podolean
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 16) pp:2473-2476
Publication Date(Web):
DOI:10.1002/adsc.200800431
Abstract
Ionic liquids have been used to support a range of magnesium- and copper-based bis(oxazoline) complexes for the enantioselective Diels–Alder reaction between N-acryloyloxazolidinone and cyclopentadiene. Compared with reaction performed in dichloromethane or diethyl ether, an enhancement in ee is observed with a large increase in reaction rate. In addition, for non-sterically hindered bis(oxazoline) ligands, that is, phenyl functionalised ligands, a reversal in configuration is found in the ionic liquid, 1-ethyl-3-methylimidazolium bis[(trifluoromethanesulfonyl)imide], compared with molecular solvents. Supported ionic liquid phase catalysts have also been developed using surface-modified silica which show good reactivity and enantioselectivity for the case of the magnesium-based bis(oxazoline) complexes. Poor ees and conversion were observed for the analogous copper-based systems. Some drop in ee was found on supporting the catalyst due a drop in the rate of reaction and, therefore, an increase in the contribution from the uncatalysed achiral reaction.
Co-reporter:Christopher Hardacre, Paul Nancarrow, David W. Rooney and Jillian M. Thompson
Organic Process Research & Development 2008 Volume 12(Issue 6) pp:1156-1163
Publication Date(Web):October 15, 2008
DOI:10.1021/op800134k
The comparison of three ionic liquid-mediated catalytic processes for the benzoylation of anisole with benzoic anhydride is presented. A detailed understanding of the mechanism by which the zeolite and metal triflate reactions in bis{trifluoromethanesulfonyl}imide-based ionic liquids has been reported previously, and these routes are considered together with an indium chloride-based ionic liquid system. Solvent extraction and vacuum/steam distillation have been assessed as possible workup procedures, and an overall preliminary economic evaluation of each overall process is reported. Although the predominant activity is associated with the in situ formation of a homogeneous acid catalyst, the low cost and facile separation of the zeolite-catalysed process leads to this route being the most economically viable overall option. The results of a continuous flow miniplant based on the zeolite catalyst are also presented and compared with the reaction using a small plug flow reactor.
Co-reporter:Rile Ge, Christopher Hardacre, Johan Jacquemin, Paul Nancarrow and David W. Rooney
Journal of Chemical & Engineering Data 2008 Volume 53(Issue 9) pp:2148-2153
Publication Date(Web):August 14, 2008
DOI:10.1021/je800335v
Co-reporter:Johan Jacquemin, Paul Nancarrow, David W. Rooney, Margarida F. Costa Gomes, Pascale Husson, Vladimir Majer, Agilio A. H. Pádua and Christopher Hardacre
Journal of Chemical & Engineering Data 2008 Volume 53(Issue 9) pp:2133-2143
Publication Date(Web):August 5, 2008
DOI:10.1021/je8002817
The density of ionic liquids (ILs) as a function of pressure and temperature has been modeled using a group contribution model. This model extends the calculations previously reported (Jacquemin et al. J. Chem. Eng. Data 2008) which used 4000 IL densities at 298.15 K and 600 IL densities as a function of temperature up to 423 K at 0.1 MPa to pressures up to 207 MPa by using described data in the literature and presented in this study. The densities of two different ionic liquids (butyltrimethylammonium bis(trifluoromethylsulfonyl)imide, [N1114][NTf2], and 1-butyl-1-methyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide, [C4mPyrro][NTf2]) were measured as a function of temperature from (293 to 415) K and over an extended pressure range from (0.1 to 40) MPa using a vibrating-tube densimeter. The model is able to predict the ionic liquid densities of over 5080 experimental data points to within 0.36 %. In addition, this methodology allows the calculation of the mechanical coefficients using the calculated density as a function of temperature and pressure with an estimated uncertainty of ± 20 %.
Co-reporter:Christopher Hardacre, John D. Holbrey, Claire L. Mullan, Mark Nieuwenhuyzen, Tristan G. A. Youngs and Daniel T. Bowron
The Journal of Physical Chemistry B 2008 Volume 112(Issue 27) pp:8049-8056
Publication Date(Web):June 13, 2008
DOI:10.1021/jp801801j
The liquid structure of 1-methyl-4-cyanopyridinium bis{(trifluoromethyl)sulfonyl}imide, a prototypical ionic liquid containing an electron-withdrawing group on the cation, has been investigated at 368 K. Experimental neutron scattering combined with empirical potential structure refinement analysis of the data and classical molecular dynamics simulations have been used to probe the liquid structure in detail. Both techniques generated highly consistent results that provide valuable validation of the force fields and refinement approaches. A significant degree of apparent charge ordering is found in the liquid structure, although the nonspherical shape of the ions results in interpenetration of cations into the first shell of adjacent cations, with much shorter closest contact distances than the averaged center-of-mass cation−cation and cation−anion separations.
Co-reporter:Johan Jacquemin, Rile Ge, Paul Nancarrow, David W. Rooney, Margarida F. Costa Gomes, Agilio A. H. Pádua and Christopher Hardacre
Journal of Chemical & Engineering Data 2008 Volume 53(Issue 3) pp:716-726
Publication Date(Web):February 27, 2008
DOI:10.1021/je700707y
The prediction of molar volumes and densities of several ionic liquids has been achieved using a group contribution model as a function of temperature between (273 and 423) K at atmospheric pressure. It was observed that the calculation of molar volumes or densities could be performed using the “ideal” behavior of the molar volumes of mixtures of ionic liquids. This model is based on the observations of Canongia Lopes et al. (J. Phys. Chem. B 2005, 109, 3519–3525) which showed that this ideal behavior is independent of the temperature and allows the molar volume of a given ionic liquid to be calculated by the sum of the effective molar volume of the component ions. Using this assumption, the effective molar volumes of ions constituting more than 220 different ionic liquids were calculated as a function of the temperature at 0.1 MPa using more than 2150 data points. These calculated results were used to build up a group contribution model for the calculation of ionic liquid molar volumes and densities with an estimated repeatability and uncertainty of 0.36 % and 0.48 %, respectively. The impact of impurities (water and halide content) in ionic liquids as well as the method of determination were also analyzed and quantified to estimate the overall uncertainty.
Co-reporter:Christopher Hardacre, John D. Holbrey, Mark Nieuwenhuyzen and Tristan G. A. Youngs
Accounts of Chemical Research 2007 Volume 40(Issue 11) pp:1146
Publication Date(Web):June 21, 2007
DOI:10.1021/ar700068x
This Account describes experimental data used to understand the structure of ionic liquids and solute–solvent interactions of both molecular solutes and dissolved metal complexes. In general, the structures of the ionic liquids determined from experimental data show good agreement with both simulated structures and solid-state structures. For all ionic liquids studied, strong charge ordering is found leading to long-range order even in the presence of a solute. For dissolved metal complexes, the ionic liquid is not innocent and a clear dependence on the speciation is observed with variations in both the cation and anion.
Co-reporter:V. Berberian;C. C. R. Allen;N. D. Sharma;D. R. Boyd;C. Hardacre
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 4-5) pp:
Publication Date(Web):20 MAR 2007
DOI:10.1002/adsc.200600437
A series of cis-dihydrodiol metabolites, available from the bacterial dioxygenase-catalysed oxidation of monosubstituted benzene substrates using Pseudomonas putida UV4 , have been converted to the corresponding catechols using both a heterogeneous catalyst (Pd/C) and a naphthalene cis-diol dehydrogenase enzyme present in whole cells of the recombinant strain Escherichia coli DH5α(pUC129: nar B). A comparative study of the merits of both routes to 3-substituted catechols has been carried out and the two methods have been found to be complementary. A similarity in mechanism for catechol formation under both enzymatic and chemoenzymatic conditions, involving regioselective oxidation of the hydroxyl group at C-1, has been found using deuterium labelled toluene cis-dihydrodiols. The potential, of combining a biocatalytic step (dioxygenase-catalysed cis-dihydroxylation) with a chemocatalytic step (Pd/C-catalysed dehydrogenation), into a one-pot route to catechols, from the parent substituted benzene substrates, has been realised.
Co-reporter:Simon Doherty;Peter Goodrich;Julian G. Knight;Mimi T. Nguyen;Vasile I. Pârvulescu;Cristina Paun
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 6) pp:
Publication Date(Web):17 APR 2007
DOI:10.1002/adsc.200600531
Imidazolium-tagged bis(oxazolines) have been prepared and used as chiral ligands in the copper(II)-catalysed Diels–Alder reaction of N-acryloyl- and N-crotonoyloxazolidinones with cyclopentadiene and 1,3-cyclohexadiene in the ionic liquid 1-ethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]imide, [emim][NTf2]. A significant and substantial enhancement in the rate and enantioselectivity was achieved in [emim][NTf2] compared with dichloromethane. For example, complete conversion and enantioselectivities up to 95 % were obtained for the reaction between N-acryloyloxazolidinone and cyclopentadiene within 2 min in [emim][NTf2] whereas the corresponding reaction in dichloromethane required 60 min to reach completion and gave an ee of only 16 %. The enhanced rates obtained in the ionic liquid enabled a catalyst loading as low as 0.5 mol % to give complete conversion within 2 min while retaining the same level of enantioselectivity. The imidazolium-tagged catalysts can be recycled ten times without any loss in activity or enantioselectivity and showed much higher affinity for the ionic liquid phase during the recycle procedure than the analogous uncharged ligand.
Co-reporter:Eric Amigues, Christopher Hardacre, Gillian Keane, Marie Migaud and Maeve O'Neill
Chemical Communications 2006 (Issue 1) pp:72-74
Publication Date(Web):06 Oct 2005
DOI:10.1039/B509248E
Ionic liquids have been shown to offer hitherto unseen control as both a storage solvent for PCl3 and POCl3 and reaction media for fluorination and mixed anhydride formation under benign conditions.
Co-reporter:Constanza Villagrán, Leigh Aldous, M. Cristina Lagunas, Richard G. Compton, Christopher Hardacre
Journal of Electroanalytical Chemistry 2006 Volume 588(Issue 1) pp:27-31
Publication Date(Web):1 March 2006
DOI:10.1016/j.jelechem.2005.11.023
The electrochemistry of phenol and 4-tert-butyl-phenol is described in [C2mim][NTf2] and [C4mpyrr][NTf2] ionic liquids. Oxidation of phenol and phenolate is observed at Epa=+1.64and+0.24V vs. Ag in both ionic liquids. On the cathodic sweep at a potential of −2.05 V vs. Ag under an oxygen atmosphere, the production of O22- dianions triggers the formation of phenolate anions which undergo chemical oxidation to the phenoxyl radical. The phenoxyl radical then reacts with the [NTf2]− anion of the ionic liquid to form the corresponding phenyl triflate molecule.
Co-reporter:Leigh Aldous, Debbie S. Silvester, Constanza Villagrán, William R. Pitner, Richard G. Compton, M. Cristina Lagunas and Christopher Hardacre
New Journal of Chemistry 2006 vol. 30(Issue 11) pp:1576-1583
Publication Date(Web):30 Aug 2006
DOI:10.1039/B609261F
For the first time, the electrochemistry of gold has been studied in detail in a ‘second-generation’, non-haloaluminate, ionic liquid. In particular, the electrochemical behaviour of Na[AuCl4] has been investigated in 1-butyl-3-methylimidazolium bis{(tifluoromethyl)sulfonyl}imide, [C4mim][NTf2], over gold, platinum and glassy carbon working electrodes. The reduction of [AuCl4]− initially forms [AuCl2]− before deposition on the electrode as Au(0). To enable stripping of deposited gold or electrodissolution of bulk gold, the presence of chloride, trichloride or chlorine is required. Specifically trichloride and chlorine have been identified as the active species which preferentially form Au(I) and Au(III), respectively.
Co-reporter:Tristan G. A. Youngs Dr.;John D. Holbrey Dr.;Maggel Deetlefs Dr.;Mark Nieuwenhuyzen Dr.;Margarida F. Costa Gomes Dr.
ChemPhysChem 2006 Volume 7(Issue 11) pp:2279-2281
Publication Date(Web):6 NOV 2006
DOI:10.1002/cphc.200600569
Sweet simulations: Molecular dynamics simulations of the solvation environment of isolated glucose monomers in a choride-based ionic liquid (IL) reveal that the sugar prefers to bind to four chloride anions. Coordination shells involving only three anions, two of which are bridging chlorides, are also observed (see picture). The presence of these lower chloride:glucose ratios explains the unexpected high degree of solvation of glucose in these Ils.
Co-reporter:Paul N. Davey, Stewart A. Forsyth, H. Q. Nimal Gunaratne, Christopher Hardacre, Angela McKeown, S. E. Jane McMath, David W. Rooney and Kenneth R. Seddon
Green Chemistry 2005 vol. 7(Issue 4) pp:224-229
Publication Date(Web):15 Mar 2005
DOI:10.1039/B416021E
An efficient synthesis of a precursor to Lilial®, based on an aldol condensation in an ionic liquid, is described, utilising piperidine as the base catalyst. The yields obtained with this methodology are significantly increased in comparison with those reported in organic solvents to date. In the ionic liquid, the self-aldol condensation of propanal is suppressed and leads to an increased selectivity with respect to the cross-aldol condensation product without the need to use an excess of 4-tert-butylbenzaldehyde to obtain high selectivities.
Co-reporter:Héctor de la Riva, Aranzazu Pintado-Alba, Mark Nieuwenhuyzen, Christopher Hardacre and M. Cristina Lagunas
Chemical Communications 2005 (Issue 39) pp:4970-4972
Publication Date(Web):08 Sep 2005
DOI:10.1039/B508863A
EXAFS has been used to directly show the existence of Au⋯Au interactions in dissolved Au(I) complexes for the first time; the information has been used to understand the optical properties of these materials.
Co-reporter:Constanza Villagrán, Craig E. Banks, William R. Pitner, Christopher Hardacre, Richard G. Compton
Ultrasonics Sonochemistry 2005 Volume 12(Issue 6) pp:423-428
Publication Date(Web):August 2005
DOI:10.1016/j.ultsonch.2004.08.001
The selective electroreduction N-methylphthalimide to 3-hydroxy-2-methyl-isoindolin-1-one has been performed in ionic liquids using phenol as a proton donor under silent and ultrasonic conditions. A significant increase in the rate of electroreduction is shown using ultrasonic activation and in addition high current efficiencies were observed. Some decomposition of the ionic liquid was found to have occurred under exposure to ultrasound.
Co-reporter:Stewart A. Forsyth, H.Q. Nimal Gunaratne, Christopher Hardacre, Angela McKeown, David W. Rooney, Kenneth R. Seddon
Journal of Molecular Catalysis A: Chemical 2005 Volume 231(1–2) pp:61-66
Publication Date(Web):20 April 2005
DOI:10.1016/j.molcata.2004.12.022
The Heck arylation of 2-methylprop-2-en-1-ol in ionic liquids and organic solvents is reported using a range of homogeneous and heterogeneous palladium catalysts. Higher activity is observed in the ionic liquid media compared with N-methylpyrrolidinone and under solventless conditions. The ionic liquid-catalyst system may be recycled easily with little loss in activity, although significant palladium leaching from the heterogeneous catalyst was observed. In the case of trans-bis(2,3-dihydro-3-methylbenzothiazole-2-ylidene)diiodopalladium(II) reported to be highly active for this transformation, significant induction periods were observed indicating that nanoparticles may be responsible for the catalysis. Using the ionic liquid Heck reaction, a recyclable synthesis for the fragrance β-Lilial® has been developed.The Heck reaction has been utilised in an ionic liquid to selectively form β-Lilial® which currently produced in the fine chemicals industry on the kiloton scale. The ionic liquid reaction shows higher rates of reaction than organic solvents and may be recycled easily.
Co-reporter:Valentin Cimpeanu;Vasile I. Parvulescu ;Pedro Amorós Dr.;Daniel Beltrán Dr.;Jillian M. Thompson Dr. Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 18) pp:
Publication Date(Web):4 AUG 2004
DOI:10.1002/chem.200400105
Heterogeneous catalytic oxidation of a series of thioethers (2-thiomethylpyrimidine, 2-thiomethyl-4,6-dimethyl-pyrimidine, 2-thiobenzylpyrimidine, 2-thiobenzyl-4,6-dimethylpyrimidine, thioanisole, and n-heptyl methyl sulfide) was performed in ionic liquids by using MCM-41 and UVM-type mesoporous catalysts containing Ti, or Ti and Ge. A range of triflate, tetrafluoroborate, trifluoroacetate, lactate and bis(trifluoromethanesulfonyl)imide-based ionic liquids were used. The oxidations were carried out by using anhydrous hydrogen peroxide or the urea–hydrogen peroxide adduct and showed that ionic liquids are very effective solvents, achieving greater reactivity and selectivity than reactions performed in dioxane. The effects of halide and acid impurities on the reactions were also investigated. Recycling experiments on catalysts were carried out in order to evaluate Ti leaching and its effect on activity and selectivity.
Co-reporter:Kris Anderson, Peter Goodrich, Christopher Hardacre and David W. Rooney
Green Chemistry 2003 vol. 5(Issue 4) pp:448-453
Publication Date(Web):02 Jul 2003
DOI:10.1039/B305633C
The selective hydrogenation of α,β unsaturated aldehydes has been performed in a range of room temperature ionic liquids. The reaction data reported show that it is possible to enhance the selectivity of supported palladium catalysts for the reduction of the conjugated CC bond by using ionic liquids as solvents compared with conventional molecular organic solvents. The catalyst system is easily recycled without the need to isolate or filter the catalyst and may be used without further treatment.
Co-reporter:Noel A Hamill, Christopher Hardacre and S. E. Jane McMath
Green Chemistry 2002 vol. 4(Issue 2) pp:139-142
Publication Date(Web):26 Feb 2002
DOI:10.1039/B109844F
The Heck reaction, performed in room temperature ionic liquids, has been studied by in situ XAFS, which indicates that palladium clusters of 0.8–1.6 nm diameter are the main species present during reaction.
Co-reporter:Michael G. Burnett, Christopher Hardacre and Heather J. Mawhinney
Physical Chemistry Chemical Physics 2002 vol. 4(Issue 15) pp:3828-3834
Publication Date(Web):19 Jun 2002
DOI:10.1039/B201175A
The adsorption of cadmium(II) on freshly precipitated aluminium(III) hydroxide in the presence of a range of chelates has been investigated. By precipitating the metal, chelate and adsorbent together it is possible to change the pH variation of the metal-complex adsorption from anionic, “ligand-like”, binding to cationic binding. This is a general phenomenon and is explained by the formation of a ternary Al–O–Cd–L surface species. As a consequence of the preparation method, the pH edge is found to shift to lower pH values in the presence of the chelate which gives rise to an apparent increase in adsorption of Cd2+. This increase is, in general, most pronounced at [chelate]/[metal]>1. Computer modelling shows that the observed trends result from the competition between Al–O–Cd–L and Al–L for the available aluminium(III)
binding sites. The enhanced adsorption in the presence of phenylenediaminetetraacetate is anomalous since it is observed at a [chelate]/[metal]≈0.1 and cannot be interpreted by the simple competition model.
Co-reporter:Michael G. Burnett, Christopher Hardacre, James M. Mallon, Heather J. Mawhinney and R. Mark Ormerod
Physical Chemistry Chemical Physics 2000 vol. 2(Issue 6) pp:1273-1279
Publication Date(Web):22 Feb 2000
DOI:10.1039/A909612D
The
adsorption of ca. millimolar concentrations of cadmium(II) on freshly precipitated aluminium(III) hydroxide
is enhanced, at pH values of below 8, in the presence of ethylenediaminetetraacetate, EDTA, via
stabilisation
of the adsorbed cation. At levels where the hydroxide phase is in large excess to the cadmium(II),
the addition of stoichiometrically equivalent concentrations of EDTA will enhance its adsorption to approximately
95%. In the presence of EDTA, cadmium adsorption increases with pH despite the fact that 99.9% of the dissolved cadmium is present as an anionic cadmium(II) EDTA complex. The adsorption observed has been modelled using the measured values of the solubility of aluminium hydroxide and its adsorption of EDTA, the normal and the EDTA-enhanced cationic binding of cadmium(II) and the accepted equilibrium constants for EDTA complexation of cadmium(II) and aluminium(III). The structures
of the dissolved and adsorbed complexes have been inferred from XAFS and Al27 NMR spectroscopy.
Co-reporter:Bo Yang, Robbie Burch, Christopher Hardacre, Gareth Headdock, P. Hu
Journal of Catalysis (September 2013) Volume 305() pp:264-276
Publication Date(Web):1 September 2013
DOI:10.1016/j.jcat.2013.05.027
•Hydrogenation barriers and reaction rates have been calculated using a two-step model.•Pd(2 1 1) has the highest hydrogenation activity.•Pd(1 1 1) selectively produces ethylene and open surfaces form ethane.•Alloying of the Pd blocks unselective sites leading to decreased ethane production.The selective hydrogenation of acetylene to ethylene on several Pd surfaces (Pd(1 1 1), Pd(1 0 0), Pd(2 1 1), and Pd(2 1 1)-defect) and Pd surfaces with subsurface species (carbon and hydrogen) as well as a number of Pd-based alloys (Pd–M/Pd(1 1 1) and Pd–M/Pd(2 1 1) (M = Cu, Ag and Au)) are investigated using density functional theory calculations to understand both the acetylene hydrogenation activity and the selectivity of ethylene formation. All the hydrogenation barriers are calculated, and the reaction rates on these surfaces are obtained using a two-step model. Pd(2 1 1) is found to have the highest activity for acetylene hydrogenation while Pd(1 0 0) gives rise to the lowest activity. In addition, more open surfaces result in over-hydrogenation to form ethane, while the close-packed surface (Pd(1 1 1)) is the most selective. However, we also find that the presence of subsurface carbon and hydrogen significantly changes the reactivity and selectivity of acetylene toward hydrogenation on Pd surfaces. On forming surface alloys of Pd with Cu, Ag and Au, the selectivity for ethylene is also found to be changed. A new energy decomposition method is used to quantitatively analyze the factors in determining the changes in selectivity. These surface modifiers are found to block low coordination unselective sites, leading to a decreased ethane production.The selective hydrogenation of acetylene to ethylene on several Pd surfaces and Pd surfaces with subsurface carbon and hydrogen as well as a number of Pd based alloys has been investigated in detail using density functional theory calculations to understand the catalyst activity and the selectivity of ethylene formation. Whilst open faces increased the activity these resulted in over hydrogenation to form ethane. Surface modification was found to block low coordination unselective sites, leading to a decreased ethane production.Download high-res image (104KB)Download full-size image
Co-reporter:Ayten Ates, Andreas Reitzmann, Christopher Hardacre, Huseyin Yalcin
Applied Catalysis A: General (4 November 2011) Volume 407(Issues 1–2) pp:67-75
Publication Date(Web):4 November 2011
DOI:10.1016/j.apcata.2011.08.026
Co-reporter:Sandra M. Rountree, M. Cristina Lagunas, Christopher Hardacre, Paul N. Davey
Applied Catalysis A: General (28 November 2011) Volume 408(Issues 1–2) pp:54-62
Publication Date(Web):28 November 2011
DOI:10.1016/j.apcata.2011.09.005
Co-reporter:H. Daly, A. Goguet, C. Hardacre, F.C. Meunier, R. Pilasombat, D. Thompsett
Journal of Catalysis (28 July 2010) Volume 273(Issue 2) pp:257-265
Publication Date(Web):28 July 2010
DOI:10.1016/j.jcat.2010.05.021
Au/CeZrO4 catalysts are highly active for the water–gas shift reaction but tend to be unstable and deactivate with time on stream. In this study, in situ DRIFTS-GC was used to investigate the nature of the Au under a range of reaction conditions as the deactivation rate is observed to vary with feed conditions (water concentration, presence of CO2) and reaction temperature. An analysis of the Au–CO bands during the reaction showed that the rate of change in the Au0–CO bands correlated with the deactivation rate under all conditions. Although Auδ+–CO species may be highly active they are very unstable under the feed and, from a comparison of the behaviour of the Au–CO species under varied feeds and reaction temperatures, it is proposed that metallic gold is the predominant active state in these catalysts for low-temperature WGS reaction.Through varying the feed conditions of the water–gas shift reaction, it was possible to enhance the stability of the catalysts significantly. Following a pre-treatment of the catalyst under the full water–gas shift feed (2% CO, 2% CO2, 8.1% H2 and 7.5% H2O) and removal of CO2 from this feed, the catalyst was found to be very stable. The extent of deactivation can be altered by varying the feed conditions during a reaction. The discovery of a way to enhance Au catalyst stability for the low-temperature water–gas shift reaction has significant implications for the development and use of these catalysts.The predominant active state in Au/CeZrO4 catalysts for low-temperature water gas shift is thought to be Au(0) and, through varying the reaction feed conditions, it was possible to enhance the stability of the catalysts significantly.Download high-res image (45KB)Download full-size image
Co-reporter:K.T. Hindle, R. Burch, P. Crawford, C. Hardacre, P. Hu, B. Kalirai, D.W. Rooney
Journal of Catalysis (25 October 2007) Volume 251(Issue 2) pp:338-344
Publication Date(Web):25 October 2007
DOI:10.1016/j.jcat.2007.07.032
The dehydrogenation of 1,2,3,4-tetrahydrocarbazole (THCZ) to form carbazole (CZ) over supported palladium catalysts was examined in the presence of hydrogen acceptors. As expected, liquid hydrogen acceptors increased the rate of reaction but, importantly, gaseous hydrogen acceptors also have been used. Ethene, propene, and but-1-ene showed up to a fivefold increase in the rate of dehydrogenation. Moreover, compared with the analogous liquid systems, the gaseous alternatives are a potentially more economic method of enhancing the activity and provide a simpler workup. The mechanism for the increase in rate was examined by density functional theory calculations, which showed that the propene hydrogenation competes effectively with the back-hydrogenation of the intermediates formed during the THCZ dehydrogenation, resulting in a shift in the equilibrium toward to the formation of CZ.
Co-reporter:Vasile I. Pârvulescu, Bogdan Cojocaru, Viorica Pârvulescu, Ryan Richards, Zhi Li, Chris Cadigan, Pascal Granger, Pierre Miquel, Chris Hardacre
Journal of Catalysis (25 May 2010) Volume 272(Issue 1) pp:92-100
Publication Date(Web):25 May 2010
DOI:10.1016/j.jcat.2010.03.008
Silver colloids prepared by reducing AgNO3 in aqueous solution with sodium citrate were embedded in alumina following two different preparation procedures resulting in samples containing 3 and 5 wt.% silver. Characterization of these materials using TEM, XPS, XAES, CP/MAS NMR, XRD, and adsorption–desorption isotherms of nitrogen showed that embedding the pre-prepared silver colloids into the alumina via the sol–gel procedure preserved the particle size of silver. However, as XAES demonstrates, the catalysts prepared in a sol–gel with a lower amount of water led to embedded colloids with a higher population of Ag+ species. The catalytic behaviors of the resultant catalysts were well correlated with the concentration of these species. Thus, the active silver species of the catalysts containing more Ag+ species selectively converts NO to N2. However, subsequent thermal aging leads to an enhancement of the conversion of NO parallel to slight alteration of the selectivity with the appearance of low amounts of N2O despite an increase of Ag+ species. Accordingly, an optimal surface Ag0/Ag+ ratio is probably needed, independently of the size of silver particles. It was found that this optimal ratio strongly depends on the operating conditions during the synthesis route.The target reaction is the reduction of NO to nitrogen which competes with the oxidation reaction under lean conditions (excess of oxygen) that may considerably alter the selectivity. Silver colloids prepared by reducing AgNO3 in aqueous solution are embedded in alumina leading to stable nano-catalysts. The activity of the catalysts is influenced by the preparation procedure.Download high-res image (69KB)Download full-size image
Co-reporter:S.O. Shekhtman, A. Goguet, R. Burch, C. Hardacre, N. Maguire
Journal of Catalysis (25 January 2008) Volume 253(Issue 2) pp:303-311
Publication Date(Web):25 January 2008
DOI:10.1016/j.jcat.2007.10.028
CO multipulse temporal analysis of products (TAP) experiments were used to characterize a ceria-supported platinum catalyst after various oxidative and reductive pretreatments using O2, H2O, CO2, and H2. Based on the amount of CO consumed, using the final CO-saturated catalyst composition as the common state point, the oxidatively pretreated catalyst could be described using a general scale. From a kinetic analysis of the CO multipulse responses, two kinetic regimes corresponding to two types of active sites could be identified. As the temperature was raised, the number of the most active sites did not change while the amount of the less active site increased. Comparison of the number of active sites determined from the TAP data reported herein with that determined by a previous steady-state isotope transient kinetic analysis experiment showed excellent agreement. This correlation indicates that the (very fast response) TAP experiments can provide information regarding the number and type of active sites that are relevant to a catalyst under real reaction conditions.
Co-reporter:Sandra M. Rountree, Sarah F.R. Taylor, Christopher Hardacre, M. Cristina Lagunas, Paul N. Davey
Applied Catalysis A: General (22 September 2014) Volume 486() pp:94-104
Publication Date(Web):22 September 2014
DOI:10.1016/j.apcata.2014.08.032
Co-reporter:Kevin Morgan, Kieran J. Cole, Alexandre Goguet, Christopher Hardacre, Graham J. Hutchings, Noleen Maguire, Sergiy O. Shekhtman, Stuart H. Taylor
Journal of Catalysis (19 November 2010) Volume 276(Issue 1) pp:38-48
Publication Date(Web):19 November 2010
DOI:10.1016/j.jcat.2010.08.013
The mechanism of CO oxidation reactions over undoped and gold-doped CuMnOX (Hopcalite) catalysts has been examined using a temporal analysis of products (TAP) reactor. Gold doping has been found to increase the activity of the mixed oxide catalyst significantly; however, using consecutive pulsing TAP experiments, the presence of gold was not found to affect the contribution of the Langmuir–Hinshelwood mechanism. Conversely, gold doping was found to promote the Mars van Krevelen mechanism. Using CO and O2 multi-pulse TAP experiments, the gold was found to modify the catalyst surface such that it stores much more oxygen that is active for the CO oxidation. The CO multi-pulse experiments indicated that two distinct types of active oxygen species were found to be involved in the CO oxidation. One type was observed in a similar amount on both doped and undoped catalysts and was associated with mixed oxide, while the second type was only found on the gold-doped catalyst and was therefore clearly associated with the presence of gold on the catalyst surface. The latter was found to be much less active than the oxygen inherent to the oxide but was at a concentration of approximately 10 times larger leading to the enhanced activity observed on gold doping.TAP studies of the CO oxidation reactions over CuMnOX (Hopcalite) catalysts has revealed that the increased activity on doping with gold is due to promotion of the Mars van Krevelen mechanism with little change in the activity associated with the Langmuir-Hinshelwood mechanism.Download high-res image (82KB)Download full-size image
Co-reporter:Sarayute Chansai, Robbie Burch, Christopher Hardacre, John Breen, Frederic Meunier
Journal of Catalysis (19 November 2010) Volume 276(Issue 1) pp:49-55
Publication Date(Web):19 November 2010
DOI:10.1016/j.jcat.2010.08.014
A mechanistic study of the H2-assisted Selective Catalytic Reduction (SCR) of NOx with octane as reductant over a Ag/Al2O3 catalyst was carried out using a modified DRIFTS cell coupled to a mass spectrometer. Using fast transient cycling switching of H2, with a time resolution of a few seconds, it was possible to differentiate potential reaction intermediates from other moieties that are clearly spectator species. Using such a periodic operation mode, effects were uncovered that are normally hidden in conventional transient studies which typically consist of a single transient. In experiments based on a single transient addition of H2 to, or removal of H2 from, the SCR feed, it was found that the changes in the concentrations of gaseous species (products and reactants) were not matched by changes at comparable timescales of the concentration of surface species observed by IR. This observation indicates that the majority of surface species observed by DRIFTS under steady-state reaction conditions are spectators. In contrast, under fast cycling experimental conditions, it was found that a surface isocyanate species had a temporal response that matched that of 15N2. This suggests that some of the isocyanate species observed by infrared spectroscopy could be important intermediates in the hydrogen-assisted SCR reaction although it is emphasised that this may be dependent on the way in which the infrared spectra are obtained. It is concluded that the use of fast transient cycling switching techniques may provide useful mechanistic information under certain circumstances.Examination of the H2-assisted Selective Catalytic Reduction of NOX with octane over a Ag/Al2O3 catalyst using fast transient cycling switching of H2, monitored by DRIFTS and mass spectrometry, suggests that some of the isocyanate species could be important intermediates in this reaction.Download high-res image (68KB)Download full-size image
Co-reporter:Vasile I. Parvulescu, Viorica Parvulescu, Dragos Ciuparu, Christopher Hardacre, Hermenegildo Garcia
Journal of Catalysis (14 February 2011) Volume 278(Issue 1) pp:111-122
Publication Date(Web):14 February 2011
DOI:10.1016/j.jcat.2010.11.021
In constant, search for micro/mesoporous materials, gallium phosphates, have attracted continued interest due to the large pore size reported for some of these solids in comparison with analogous aluminum phosphates. However up to now, the porosity of gallium phosphates collapsed upon template removal or exposure to the ambient moisture. In the present work, we describe high-surface thermally stable mesoporous gallium phosphates synthesized from gallium propoxide and PCl3 and different templating agents such as amines (dipropylamine, piperidine and aminopiperidine) and quaternary ammonium salts (C16H33(CH3)3NBr and C16PyCl). These highly reactive precursors have so far not been used as gallium and phosphate sources for the synthesis of gallophosphates. Conceptually, our present synthetic procedure is based on the fast formation of gallium phosphate nanoparticles via the reaction of gallium propoxide with PCl3 and subsequent construction of the porous material with nanoparticles as building blocks. The organization of the gallophosphate nanoparticles in stable porous structures is effected by the templates. Different experimental procedures varying the molar composition of the sol–gel, pH and the pretreatment of gallium precursor were assayed, most of them leading to satisfactory materials in terms of thermal stability and porosity. In this way, a series of gallium phosphates with surface are above 200 m2 g−1, and narrow pore size from 3 to 6 nm and remarkable thermal stability (up to 550 °C) have been prepared. In some cases, the structure tends to show some periodicity and regularity as determined by XRD. The remarkable stability has allowed us to test the catalytic activity of gallophosphates for the aerobic oxidation of alkylaromatics with notable good results. Our report reopens the interest for gallophosphates in heterogeneous catalysis.Graphical abstractMesoporous gallium phosphates synthesized by hydrolysis of gallium alcoxide in the presence of PCl3 assisted by surfactants form robust structures exhibiting the potential of the aerobic oxidation of toluene.Download high-res image (47KB)Download full-size imageResearch highlights► A new procedure for the synthesis of porous gallophosphates is described. ► Thermally stable porous gallophosphates have been obtained. ► The catalytic activity of porous gallophosphate for the aerobic oxidation of benzylic oxidation has been demonstrated.
Co-reporter:Leanne McLaughlin, Ekaterina Novakova, Robbie Burch, Christopher Hardacre
Applied Catalysis A: General (1 June 2008) Volume 340(Issue 2) pp:
Publication Date(Web):1 June 2008
DOI:10.1016/j.apcata.2008.02.014
The mechanism of the hydrogenation/hydrogenolysis of dinitrodiphenyldisulfides using sulfided NiMo/γAl2O3 catalysts has been examined in detail. Although two routes are possible, the major pathway involves an initial SS bond cleavage followed by reduction of the nitro group. Importantly, the disulfide hydrogenolysis occurs in the absence of the catalyst with the role of the catalyst thought to be to activate the hydrogen and trap the cleaved intermediate as well as facilitate the reduction of the nitro group. Monitoring the mass balance throughout the reaction demonstrates the difficulty in measuring intrinsic kinetics for gas–liquid–solid reactions. Although the mass balance is restored at the end of the reaction, up to 45% of the substrate/products is found to be adsorbed on the catalyst during the reaction.The mechanism of the hydrogenation/hydrogenolysis of dinitrodiphenyldisulfides using sulfided NiMo/γAl2O3 catalysts has been examined in detail. Although two routes are possible, the major pathway involves an initial SS bond cleavage followed by reduction of the nitro group. The disulfide hydrogenolysis occurs in the absence of the catalyst with the role of the catalyst thought to be to activate the hydrogen and trap the cleaved intermediate as well as facilitate the reduction of the nitro group.
Co-reporter:Haresh G. Manyar, Daniel Weber, Helen Daly, Jillian M. Thompson, David W. Rooney, Lynn F. Gladden, E. Hugh Stitt, J. Jose Delgado, Serafin Bernal, Christopher Hardacre
Journal of Catalysis (1 July 2009) Volume 265(Issue 1) pp:80-88
Publication Date(Web):1 July 2009
DOI:10.1016/j.jcat.2009.04.013
A Ru/SiO2 catalyst was investigated for the liquid-phase hydrogenation of butan-2-one to butan-2-ol with water as a medium. Although excellent reactivity was observed, a gradual deactivation of the catalyst was found on recycle of the catalyst. The spent catalyst was characterized by using XRD, XPS, TEM, TPR, CO chemisorption, FTIR and ICP analyses. Formation of Ru(OH)x surface species is proposed to be the main cause of catalyst deactivation with no significant Ru leaching into the reaction mixture. Following catalyst regeneration, up to 85% of the initial catalytic activity could be recovered successfully. Moreover, adsorption of secondary aliphatic alcohols on the catalyst was found to significantly reduce the formation of Ru(OH)x during the reaction, thus protecting the catalyst from deactivation.Deactivation of a Ru/SiO2 catalyst for butan-2-one hydrogenation in water has been found to be associated with the formation of Ru(OH)x; however, this effect may be mitigated by the presence of secondary alcohols which form a hydrophobic surface coating.Download high-res image (101KB)Download full-size image
Co-reporter:Ekaterina K. Novakova, Leanne McLaughlin, Robbie Burch, Paul Crawford, Ken Griffin, Christopher Hardacre, Peijun Hu, David W. Rooney
Journal of Catalysis (1 July 2007) Volume 249(Issue 1) pp:93-101
Publication Date(Web):1 July 2007
DOI:10.1016/j.jcat.2007.04.002
For the first time, the hydrogenation/hydrogenolysis of a range of disulfides has been achieved over a supported palladium catalyst using hydrogen under relatively benign conditions. These unexpected results demonstrate that it is possible to avoid the poisoning of the catalyst by either the nitrogen-containing groups or the sulfur species, allowing both efficient reaction and recycling of the catalyst under the proper conditions (e.g., at low temperatures). A slight loss in activity was found on recycling; however, the catalyst activity can be recovered using hydrogen pretreatment. The reaction mechanism for the hydrogenolysis and hydrogenation of ortho-, meta-, and para-dinitrodiphenyldisulfide to the corresponding aminothiophenol has been elucidated. Density functional theory calculations were used to investigate the adsorption mode of the dinitrodiphenyldisulfides; a clear dependence on adsorption geometry was found regarding whether the molecule is cleaved at the S–S bond before the reduction of the nitro group or vice versa. This study demonstrates the versatility of these catalysts for the hydrogenation/hydrogenolysis of sulfur-containing molecules, which normally are considered poisons, and will extend their use to a new family of substrates.
Co-reporter:Jia-Mei Jin, Tian Sheng, Xiao Lin, Richard Kavanagh, Philip Hamer, Peijun Hu, Christopher Hardacre, Alex Martinez-Bonastre, Jonathan Sharman, David Thompsett and Wen-Feng Lin
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 20) pp:NaN9440-9440
Publication Date(Web):2014/04/01
DOI:10.1039/C4CP00859F
The most active binary PtSn catalyst for direct ethanol fuel cell applications has been studied at 20 °C and 60 °C, using variable temperature electrochemical in situ FTIR. In comparison with Pt, binary PtSn inhibits ethanol dissociation to CO(a), but promotes partial oxidation to acetaldehyde and acetic acid. Increasing the temperature from 20 °C to 60 °C facilitates both ethanol dissociation to CO(a) and then further oxidation to CO2, leading to an increased selectivity towards CO2; however, acetaldehyde and acetic acid are still the main products. Potential-dependent phase diagrams for surface oxidants of OH(a) formation on Pt(111), Pt(211) and Sn modified Pt(111) and Pt(211) surfaces have been determined using density functional theory (DFT) calculations. It is shown that Sn promotes the formation of OH(a) with a lower onset potential on the Pt(111) surface, whereas an increase in the onset potential is found upon modification of the (211) surface. In addition, Sn inhibits the Pt(211) step edge with respect to ethanol C–C bond breaking compared with that found on the pure Pt, which reduces the formation of CO(a). Sn was also found to facilitate ethanol dehydrogenation and partial oxidation to acetaldehyde and acetic acid which, combined with the more facile OH(a) formation on the Pt(111) surface, gives us a clear understanding of the experimentally determined results. This combined electrochemical in situ FTIR and DFT study provides, for the first time, an insight into the long-term puzzling features of the high activity but low CO2 production found on binary PtSn ethanol fuel cell catalysts.
Co-reporter:Marta Falkowska, Sarayute Chansai, Haresh G. Manyar, Lynn F. Gladden, Daniel T. Bowron, Tristan G. A. Youngs and Christopher Hardacre
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 26) pp:NaN17243-17243
Publication Date(Web):2016/03/29
DOI:10.1039/C6CP01494A
Total neutron scattering has been used to follow the hydrogenation of toluene-d8 to methylcyclohexane-d14 over 3 wt% platinum supported on highly ordered mesoporous silica (MCM-41) at 298 K and under 150 mbar D2 pressure. The detailed kinetic information so revealed indicates that liquid reorganisation inside pores is the slowest step of the whole process. Additionally, the results were compared with the reaction performed under 250 mbar D2 pressure as well as with toluene-h8 hydrogenation using D2 at 150 mbar.
Co-reporter:Alex R. Neale, Peilin Li, Johan Jacquemin, Peter Goodrich, Sarah C. Ball, Richard G. Compton and Christopher Hardacre
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 16) pp:NaN11262-11262
Publication Date(Web):2016/03/17
DOI:10.1039/C5CP07160G
This paper reports on the solubility and diffusivity of dissolved oxygen in a series of ionic liquids (ILs) based on the bis{(trifluoromethyl)sulfonyl}imide anion with a range of related alkyl and ether functionalised cyclic alkylammonium cations. Cyclic voltammetry has been used to observe the reduction of oxygen in ILs at a microdisk electrode and chronoamperometric measurements have then been applied to simultaneously determine both the concentration and the diffusion coefficient of oxygen in different ILs. The viscosity of the ILs and the calculated molar volume and free volume are also reported. It is found that, within this class of ILs, the oxygen diffusivity generally increases with decreasing viscosity of the neat IL. An inverse relationship between oxygen solubility and IL free volume is reported for the two IL families implying that oxygen is not simply occupying the available empty space. In addition, it is reported that the introduction of an ether-group into the IL cation structure promotes the diffusivity of dissolved oxygen but reduces the solubility of the gas.
Co-reporter:Nicolas Legagneux, Jean-Marie Basset, Amélie Thomas, Frédéric Lefebvre, Alexandre Goguet, Jacinto Sá and Christopher Hardacre
Dalton Transactions 2009(Issue 12) pp:NaN2240-2240
Publication Date(Web):2009/02/04
DOI:10.1039/B816806G
Dodecatungsto-silicic H4SiW12O40 and -phosphoric acids H3PW12O40 were deposited on silica by a classical impregnation technique. The resulting materials were studied by in situ Raman and infrared spectroscopy, XPS and by solid-state 1H MAS NMR as a function of their dehydroxylation temperature. The data show that in the case of H3PW12O40 three silanol groups are protonated while in the case of H4SiW12O40 at least one acidic proton remains. Upon heating this proton reacts leading to a disordered structure and a broadening of the W–O Raman bands.
Co-reporter:Kathryn Ralphs, Christopher Hardacre and Stuart L. James
Chemical Society Reviews 2013 - vol. 42(Issue 18) pp:NaN7718-7718
Publication Date(Web):2013/06/11
DOI:10.1039/C3CS60066A
Mechanochemical synthesis has the potential to provide more sustainable preparative routes to catalysts than the current multistep solvent-based routes. In this review, the mechanochemical synthesis of catalysts is discussed, with emphasis placed on catalysts for environmental, energy and chemical synthesis applications. This includes the formation of mixed-metal oxides as well as the process of dispersing metals onto solid supports. In most cases the process involves no solvent. Encouragingly, there are several examples where the process is advantageous compared with the more normal solvent-based methods. This can be because of process cost or simplicity, or, notably, where it provides more active/selective catalysts than those made by conventional wet chemical methods. The need for greater, and more systematic, exploration of this currently unconventional approach to catalyst synthesis is highlighted.
Co-reporter:Tristan G. A. Youngs, John D. Holbrey, Claire L. Mullan, Sarah E. Norman, M. Cristina Lagunas, Carmine D'Agostino, Mick D. Mantle, Lynn F. Gladden, Daniel T. Bowron and Christopher Hardacre
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1605-1605
Publication Date(Web):2011/06/09
DOI:10.1039/C1SC00241D
β-D-glucose dissolved in the ionic liquid 1-ethyl-3-methylimidazolium acetate in a 6:1 molar ratio (ionic liquid:glucose) has been studied by neutron scattering, NMR and molecular dynamics simulations. Good agreement was found between simulated neutron scattering profiles generated for isotopically substituted liquid systems and those experimentally determined as well as between simulated and experimental diffusion coefficients obtained by Pulsed Field Gradient NMR spectroscopy. The overriding glucose–ionic liquid interactions in the liquid are hydrogen-bonding between acetate oxygens and sugarhydroxyl groups. The ionic liquid cation was found to play only a minor role in the solvation of the sugar and does not participate in hydrogen-bonding with the sugar to any significant degree. NOESY experiments lend further evidence that there is no direct interaction between sugarhydroxyl groups and acidic hydrogens on the ionic liquid cation.
Co-reporter:Kerri Crossey, Christopher Hardacre and Marie E. Migaud
Chemical Communications 2012 - vol. 48(Issue 98) pp:NaN11971-11971
Publication Date(Web):2012/10/24
DOI:10.1039/C2CC36367D
A range of nucleoside phosphoramidites incorporating small amino substituents have been readily synthesised using ionic liquid stabilised phosphorodiamidites coupled with mechanochemistry.
Co-reporter:Kevin Morgan, Robbie Burch, Muhammad Daous, Juan José Delgado, Alexandre Goguet, Christopher Hardacre, Lachezar A. Petrov and David W. Rooney
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 3) pp:NaN737-737
Publication Date(Web):2014/01/02
DOI:10.1039/C3CY00915G
Methods to control the dispersion of gold in supported heterogeneous catalysts are very valuable due to the strong nanoparticle size dependence on their activity and selectivity towards many reactions. Additionally, the ability to disperse large, inactive gold nanoparticles to smaller nanoparticles provides an opportunity to reactivate, stabilise and increase the lifetime of gold catalysts making them more practical for industrial applications. Previously it has been demonstrated that the use of gas phase iodomethane (J. Am. Chem. Soc., 2009, 131, 6973; Angew. Chem., Int. Ed., 2011, 50, 8912) was able to re-disperse gold from >20 nm particles to dimers and trimers. In the current work, we show that this technique can be applied using less hazardous halohydrocarbons treatments, both in the gas phase and the liquid phase. The ability of these individual halohydrocarbons to re-disperse gold as well as the extent to which leaching occurs is assessed.
Co-reporter:Kathryn Ralphs, Carmine D'Agostino, Robbie Burch, Sarayute Chansai, Lynn F. Gladden, Christopher Hardacre, Stuart L. James, Jonathan Mitchell and Sarah F. R. Taylor
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 2) pp:NaN539-539
Publication Date(Web):2013/12/03
DOI:10.1039/C3CY00945A
The surface modification of a mechanochemically prepared Ag/Al2O3 catalyst compared with catalysts prepared by standard wet impregnated methods has been probed using two-dimensional T1–T2 NMR correlations, H2O temperature programmed desorption (TPD) and DRIFTS. The catalysts were examined for the selective catalytic reduction of NOx using n-octane in the presence and absence of H2. Higher activities were observed for the ball milled catalysts irrespective of whether H2 was added. This higher activity is thought to be related to the increased affinity of the catalyst surface towards the hydrocarbon relative to water, following mechanochemical preparation, resulting in higher concentrations of the hydrocarbon and lower concentrations of water at the surface. DRIFTS experiments demonstrated that surface isocyanate was formed significantly quicker and had a higher surface concentration in the case of the ball milled catalyst which has been correlated with the stronger interaction of the n-octane with the surface. This increased interaction may also be the cause of the reduced activation barrier measured for this catalyst compared with the wet impregnated system. The decreased interaction of water with the surface on ball milling is thought to reduce the effect of site blocking whilst still providing a sufficiently high surface concentration of water to enable effective hydrolysis of the isocyanate to form ammonia and, thereafter, N2.
Co-reporter:Octavian Dumitru Pavel, Peter Goodrich, Liliana Cristian, Simona M. Coman, Vasile I. Pârvulescu and Christopher Hardacre
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 5) pp:NaN2704-2704
Publication Date(Web):2015/02/23
DOI:10.1039/C5CY00011D
The immobilization of a ruthenium complex (Ru2Cl4(az-tpy)2) within a range of supported ionic liquids ([C4C1im]Cl, [C4C1im][NTf2], [C6C1im]Cl, [C4C1pyrr]Br, [C4C1im]Br, [C4C1pyrr]Cl) dispersed silica (SILP) operates as an efficient heterogeneous catalyst in oxidation of long chain linear primary amines to corresponding nitriles. This reaction follows a “green” route using a cheap and easy to handles oxidant (oxygen or air). The conversion was found to be strongly influenced by the alkyl chain length of the amine substrate and the choice of oxidant. No condensation reaction was observed between the starting amines and the selectivity to nitrile is 100%. Moving from a composition of 20 atm N2/5 atm O2 to 5 atm N2/20 atm O2 led to enhancements in the conversion (n-alkylamines) and selectivity (benzonitrile) which have been correlated with an increase of the solubilized oxygen. This was further supported by using different inert gas (nitrogen, helium, argon)/oxygen mixtures indicating that the O2 solubility in the SILP system, has an important effect on conversions and TON in this reaction using SILP catalysts. Experiments performed in the presence of CO2 led to a different behaviour due to the formation of amine-CO2 adducts. The application of the Weisz–Prater criterion confirmed the absence of any diffusional constraints.
Co-reporter:Carmine D'Agostino, Sarayute Chansai, Isabelle Bush, Chensong Gao, Mick D. Mantle, Christopher Hardacre, Stuart L. James and Lynn F. Gladden
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 6) pp:NaN1666-1666
Publication Date(Web):2015/10/30
DOI:10.1039/C5CY01508A
The selective catalytic reduction (SCR) of NOx in the presence of different reducing agents over Ag/Al2O3 prepared by wet impregnation was investigated by probing catalyst activity and using NMR relaxation time analysis to probe the strength of surface interaction of the various reducing agent species and water. The results reveal that the strength of surface interaction of the reducing agent relative to water, the latter present in engine exhausts as a fuel combustion product and, in addition, produced during the SCR reaction, plays an important role in determining catalyst performance. Reducing agents with weak strength of interaction with the catalyst surface, such as hydrocarbons, show poorer catalytic performance than reducing agents with a higher strength of interaction, such as alcohols. This is attributed to the greater ability of oxygenated species to compete with water in terms of surface interaction with the catalyst surface, hence reducing the inhibiting effect of water molecules blocking catalyst sites. The results support the observations of earlier work in that the light off-temperature and maximum NOx conversion and temperature at which that occurs are sensitive to the reducing agent present during reaction, and the proposal that improved catalyst performance is caused by increased adsorption strength of the reducing agent, relative to water, at the catalyst surface. Importantly, the NMR relaxation time analysis approach to characterising the strength of adsorption more readily describes the trends in catalytic behaviour than does a straightforward consideration of the polarity (i.e., relative permittivity) of the reducing agents studied here. In summary, this paper describes a simple approach to characterising the interaction energy of water and reducing agent so as to aid the selection of reducing agent and catalyst to be used in SCR conversions.
Co-reporter:Alexandre Goguet, Christopher Hardacre, Burapat Inceesungvorn, Kevin Morgan and Sergiy O. Shekhtman
Catalysis Science & Technology (2011-Present) 2011 - vol. 1(Issue 5) pp:NaN767-767
Publication Date(Web):2011/02/14
DOI:10.1039/C0CY00075B
All TAP micro-reactor configurations contain inert particles which are used so that the catalyst zone can be maintained under isothermal conditions. Even on “inert” particles adsorption will occur to some degree; however, the extent to which this occurs has a critical influence on the analysis of the TAP data. In many cases the assumption that there is no interaction between probe molecules and inert particles is required as reversible adsorption over inert material is problematic when the TAP model has to be solved. Moreover, as the TAP pulse response experiments are designed to be conducted within the Knudsen diffusion regime, central to TAP data analysis is the characterization of the diffusional transport of reagent molecules through the micro-reactor, which is achieved via “diffusion only” experiments over an inert one zone packing. Therefore, if there are any processes occurring in addition to Knudsen diffusion over the inert material, such as reversible adsorption, it is important to factor these into the analysis. If these additional processes are not included, the entire data analysis would be questionable. The current work discloses the development of a function which accounts for the adsorption over the inert material, so that the TAP data analysis can be accurately determined. This newly developed analysis method has been exemplified using the selective reduction of oxygen in a hydrogen rich ethylene feed over silver catalysts as a case study.
Co-reporter:Haresh G. Manyar, Richard Morgan, Kevin Morgan, Bo Yang, P. Hu, Jakub Szlachetko, Jacinto Sá and Christopher Hardacre
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 6) pp:NaN1500-1500
Publication Date(Web):2013/04/09
DOI:10.1039/C3CY00031A
The change in the Pt electronic structure following the adsorption of an α,β-unsaturated aldehyde and ketone was followed by in situ HERFD-XANES in the liquid phase. The resulting shift in the Pt Fermi energy is in good agreement with the molecule adsorption energy trends calculated by DFT and provides insight into the reaction selectivity.
Co-reporter:Kevin Morgan, Burapat Inceesungvorn, Alexandre Goguet, Christopher Hardacre, Frederic C. Meunier and Sergiy O. Shekhtman
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 10) pp:NaN2133-2133
Publication Date(Web):2012/06/25
DOI:10.1039/C2CY20295F
Catalysts currently employed for the polymerization of ethylene have previously been found to deactivate in the presence of oxygen. It is, therefore, important that oxygen is removed from the ethylene feedstock prior to the polymerization. The Ag/γ–Al2O3 catalyst exhibits excellent activity and selectivity toward oxygen reduction with hydrogen in the presence of ethylene. TAP vacuum pulse experiments have been utilised to understand the catalytic behaviour of the Ag/γ–Al2O3 catalyst. TAP multi-pulse experiments have determined the types of active sites that are found on the Ag/γ–Al2O3 catalyst, and the intrinsic activity of these sites. The lifetime of the reactive adsorbed oxygen intermediate has also been determined through TAP consecutive pulse experiments. Multi-pulse and consecutive pulse data have been combined with ethylene adsorption/desorption rate constants to provide an overview of the Ag/γ–Al2O3 catalyst system.
Co-reporter:V. K. Puthiyapura, D. J. L. Brett, A. E. Russell, W. F. Lin and C. Hardacre
Chemical Communications 2015 - vol. 51(Issue 69) pp:NaN13415-13415
Publication Date(Web):2015/07/20
DOI:10.1039/C5CC04188K
Pt and PtSn catalysts were studied for n-butanol electro-oxidation at various temperatures. PtSn showed a higher activity towards butanol electro-oxidation compared to Pt in acidic media. The onset potential for n-butanol oxidation on PtSn is ∼520 mV lower than that found on Pt, and significantly lower activation energy was found for PtSn compared with that for Pt.
Co-reporter:Christopher Hardacre, Haifeng Huang, Stuart L. James, Marie E. Migaud, Sarah E. Norman and William R. Pitner
Chemical Communications 2011 - vol. 47(Issue 20) pp:NaN5848-5848
Publication Date(Web):2011/04/18
DOI:10.1039/C1CC11025J
Ionic liquids have been used in combination with ball milling on a range of chlorophosphoramidite reagents to phosphitylate nucleosides and 2-deoxynucleosides. The enhanced stability offered by the ionic liquid mediated processes combined with efficient mass transfer induced by ball milling has enabled excellent yields to be obtained even when using small dialkyl amino groups as well as the more commonly used diisopropylamino protection.
Co-reporter:Burapat Inceesungvorn, Frederic C. Meunier, Chris Hardacre, Robbie Burch and Ken Griffin
Chemical Communications 2008(Issue 46) pp:NaN6214-6214
Publication Date(Web):2008/10/21
DOI:10.1039/B813470G
The selective reduction of molecular oxygen with excess H2 in the presence of alkenes was achieved successfully for the first time: silver supported on alumina catalysts exhibited full conversion of O2 at temperature as low as 50 °C, while the conversion of ethene or propene remained essentially zero up to 250 °C.
Co-reporter:Sofiane Saouane, Sarah E. Norman, Christopher Hardacre and Francesca P. A. Fabbiani
Chemical Science (2010-Present) 2013 - vol. 4(Issue 3) pp:NaN1280-1280
Publication Date(Web):2013/01/02
DOI:10.1039/C2SC21959J
The solid-state polymorphism of the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate, [bmim][PF6], has been investigated via low-temperature and high-pressure crystallisation experiments. The samples have been characterised by single-crystal X-ray diffraction, optical microscopy and Raman spectroscopy. The solid-state phase behaviour of the compound is confirmed and clarified with respect to previous phase diagrams. The structures of the previously reported γ-form, which essentially exhibits a G′T cation conformation, as well as those of the elusive β- and α-forms, are reported. Crystals of the β-phase are twinned and the structure is heavily disordered; the cation conformation in this form is predominantly TT, though significant contributions from other less frequently encountered conformers are also observed at low temperature and high pressure. The cation conformation in the α-form is GT; the presence of the G′T conformer at 193 K in this phase can be eliminated on cooling to 100 K. Whilst X-ray structural data are overall in good agreement with previous interpretations based on Raman and NMR studies, they also reveal a more subtle interplay of intermolecular interactions, which give rise to a wider range of conformers than previously considered.
Co-reporter:Tristan G. A. Youngs, Haresh Manyar, Daniel T. Bowron, Lynn F. Gladden and Christopher Hardacre
Chemical Science (2010-Present) 2013 - vol. 4(Issue 9) pp:NaN3489-3489
Publication Date(Web):2013/06/17
DOI:10.1039/C3SC51477C
Using benzene hydrogenation over Pt/SiO2 as an industrially-relevant example, we show that state-of-the-art neutron total scattering methods spanning a wide Q-range now permit relevant time-resolved catalytic chemistry to be probed directly in situ within the pore of the catalyst. The method gives access to the reaction rates on both nanometric and atomic length scales, whilst simultaneously providing an atomistic structural viewpoint on the reaction mechanism itself.
Co-reporter:Haresh G. Manyar, Cristina Paun, Rashidah Pilus, David W. Rooney, Jillian M. Thompson and Christopher Hardacre
Chemical Communications 2010 - vol. 46(Issue 34) pp:NaN6281-6281
Publication Date(Web):2010/07/27
DOI:10.1039/C0CC01365J
Selective hydrogenation of carboxylic acids to alcohols and alkanes has been achieved under remarkably mild reaction temperatures and H2 pressures (333 K, 0.5 MPa) using Pt/TiO2 catalyst.

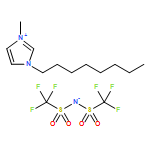
![1,1'-[oxybis(methylene)]bis(4-chlorobenzene)](http://img.cochemist.com/ccimg/56500/56428-00-3.png)
![1,1'-[oxybis(methylene)]bis(4-chlorobenzene)](http://img.cochemist.com/ccimg/56500/56428-00-3_b.png)




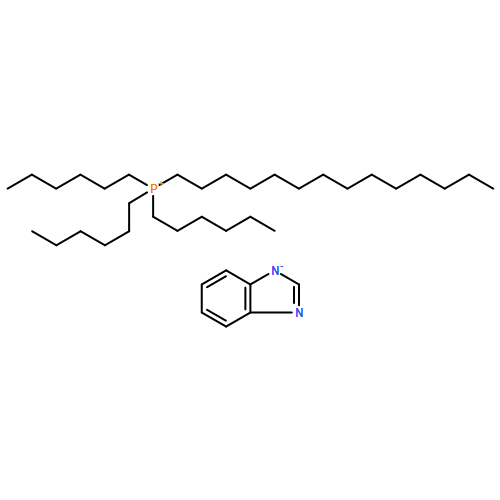



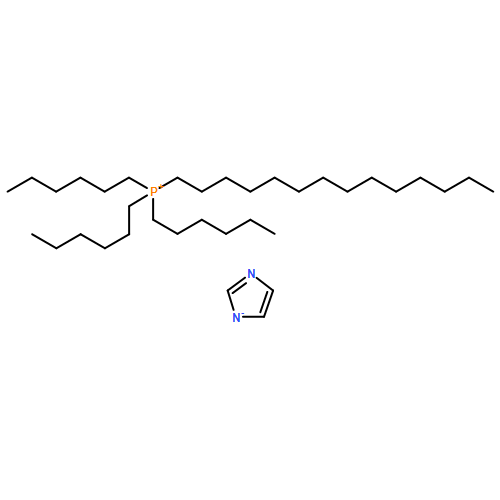


![1,2-Benzenediol, 3-[(1S)-2,2,2-trifluoro-1-hydroxyethyl]-](http://img.cochemist.com/ccimg/499300/499202-24-3.png)
![1,2-Benzenediol, 3-[(1S)-2,2,2-trifluoro-1-hydroxyethyl]-](http://img.cochemist.com/ccimg/499300/499202-24-3_b.png)
![3,5-CYCLOHEXADIENE-1,2-DIOL, 3-[(1R)-2,2,2-TRIFLUORO-1-HYDROXYETHYL]-, (1S,2R)- (9CI)](http://img.cochemist.com/ccimg/499300/499202-15-2.png)
![3,5-CYCLOHEXADIENE-1,2-DIOL, 3-[(1R)-2,2,2-TRIFLUORO-1-HYDROXYETHYL]-, (1S,2R)- (9CI)](http://img.cochemist.com/ccimg/499300/499202-15-2_b.png)




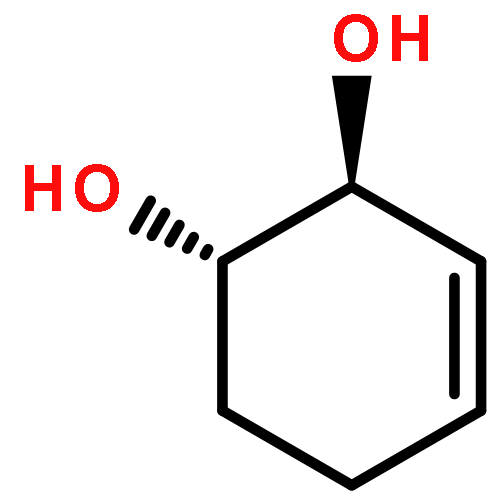






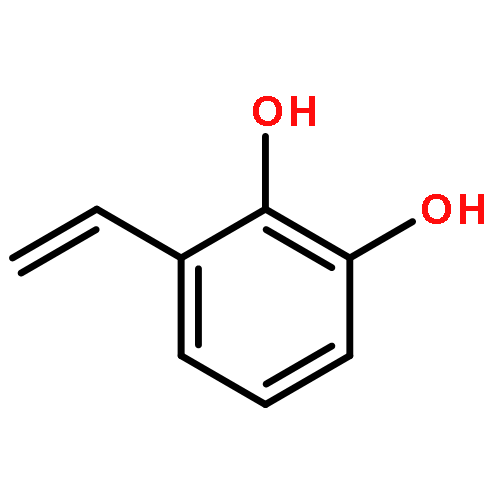
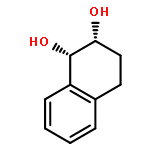
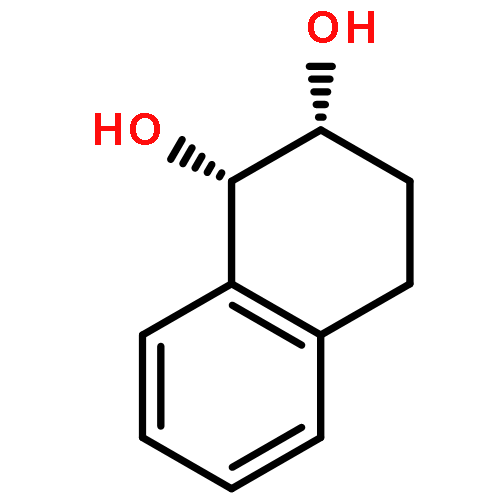
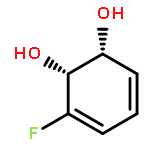
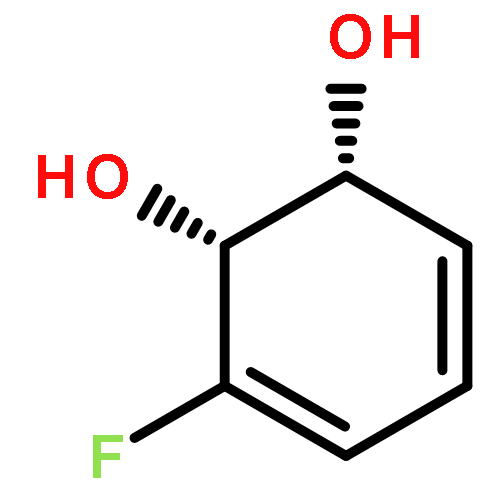
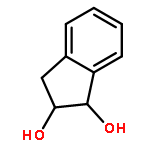
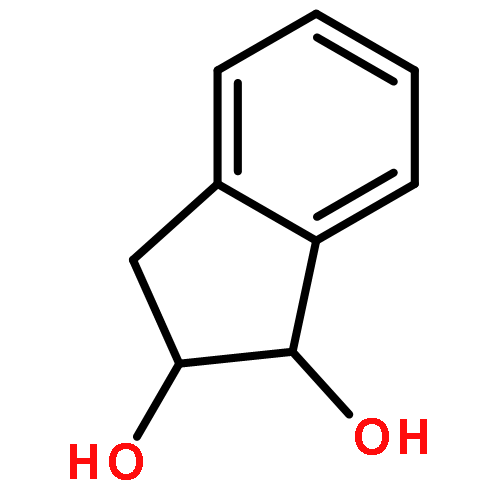




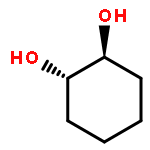
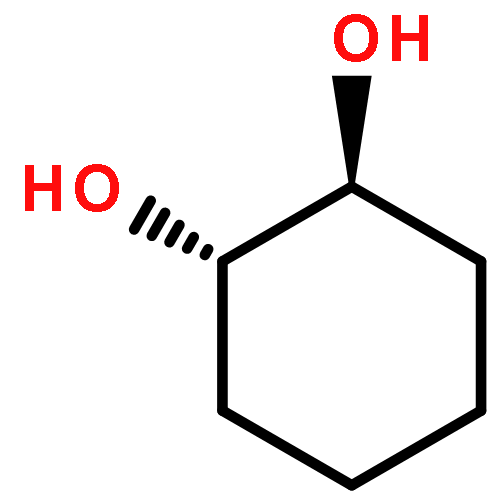
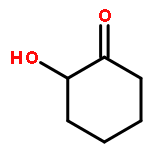








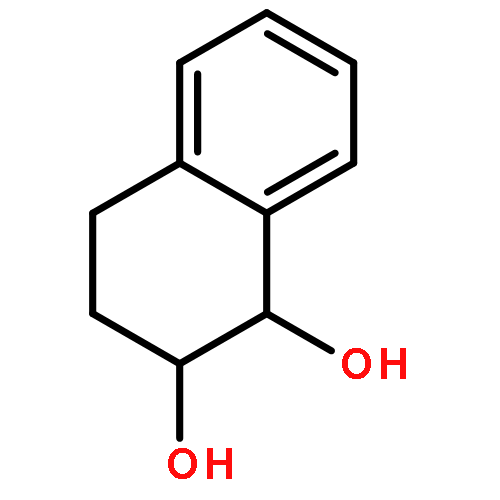



















![[1,1'-Biphenyl]-2,3-diol](http://img.cochemist.com/ccimg/1200/1133-63-7.png)
![[1,1'-Biphenyl]-2,3-diol](http://img.cochemist.com/ccimg/1200/1133-63-7_b.png)