Co-reporter:Yasemin Sahbaz, Tri-Hung Nguyen, Leigh Ford, Claire L. McEvoy, Hywel D. Williams, Peter J. Scammells, and Christopher J. H. Porter
Molecular Pharmaceutics November 6, 2017 Volume 14(Issue 11) pp:3669-3669
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.molpharmaceut.7b00442
This study aimed to transform weakly acidic poorly water-soluble drugs (PWSD) into ionic liquids (ILs) to promote solubility in, and the utility of, lipid-based formulations. Ionic liquids (ILs) were formed directly from tolfenamic acid (Tolf), meclofenamic acid, diclofenac, and ibuprofen by pairing with lipophilic counterions. The drug-ILs were obtained as liquids or low melting solids and were significantly more soluble (either completely miscible or highly soluble) in lipid based, self-emulsifying drug delivery systems (SEDDS) when compared to the equivalent free acid. In vivo assessment of a SEDDS lipid solution formulation of Tolf didecyldimethylammonium salt and the same formulation of Tolf free acid at low dose (18 mg/kg, where the free acid was soluble in the SEDDS), resulted in similar absorption profiles and overall exposure. At high dose (100 mg/kg), solution SEDDS formulations of the Tolf ILs (didecyldimethylammonium, butyldodecyldimethylammonium or didecylmethylammonium salts) were possible, but the lower lipid solubility of Tolf free acid dictated that administration of the free acid was only possible as a suspension in the SEDDS formulation or as an aqueous suspension. Under these conditions, total drug plasma exposure was similar for the IL formulations and the free acid, but the plasma profiles were markedly different, resulting in flatter, more prolonged exposure profiles and reduced Cmax for the IL formulations. Isolation of a weakly acidic drug as an IL may therefore provide advantage as it allows formulation as a solution SEDDS rather than a lipid suspension, and in some cases may provide a means of slowing or sustaining absorption. The current studies compliment previous studies with weakly basic PWSD and demonstrate that transformation into highly lipophilic ILs is also possible for weakly acidic compounds.Keywords: drug delivery; in vitro digestion; ionic liquid; lipid formulation; lipolysis; poorly water-soluble drug; SEDDS;
Co-reporter:Shane M. Devine;Christopher A. MacRaild;Raymond S. Norton;Peter J. Scammells
MedChemComm (2010-Present) 2017 vol. 8(Issue 1) pp:13-20
Publication Date(Web):2017/01/26
DOI:10.1039/C6MD00495D
Malaria continues to frustrate humanity's attempts to eradicate this deadly disease. Although gains have been made over the last 15 years, drug resistance to malaria continues to be a major concern. The lack of new antimalarials with novel mechanisms of action continues to challenge the scientific community to find innovative targets to combat this persistent disease. One such target, apical membrane antigen 1 (AMA1), is an essential protein that helps the parasite invade host erythrocytes. Recently, a number of efforts have focused on the druggability of this target, aiming to block the interactions of AMA1 that mediate invasion of host cells. This review covers recent progress in drug discovery targeting this crucial protein–protein interaction in malaria.
Co-reporter:Manuela Jörg, Alisa Glukhova, Alaa Abdul-Ridha, Elizabeth A. Vecchio, Anh T. N. Nguyen, Patrick M. Sexton, Paul J. White, Lauren T. May, Arthur Christopoulos, and Peter J. Scammells
Journal of Medicinal Chemistry 2016 Volume 59(Issue 24) pp:11182-11194
Publication Date(Web):December 1, 2016
DOI:10.1021/acs.jmedchem.6b01561
The A1 adenosine receptor (A1AR) is an important G protein-coupled receptor that regulates a range of physiological functions. Herein we report the discovery of novel irreversible agonists acting at the A1AR, which have the potential to serve as useful research tools for studying receptor structure and function. A series of novel adenosine derivatives bearing electrophilic substituents was synthesized, and four compounds, 8b, 15a, 15b, and 15d, were shown to possess similar potency and efficacy to the reference high efficacy agonist, NECA, in an assay of ERK1/2 phosphorylation assay. Insensitivity to antagonist addition in a real-time, label-free, xCELLigence assay was subsequently used to identify compounds that likely mediated their agonism through an irreversible interaction with the A1AR. Of these compounds, 15b and 15d were more directly validated as irreversible agonists of the A1AR using membrane-based [3H]DPCPX and [35S]GTPγS binding experiments.
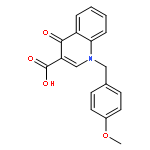
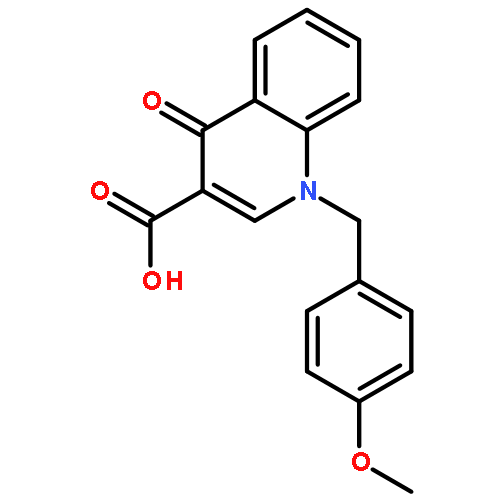
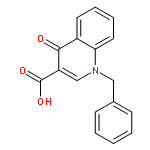
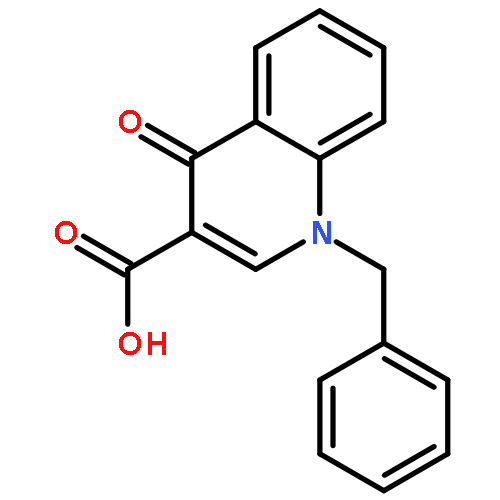
Co-reporter:Shailesh N. Mistry; Manuela Jörg; Herman Lim; Natalie B. Vinh; Patrick M. Sexton; Ben Capuano; Arthur Christopoulos; J. Robert Lane;Peter J. Scammells
Journal of Medicinal Chemistry 2016 Volume 59(Issue 1) pp:388-409
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.jmedchem.5b01562
Positive allosteric modulators (PAMs) of the M1 muscarinic acetylcholine receptor (M1 mAChR) are a promising strategy for the treatment of the cognitive deficits associated with diseases including Alzheimer’s and schizophrenia. Herein, we report the design, synthesis, and characterization of a novel family of M1 mAChR PAMs. The most active compounds of the 4-phenylpyridin-2-one series exhibited comparable binding affinity to the reference compound, 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (BQCA) (1), but markedly improved positive cooperativity with acetylcholine, and retained exquisite selectivity for the M1 mAChR. Furthermore, our pharmacological characterization revealed ligands with a diverse range of activities, including modulators that displayed both high intrinsic efficacy and PAM activity, those that showed no detectable agonism but robust PAM activity and ligands that displayed robust allosteric agonism but little modulatory activity. Thus, the 4-phenylpyridin-2-one scaffold offers an attractive starting point for further lead optimization.
Co-reporter:Shailesh N. Mistry, Herman Lim, Manuela Jörg, Ben Capuano, Arthur Christopoulos, J. Robert Lane, and Peter J. Scammells
ACS Chemical Neuroscience 2016 Volume 7(Issue 5) pp:647
Publication Date(Web):February 18, 2016
DOI:10.1021/acschemneuro.6b00018
Benzoquinazolinone 1 is a positive allosteric modulator (PAM) of the M1 muscarinic acetylcholine receptor (mAChR), which is significantly more potent than the prototypical PAM, 1-(4-methoxybenzyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (BQCA). In this study, we explored the structural determinants that underlie the activity of 1 as a PAM of the M1 mAChR. We paid particular attention to the importance of the tricyclic scaffold of compound 1, for the activity of the molecule. Complete deletion of the peripheral fused benzene ring caused a significant decrease in affinity and binding cooperativity with acetylcholine (ACh). This loss of affinity was rescued with the addition of either one or two methyl groups in the 7- and/or 8-position of the quinazolin-4(3H)-one core. These results demonstrate that the tricyclic benzo[h]quinazolin-4(3H)-one core could be replaced with a quinazolin-4(3H)-one core and maintain functional affinity. As such, the quinazolin-4(3H)-one core represents a novel scaffold to further explore M1 mAChR PAMs with improved physicochemical properties.Keywords: M1 muscarinic acetylcholine receptor; positive allosteric modulator
Co-reporter:Manuela Jörg; Lauren T. May; Frankie S. Mak; Kiew Ching K. Lee; Neil D. Miller; Peter J. Scammells;Ben Capuano
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:718-738
Publication Date(Web):December 9, 2014
DOI:10.1021/jm501254d
A relatively new strategy in drug discovery is the development of dual acting ligands. These molecules are potentially able to interact at two orthosteric binding sites of a heterodimer simultaneously, possibly resulting in enhanced subtype selectivity, higher affinity, enhanced or modified physiological response, and reduced reliance on multiple drug administration regimens. In this study, we have successfully synthesized a series of classical heterobivalent ligands as well as a series of more integrated and “drug-like” dual acting molecules, incorporating ropinirole as a dopamine D2 receptor agonist and ZM 241385 as an adenosine A2A receptor antagonist. The best compounds of our series maintained the potency of the original pharmacophores at both receptors (adenosine A2A and dopamine D2). In addition, the integrated dual acting ligands also showed promising results in preliminary blood–brain barrier permeability tests, whereas the classical heterobivalent ligands are potentially more suited as pharmacological tools.
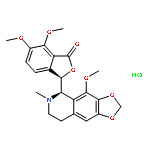
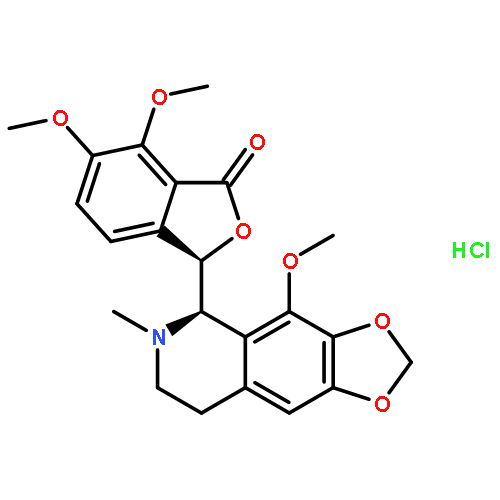
Co-reporter:Aaron DeBono; Ben Capuano;Peter J. Scammells
Journal of Medicinal Chemistry 2015 Volume 58(Issue 15) pp:5699-5727
Publication Date(Web):March 26, 2015
DOI:10.1021/jm501180v
Many nitrogen-moiety containing alkaloids derived from plant origins are bioactive and play a significant role in human health and emerging medicine. Noscapine, a phthalideisoquinoline alkaloid derived from Papaver somniferum, has been used as a cough suppressant since the mid 1950s, illustrating a good safety profile. Noscapine has since been discovered to arrest cells at mitosis, albeit with moderately weak activity. Immunofluorescence staining of microtubules after 24 h of noscapine exposure at 20 μM elucidated chromosomal abnormalities and the inability of chromosomes to complete congression to the equatorial plane for proper mitotic separation ( Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1601−1606 ). A number of noscapine analogues possessing various modifications have been described within the literature and have shown significantly improved antiprolific profiles for a large variety of cancer cell lines. Several semisynthetic antimitotic alkaloids are emerging as possible candidates as novel anticancer therapies. This perspective discusses the advancing understanding of noscapine and related analogues in the fight against malignant disease.
Co-reporter:Shailesh N. Mistry; Jeremy Shonberg; Christopher J. Draper-Joyce; Carmen Klein Herenbrink; Mayako Michino; Lei Shi; Arthur Christopoulos; Ben Capuano; Peter J. Scammells;J. Robert Lane
Journal of Medicinal Chemistry 2015 Volume 58(Issue 17) pp:6819-6843
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.jmedchem.5b00585
Recently, we have demonstrated that N-((trans)-4-(2-(7-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide (SB269652) (1) adopts a bitopic pose at one protomer of a dopamine D2 receptor (D2R) dimer to negatively modulate the binding of dopamine at the other protomer. The 1H-indole-2-carboxamide moiety of 1 extends into a secondary pocket between the extracellular ends of TM2 and TM7 within the D2R protomer. To target this putative allosteric site, we generated and characterized fragments that include and extend from the 1H-indole-2-carboxamide moiety of 1. N-Isopropyl-1H-indole-2-carboxamide (3) displayed allosteric pharmacology and sensitivity to mutations of the same residues at the top of TM2 as was observed for 1. Using 3 as an “allosteric lead”, we designed and synthesized an extensive fragment library to generate novel SAR and identify N-butyl-1H-indole-2-carboxamide (11d), which displayed both increased negative cooperativity and affinity for the D2R. These data illustrate that fragmentation of extended compounds can expose fragments with purely allosteric pharmacology.
Co-reporter:Luke S. Schembri; Leigh A. Stoddart; Stephen J. Briddon; Barrie Kellam; Meritxell Canals; Bim Graham;Peter J. Scammells
Journal of Medicinal Chemistry 2015 Volume 58(Issue 24) pp:9754-9767
Publication Date(Web):December 3, 2015
DOI:10.1021/acs.jmedchem.5b01664
Fluorescently labeled ligands are useful pharmacological research tools for studying receptor localization, trafficking, and signaling processes via fluorescence imaging. They are also employed in fluorescent binding assays. This study is centered on the design, synthesis, and pharmacological evaluation of fluorescent probes for the opioid receptors, for which relatively few non-peptidic fluorescent probes currently exist. The known μ-opioid receptor (MOR) partial agonist, buprenorphine, was structurally elaborated to include an amidoalkylamine linker moiety that was coupled with a range of fluorophores to afford new fluorescent probes. All compounds proved to be selective MOR antagonists. Confocal fluorescence microscopy studies revealed that the probe incorporating a sulfonated cyanine-5 fluorophore was the most appropriate for imaging studies. This ligand was subsequently employed in an automated fluorescence-based competition binding assay, allowing the pKi values of several well-known opioid ligands to be determined. Thus, this new probe will prove useful in future studies of MOR receptor pharmacology.
Co-reporter:Yasemin Sahbaz; Hywel D. Williams; Tri-Hung Nguyen; Jessica Saunders; Leigh Ford; Susan A. Charman; Peter J. Scammells;Christopher J. H. Porter
Molecular Pharmaceutics 2015 Volume 12(Issue 6) pp:1980-1991
Publication Date(Web):April 23, 2015
DOI:10.1021/mp500790t
Absorption after oral administration is a requirement for almost all drug products but is a challenge for drugs with intrinsically low water solubility. Here, the weakly basic, poorly water-soluble drugs (PWSDs) itraconazole, cinnarizine, and halofantrine were converted into lipophilic ionic liquids to facilitate incorporation into lipid-based formulations and integration into lipid absorption pathways. Ionic liquids were formed via metathesis reactions of the hydrochloride salt of the PWSDs with a range of lipophilic counterions. The resultant active pharmaceutical ingredient–ionic liquids (API–ILs) were liquids or low melting point solids and either completely miscible or highly soluble in lipid based, self-emulsifying drug delivery systems (SEDDS) comprising mixtures of long or medium chain glycerides, surfactants such as Kolliphor-EL and cosolvents such as ethanol. They also readily incorporated into the colloids formed in intestinal fluids during lipid digestion. Itraconazole docusate or cinnarizine decylsulfate API-ILs were subsequently dissolved in long chain lipid SEDDS at high concentration, administered to rats and in vivo exposure assessed. The data were compared to control formulations based on the same SEDDS formulations containing the same concentrations of drug as the free base, but in this case as a suspension (since the solubility of the free base in the SEDDS was much lower than the API–ILs). For itraconazole, comparison was also made to a physical mixture of itraconazole free base and sodium docusate in the same SEDDS formulation. For both drugs plasma exposure was significantly higher for the API-IL containing formulations (2-fold for cinnarizine and 20-fold for itraconazole), when compared to the suspension formulations (or the physical mixture in the case of itraconazole) at the same dose. The liquid SEDDS formulations, made possible by the use of the API–ILs, also provide advantages in dose uniformity, capsule filling, and stability compared to similar suspension formulations. The data suggest that the formation of lipophilic ionic liquids provides a means of increasing dissolved-drug loading in lipid based formulations and thereby promoting the exposure of poorly water-soluble drugs after oral administration.
Co-reporter:Natalie B. Vinh;Dr. Shane M. Devine;Dr. Lenka Munoz;Dr. Renae M. Ryan;Dr. Bing H. Wang; Henry Krum;Dr. David K. Chalmers;Dr. Jamie S. Simpson; Peter J. Scammells
ChemistryOpen 2015 Volume 4( Issue 1) pp:56-64
Publication Date(Web):
DOI:10.1002/open.201402076
Abstract
p38α mitogen-activated protein kinase (MAPK) plays a role in several cellular processes and consequently has been a therapeutic target in inflammatory diseases, cancer, and cardiovascular disease. A number of known p38α MAPK inhibitors contain vicinal 4-fluorophenyl/4-pyridyl rings connected to either a 5- or 6-membered heterocycle. In this study, a small library of substituted thiophene-based compounds bearing the vicinal 4-fluorophenyl/4-pyridyl rings was designed using computational docking as a visualisation tool. Compounds were synthesised and evaluated in a fluorescence polarisation binding assay. The synthesised analogues had a higher binding affinity to the active phosphorylated form of p38α MAPK than the inactive nonphosphorylated form of the protein. 4-(2-(4-fluorophenyl)thiophen-3-yl)pyridine had a Ki value of 0.6 μm to active p38α MAPK highlighting that substitution of the core ring to a thiophene retains affinity to the enzyme and can be utilised in p38α MAPK inhibitors. This compound was further elaborated using a substituted phenyl ring in order to probe the second hydrophobic pocket. Many of these analogues exhibited low micromolar affinity to active p38α MAPK. The suppression of neonatal rat fibroblast collagen synthesis was also observed suggesting that further development of these compounds may lead to potential therapeutics having cardioprotective properties.
Co-reporter:Hywel D. Williams, Yasemin Sahbaz, Leigh Ford, Tri-Hung Nguyen, Peter J. Scammells and Christopher J. H. Porter
Chemical Communications 2014 vol. 50(Issue 14) pp:1688-1690
Publication Date(Web):17 Dec 2013
DOI:10.1039/C3CC48650H
Ionic liquids (ILs) have been exploited to improve the absorption of poorly water-soluble drugs. Custom-made ILs solubilized very high quantities of the poorly water-soluble drugs, danazol and itraconazole, and maintained drug solubilization under simulated gastro-intestinal conditions. A danazol-containing self-emulsifying IL formulation gave rise to 4.3-fold higher exposure than the crystalline drug and prolonged exposure compared with a lipid formulation.
Co-reporter:Briana J. Davie ; Celine Valant ; Jonathan M. White ; Patrick M. Sexton ; Ben Capuano ; Arthur Christopoulos ;Peter J. Scammells
Journal of Medicinal Chemistry 2014 Volume 57(Issue 12) pp:5405-5418
Publication Date(Web):May 23, 2014
DOI:10.1021/jm500556a
Activation of the M1 muscarinic acetylcholine receptor (mAChR) is a prospective treatment for alleviating cognitive decline experienced in central nervous system (CNS) disorders. Current therapeutics indiscriminately enhance the activity of the endogenous neurotransmitter ACh, leading to side effects. BQCA is a positive allosteric modulator and allosteric agonist at the M1 mAChR that has high subtype selectivity and is a promising template from which to generate higher affinity, more pharmacokinetically viable drug candidates. However, to efficiently guide rational drug design, the binding site of BQCA needs to be conclusively elucidated. We report the synthesis and pharmacological validation of BQCA analogues designed to bind irreversibly to the M1 mAChR. One analogue in particular, 11, can serve as a useful structural probe to confirm the location of the BQCA binding site; ideally, by co-crystallization with the M1 mAChR. Furthermore, this ligand may also be used as a pharmacological tool with a range of applications.
Co-reporter:Shailesh N. Mistry ; Nyssa Drinkwater ; Chiara Ruggeri ; Komagal Kannan Sivaraman ; Sasdekumar Loganathan ; Sabine Fletcher ; Marcin Drag ; Alessandro Paiardini ; Vicky M. Avery ; Peter J. Scammells ;Sheena McGowan
Journal of Medicinal Chemistry 2014 Volume 57(Issue 21) pp:9168-9183
Publication Date(Web):October 9, 2014
DOI:10.1021/jm501323a
Plasmodium parasites, the causative agents of malaria, have developed resistance to most of our current antimalarial therapies, including artemisinin combination therapies which are widely described as our last line of defense. Antimalarial agents with a novel mode of action are urgently required. Two Plasmodium falciparum aminopeptidases, PfA-M1 and PfA-M17, play crucial roles in the erythrocytic stage of infection and have been validated as potential antimalarial targets. Using compound-bound crystal structures of both enzymes, we have used a structure-guided approach to develop a novel series of inhibitors capable of potent inhibition of both PfA-M1 and PfA-M17 activity and parasite growth in culture. Herein we describe the design, synthesis, and evaluation of a series of hydroxamic acid-based inhibitors and demonstrate the compounds to be exciting new leads for the development of novel antimalarial therapeutics.
Co-reporter:Briana J. Davie, Patrick M. Sexton, Ben Capuano, Arthur Christopoulos, and Peter J. Scammells
ACS Chemical Neuroscience 2014 Volume 5(Issue 10) pp:902
Publication Date(Web):September 4, 2014
DOI:10.1021/cn500173x
The field of G protein-coupled receptor drug discovery has benefited greatly from the structural and functional insights afforded by photoactivatable ligands. One G protein-coupled receptor subfamily for which photoactivatable ligands have been developed is the muscarinic acetylcholine receptor family, though, to date, all such ligands have been designed to target the orthosteric (endogenous ligand) binding site of these receptors. Herein we report the synthesis and pharmacological investigation of a novel photoaffinity label, MIPS1455 (4), designed to bind irreversibly to an allosteric site of the M1 muscarinic acetylcholine receptor; a target of therapeutic interest for the treatment of cognitive deficits. MIPS1455 may be a valuable molecular tool for further investigating allosteric interactions at this receptor.Keywords: allosteric; BQCA; cognition; Muscarinic; photoactivatable; photoaffinity
Co-reporter:Manuela Jörg, Agnieszka A. Kaczor, Frankie S. Mak, Kiew Ching K. Lee, Antti Poso, Neil D. Miller, Peter J. Scammells and Ben Capuano
MedChemComm 2014 vol. 5(Issue 7) pp:891-898
Publication Date(Web):06 May 2014
DOI:10.1039/C4MD00066H
Herein, we report the development of novel functionalized congeners of ropinirole toward the design of pharmacological tools to probe structural requirements at the dopamine D2 receptor. Subsequently, we have used the functionalized amine congener 11 and synthesized and pharmacologically evaluated a series of homobivalent ligands of ropinirole with designated spacer lengths ranging from 14 to 30 atoms. The most potent homobivalent ligands (22-, 26- and 30-atom spacers) showed approximately 20- to 80-fold greater potency (EC50 = 3.9, 6.2 and 14 nM, respectively) than ropinirole (304 nM) in a [35S]GTPγS functional assay. Molecular modeling studies suggest that the observed increase in potency of the homobivalent ligands is possibly due to a bitopic binding mode involving the orthosteric site and an allosteric interaction at the dopamine D2 receptor protomer rather than bridging interactions at two orthosteric sites across a dopamine D2 receptor dimer. This research has the potential to advance the development of structurally related bitopic ligands, biomarkers such as radioligands and fluorescently labeled probes, and furnish new homo- and heterobivalent ligands towards a better understanding of the dopamine D2 receptor and potential novel treatment for Parkinson's disease.
Co-reporter:Shane M. Devine, Lauren T. May and Peter J. Scammells
MedChemComm 2014 vol. 5(Issue 2) pp:192-196
Publication Date(Web):20 Dec 2013
DOI:10.1039/C3MD00364G
A series of N6-substituted 2-aminoadenosine-5′-N-methylcarboxamides were synthesized from the versatile intermediate, O6-(benzotriazol-1-yl)-2-amino-2′,3′-O-isopropylideneinosine-5′-N-methylcarboxamide (1). These compounds were evaluated as A3 adenosine receptor agonists in terms of their potency and receptor sub-type selectivity in a cAMP accumulation assay and a number of potent and selective compounds were identified. One such compound, 2-amino-N6-(3-chlorobenzyl)adenosine-5′-N-methylcarboxamide (3i), was over 500-fold selective for the A3AR over the other three adenosine receptor subtypes. This represents a significantly greater selectivity profile compared with the prototypical IB-MECA.
Co-reporter:Manuela Jörg;Stephen Headey;Peter J. Scammells;Ben Capuano
Magnetic Resonance in Chemistry 2014 Volume 52( Issue 11) pp:715-718
Publication Date(Web):
DOI:10.1002/mrc.4114
Keywords:
- NMR;
- 1H;
- 13C;
- oxindole;
- ropinirole;
- concentration-dependent effects;
- solvent-dependent effects
Co-reporter:Aaron J. DeBono;Dr. Sarah J. Mistry;Jinhan Xie;Divya Muthiah;Jackson Phillips;Dr. Sabatino Ventura;Dr. Richard Callaghan; Colin W. Pouton;Dr. Ben Capuano; Peter J. Scammells
ChemMedChem 2014 Volume 9( Issue 2) pp:399-410
Publication Date(Web):
DOI:10.1002/cmdc.201300395
Abstract
Noscapine, a phthalideisoquinoline alkaloid derived from Papaver somniferum, is a well-known antitussive drug that has a relatively safe in vitro toxicity profile. Noscapine is also known to possess weak anticancer efficacy, and since its discovery, efforts have been made to design derivatives with improved potency. Herein, the synthesis of a series of noscapine analogues, which have been modified in the 6′, 9′, 1 and 7-positions, is described. In a previous study, replacement of the naturally occurring N-methyl group in the 6′-position with an N-ethylaminocarbonyl was shown to promote cell-cycle arrest and cytotoxicity against three cancer cell lines. Here, this modification has been combined with other structural changes that have previously been shown to improve anticancer activity, namely halo substitution in the 9′-position, regioselective O-demethylation to reveal a free phenol in the 7-position, and reduction of the lactone to the corresponding cyclic ether in the 1-position. The incorporation of new aryl substituents in the 9′-position was also investigated. The study identified interesting new compounds able to induce G2/M cell-cycle arrest and that possess cytotoxic activity against the human prostate carcinoma cell line PC3, the human breast adenocarcinoma cell line MCF-7, and the human pancreatic epithelioid carcinoma cell line PANC-1. In particular, the ethyl urea cyclic ether noscapinoids and a compound containing a 6′-ethylaminocarbonyl along with 9′-chloro, 7-hydroxy and lactone moieties exhibited the most promising biological activities, with EC50 values in the low micromolar range against all three cancer cell lines, and these derivatives warrant further investigation.
Co-reporter:Shailesh N. Mistry ; Celine Valant ; Patrick M. Sexton ; Ben Capuano ; Arthur Christopoulos ;Peter J. Scammells
Journal of Medicinal Chemistry 2013 Volume 56(Issue 12) pp:5151-5172
Publication Date(Web):May 29, 2013
DOI:10.1021/jm400540b
Established therapy in Alzheimer’s disease involves potentiation of the endogenous orthosteric ligand, acetylcholine, at the M1 muscarinic receptors found in higher concentrations in the cortex and hippocampus. Adverse effects, due to indiscriminate activation of other muscarinic receptor subtypes, are common. M1 muscarinic positive allosteric modulators/allosteric agonists such as BQCA offer an attractive solution, being exquisitely M1-selective over other muscarinic subtypes. A common difficulty with allosteric ligands is interpreting SAR, based on composite potency values derived in the presence of fixed concentration of agonist. In reality these values encompass multiple pharmacological parameters, each potentially and differentially sensitive to structural modification of the ligand. We report novel BQCA analogues which appear to augment ligand affinity for the receptor (pKB), intrinsic efficacy (τB), and both binding (α) and functional (β) cooperativity with acetylcholine. Ultimately, development of such enriched SAR surrounding allosteric modulators will provide insight into their mode of action.
Co-reporter:Briana J. Davie, Arthur Christopoulos, and Peter J. Scammells
ACS Chemical Neuroscience 2013 Volume 4(Issue 7) pp:1026
Publication Date(Web):May 9, 2013
DOI:10.1021/cn400086m
Since the cholinergic hypothesis of memory dysfunction was first reported, extensive research efforts have focused on elucidating the mechanisms by which this intricate system contributes to the regulation of processes such as learning, memory, and higher executive function. Several cholinergic therapeutic targets for the treatment of cognitive deficits, psychotic symptoms, and the underlying pathophysiology of neurodegenerative disorders, such as Alzheimer’s disease and schizophrenia, have since emerged. Clinically approved drugs now exist for some of these targets; however, they all may be considered suboptimal therapeutics in that they produce undesirable off-target activity leading to side effects, fail to address the wide variety of symptoms and underlying pathophysiology that characterize these disorders, and/or afford little to no therapeutic effect in subsets of patient populations. A promising target for which there are presently no approved therapies is the M1 muscarinic acetylcholine receptor (M1 mAChR). Despite avid investigation, development of agents that selectively activate this receptor via the orthosteric site has been hampered by the high sequence homology of the binding site between the five muscarinic receptor subtypes and the wide distribution of this receptor family in both the central nervous system (CNS) and the periphery. Hence, a plethora of ligands targeting less structurally conserved allosteric sites of the M1 mAChR have been investigated. This Review aims to explain the rationale behind allosterically targeting the M1 mAChR, comprehensively summarize and critically evaluate the M1 mAChR allosteric ligand literature to date, highlight the challenges inherent in allosteric ligand investigation that are impeding their clinical advancement, and discuss potential methods for resolving these issues.Keywords: allosteric ligands; Alzheimer’s disease; cognitive deficits; M1 mAChR; memory; schizophrenia
Co-reporter:Manuela Jörg, Jeremy Shonberg, Frankie S. Mak, Neil D. Miller, Elizabeth Yuriev, Peter J. Scammells, Ben Capuano
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3427-3433
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.070
Growing evidence has suggested a role in targeting the adenosine A2A receptor for the treatment of Parkinson’s disease. The literature compounds KW 6002 (2) and ZM 241385 (5) were used as a starting point from which a series of novel ligands targeting the adenosine A2A receptor were synthesized and tested in a recombinant human adenosine A2A receptor functional assay. In order to further explore these molecules, we investigated the biological effects of assorted linkers attached to different positions on selected adenosine A2A receptor antagonists, and assessed their potential binding modes using molecular docking studies. The results suggest that linking from the phenolic oxygen of selected adenosine A2A receptor antagonists is relatively well tolerated due to the extension towards extracellular space, and leads to the potential of attaching further functionality from this position.
Co-reporter:Jeremy Shonberg, J. Robert Lane, Peter J. Scammells and Ben Capuano
MedChemComm 2013 vol. 4(Issue 9) pp:1290-1296
Publication Date(Web):18 Jul 2013
DOI:10.1039/C3MD00154G
Bivalent ligands represent useful tools to investigate the phenomenon of GPCR dimerization. We synthesized bivalent ligands based on (R)-apomorphine with variations in spacer length, and assessed these compounds in functional and binding assays at the dopamine D2 receptor. The results present novel SAR for bivalent ligands targeting the D2R, and identify a relationship for spacer length with ligand potency, efficacy and affinity.
Co-reporter:Nicholas Barlow; Stephen P. Baker; Peter J. Scammells
ChemMedChem 2013 Volume 8( Issue 12) pp:2036-2046
Publication Date(Web):
DOI:10.1002/cmdc.201300286
Abstract
Heterobivalent ligands that possess pharmacophores designed to interact with both the A1 adenosine receptor (A1AR) and the β2 adrenergic receptor (β2AR) were prepared. More specifically, these ligands contain an adenosine moiety that is linked via its N6-position to the amino group of the saligenin-substituted ethanolamine moiety present in the well-known β2AR agonist, salbutamol. The affinities of these ligands were determined at both receptors and found to vary with linker length and composition. With all compounds, affinity and functional potencies were found to have selectivity for the A1AR over the β2AR. In all cases, cAMP accumulation (a β2AR-mediated response) was mainly observed when the A1AR was blocked or its function decreased by pertussis toxin or chronic agonist treatment. This suggests that heterobivalent compounds for receptors that mediate opposite responses might be useful for elucidating the mechanisms of receptor cross-talk and how this interaction, in terms of responsiveness, may change under pathophysiological conditions.
Co-reporter:Nicholas E. Hausler ; Shane M. Devine ; Fiona M. McRobb ; Lyndon Warfe ; Colin W. Pouton ; John M. Haynes ; Steven E. Bottle ; Paul J. White ;Peter J. Scammells
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3521-3534
Publication Date(Web):March 20, 2012
DOI:10.1021/jm300206u
A series of adenosine-5′-N-alkylcarboxamides and N6-(2,2-diphenylethyl)adenosine-5′-N-alkylcarboxamides bearing antioxidant moieties in the 2-position were synthesized from the versatile intermediate, O6-(benzotriazol-1-yl)-2-fluoro-2′,3′-O-isopropylideneinosine-5′-N-alkylcarboxamide (1). These compounds were evaluated as A2A adenosine receptor (A2AR) agonists in a cAMP accumulation assay, and a number of potent and selective agonists were identified. Three of these compounds were evaluated further in an ischemic injury cell survival assay and a reactive oxygen species (ROS) production assay whereby 15b and 15c were shown to reduce ROS activity and cell death due to ischemia.
Co-reporter:Celine Valant ; Luigi Aurelio ; Shane M. Devine ; Trent D. Ashton ; Jonathan M. White ; Patrick M. Sexton ; Arthur Christopoulos ;Peter J. Scammells
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2367-2375
Publication Date(Web):February 8, 2012
DOI:10.1021/jm201600e
A series of novel 2-amino-3-benzoylthiophenes (2A3BTs) were screened using a functional assay of A1R mediated phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in intact CHO cells to identify potential agonistic effects as well as the ability to allosterically modulate the activity of the orthosteric agonist, R-PIA. Two derivatives, 8h and 8i, differing only in terms of the absence or presence of an electron-withdrawing group on the benzoyl moiety of the 2A3BT scaffold, were identified as biased allosteric agonists and positive allosteric modulators of agonist function at the adenosine A1 receptor (A1R) in two different functional assays. Our findings indicate that subtle structural variations can promote functionally distinct receptor conformational states.
Co-reporter:Gaik B. Kok and Peter J. Scammells
RSC Advances 2012 vol. 2(Issue 30) pp:11318-11325
Publication Date(Web):25 Sep 2012
DOI:10.1039/C2RA21693K
Significantly improved reaction conditions for the synthesis of oxycodone (1), via the conventional two-step process involving oxidation of thebaine (3) into 14-hydroxycodeinone (5) and the subsequent reduction of 5via catalytic hydrogenation, are reported. Employing the hydrochloride salt of thebaine (3) in the oxidation step, in place of the more traditionally used free base form of this opiate, now provides 5 in high yield and purity. For the reduction step, aqueous acetic acid has typically been employed as the solvent. However, this was found to generate varying amounts of 14-hydroxydihydrocodeine (16) as the dominant by-product. Instead, using 5% Pd/BaSO4 as the catalyst and MeOH as the solvent completely eliminated the formation of 16, giving oxycodone (1) in high yield and purity, without the need to purify the intermediates. These improved conditions have also proved effective in the synthesis of other 14-hydroxyopiates, such as oxymorphone (2) and N-noroxymorphone (9). In the latter case, a high overall yield was achieved by starting from N-nororipavine (10), without the need to employ protecting groups.
Co-reporter:Joshua I. Gosling; Stephen P. Baker;Dr. John M. Haynes; Michael Kassiou; Colin W. Pouton;Lyndon Warfe;Dr. Paul J. White; Peter J. Scammells
ChemMedChem 2012 Volume 7( Issue 7) pp:1191-1201
Publication Date(Web):
DOI:10.1002/cmdc.201200208
Abstract
A concise synthesis of a series of N6-substituted adenosines with bicyclo[3.2.1]octan-6-yl and polycyclic N6-substituents has been developed. The adenosine A1 receptor (A1R) affinity and potency of these compounds was initially assessed using competitive binding assays and cyclic adenosine monophosphate (cAMP) accumulation assays in DDT1 MF-2 cells. The potency and receptor subtype selectivity of selected examples was further evaluated by measuring their effects on cAMP accumulation at all human adenosine receptor subtypes expressed in CHO cells. The results of these assays indicated that all of the synthesised N6-substituted adenosines are full agonists at A1R and activate this receptor selectively over the other adenosine receptor subtypes. The two standout compounds in terms of potency were N6-(3-thiabicyclo[3.2.1]octan-6-yl)adenosine and N6-(cubanylmethyl)adenosine with EC50 values at human A1R of 2.3 nM and 1.1 nM, respectively. The cubanylmethyl derivative in particular proved to be highly receptor subtype selective. These two compounds were further evaluated in a simulated ischaemia model in cultured cardiomyoblasts, where they were found to impart protective effects under hypoxic conditions that resulted in a significant reduction in cell death.
Co-reporter:Aaron J. DeBono;Jin Han Xie;Dr. Sabatino Ventura; Colin W. Pouton;Dr. Ben Capuano; Peter J. Scammells
ChemMedChem 2012 Volume 7( Issue 12) pp:2122-2133
Publication Date(Web):
DOI:10.1002/cmdc.201200365
Abstract
Noscapine is a phthalideisoquinoline alkaloid isolated from the opium poppy Papaver somniferum. It has long been used as an antitussive agent, but has more recently been found to possess microtubule-modulating properties and anticancer activity. Herein we report the synthesis and pharmacological evaluation of a series of 6′-substituted noscapine derivatives. To underpin this structure–activity study, an efficient synthesis of N-nornoscapine and its subsequent reduction to the cyclic ether derivative of N-nornoscapine was developed. Reaction of the latter with a range of alkyl halides, acid chlorides, isocyanates, thioisocyanates, and chloroformate reagents resulted in the formation of the corresponding N-alkyl, N-acyl, N-carbamoyl, N-thiocarbamoyl, and N-carbamate derivatives, respectively. The ability of these compounds to inhibit cell proliferation was assessed in cell-cycle cytotoxicity assays using prostate cancer (PC3), breast cancer (MCF-7), and colon cancer (Caco-2) cell lines. Compounds that showed activity in the cell-cycle assay were further evaluated in cell viability assays using PC3 and MCF-7 cells.
Co-reporter:Luigi Aurelio, Bernard L. Flynn and Peter J. Scammells
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 13) pp:4886-4902
Publication Date(Web):29 Mar 2011
DOI:10.1039/C0OB01156H
2-Amino-3-acylthiophenes are known to allosterically modulate the A1adenosine receptor and are also used as intermediates in the synthesis of therapeutic agents and pharmacophores such as thienoazepines and thienopyrimidines. The N-alkylation of 2-aminothiophenes has been notoriously difficult to accomplish under mild conditions and there are very few examples of N-alkylated 2-aminothiophenes in the literature, all of which use forcing conditions to effect the alkylation. Here we describe the synthesis of such compounds under mild conditions utilising 2-carbamoylamino and 2-acylamino-3-acylthiophenes with caesium carbonate, and tetrabutylammonium iodide in DMF.
Co-reporter:Gaik B. Kok and Peter J. Scammells
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 4) pp:1008-1011
Publication Date(Web):24 Dec 2010
DOI:10.1039/C0OB01021A
Further investigations into the direct synthesis of N-nororipavine from oripavine using iron powder under non-classical Polonovski conditions have been conducted. The stoichiometry, solvents and iron oxidation rates were found to have a dramatic effect on the rate of N-demethylation as well as product yield. Herein, we also present high-yield access to the N-demethylated product simply by employing stainless steel rather than iron powder as redox catalyst. To our knowledge, this is the first time stainless steel has been used to moderate the redox chemistry of iron in organic synthesis.
Co-reporter:Shane M. Devine ;Peter J. Scammells
European Journal of Organic Chemistry 2011 Volume 2011( Issue 6) pp:1092-1098
Publication Date(Web):
DOI:10.1002/ejoc.201001395
Abstract
An efficient synthesis of 2-halo-O6-(benzotriazol-1-yl)-substituted purine nucleosides has been accomplished via (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP)-mediated coupling and subsequent halogenation via diazotization of the 2-amino group of various protected guanosines and directly from guanosine itself. These products are amenable to substitution and coupling reactions in the 2- and 6-positions and, accordingly, provide efficient access to highly functionalized purine nucleosides.
Co-reporter:Leigh Ford;Farzad Atefi;Robert D. Singer;Peter J. Scammells
European Journal of Organic Chemistry 2011 Volume 2011( Issue 5) pp:942-950
Publication Date(Web):
DOI:10.1002/ejoc.201001468
Abstract
Alkylpyridinium (10a, R = Me and 10b, R = Et) and tetraalkylphosphonium (19) ionic liquids which possess ether functionality were prepared and evaluated as solvents and co-solvents for Grignard reactions. Interestingly, reduction of the starting aldehydes to the corresponding primary alcohol was the favored pathway when reactions were conducted in pyridinium ILs 10a, b in the absence an ethereal co-solvent. However, the same reactions in the phosphonium IL 19 afforded the expected Grignard products in good yield in cases where diethyl ether was present as a co-solvent.
Co-reporter:Leigh Ford, Jitendra R. Harjani, Farzad Atefi, M. Teresa Garcia, Robert D. Singer and Peter J. Scammells
Green Chemistry 2010 vol. 12(Issue 10) pp:1783-1789
Publication Date(Web):07 Sep 2010
DOI:10.1039/C0GC00082E
A range of ionic liquids (ILs) containing a pyridinium cation were synthesised and their biodegradability was evaluated using the CO2 headspace test (ISO 14593). ILs bearing a 1-(2-hydroxyethyl) side chain were prepared from either pyridine or nicotinic acid derivatives. These ILs showed high levels of biodegradation under aerobic conditions and can be classified as ‘readily biodegradable’. In contrast, pyridinium ILs with methyl or ethyl ether side chains showed significantly lower levels of biodegradability in the same test. Biodegradation studies on a range of novel ILs with acetal and carbamate functionalities, as well as thiazolium-based salts, also showed low levels of mineralization.
Co-reporter:Luigi Aurelio ; Celine Valant ; Bernard L. Flynn ; Patrick M. Sexton ; Jonathan M. White ; Arthur Christopoulos ;Peter J. Scammells
Journal of Medicinal Chemistry 2010 Volume 53(Issue 18) pp:6550-6559
Publication Date(Web):August 31, 2010
DOI:10.1021/jm1008538
2-Amino-3-benzoylthiophenes (2A3BTs) have been widely reported to act as allosteric enhancers (AEs) at the A1 adenosine receptor (A1AR). Herein we describe the synthesis of a series of 1-aminoindeno[1,2-c]thiophen-8-ones and a series of (2-aminoindeno[2,1-b]thiophen-3-yl)(phenyl)methanones as conformationally rigid analogues of the 2A3BTs. These compounds were screened using a functional assay of A1AR-mediated phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in intact Chinese hamster ovary (CHO) cells to identify both potential agonistic effects as well as the ability to allosterically modulate the activity of the orthosteric agonist, N6-(R-phenylisopropyl)adenosine (R-PIA). All of the 1-aminoindeno[1,2-c]thiophen-8-ones (14a−c and 17a−f) proved either to be inactive or behaved as antagonists in the functional assay. However, the (2-aminoindeno[2,1-b]thiophen-3-yl)(phenyl)methanones with para-chloro substitution (compounds 25b, 25d, and 25f) did significantly augment the R-PIA response, indicating a positive allosteric effect.
Co-reporter:Hywel D. Williams, Yasemin Sahbaz, Leigh Ford, Tri-Hung Nguyen, Peter J. Scammells and Christopher J. H. Porter
Chemical Communications 2014 - vol. 50(Issue 14) pp:NaN1690-1690
Publication Date(Web):2013/12/17
DOI:10.1039/C3CC48650H
Ionic liquids (ILs) have been exploited to improve the absorption of poorly water-soluble drugs. Custom-made ILs solubilized very high quantities of the poorly water-soluble drugs, danazol and itraconazole, and maintained drug solubilization under simulated gastro-intestinal conditions. A danazol-containing self-emulsifying IL formulation gave rise to 4.3-fold higher exposure than the crystalline drug and prolonged exposure compared with a lipid formulation.
Co-reporter:Luigi Aurelio, Bernard L. Flynn and Peter J. Scammells
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 13) pp:NaN4902-4902
Publication Date(Web):2011/03/29
DOI:10.1039/C0OB01156H
2-Amino-3-acylthiophenes are known to allosterically modulate the A1adenosine receptor and are also used as intermediates in the synthesis of therapeutic agents and pharmacophores such as thienoazepines and thienopyrimidines. The N-alkylation of 2-aminothiophenes has been notoriously difficult to accomplish under mild conditions and there are very few examples of N-alkylated 2-aminothiophenes in the literature, all of which use forcing conditions to effect the alkylation. Here we describe the synthesis of such compounds under mild conditions utilising 2-carbamoylamino and 2-acylamino-3-acylthiophenes with caesium carbonate, and tetrabutylammonium iodide in DMF.
Co-reporter:Gaik B. Kok and Peter J. Scammells
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 4) pp:NaN1011-1011
Publication Date(Web):2010/12/24
DOI:10.1039/C0OB01021A
Further investigations into the direct synthesis of N-nororipavine from oripavine using iron powder under non-classical Polonovski conditions have been conducted. The stoichiometry, solvents and iron oxidation rates were found to have a dramatic effect on the rate of N-demethylation as well as product yield. Herein, we also present high-yield access to the N-demethylated product simply by employing stainless steel rather than iron powder as redox catalyst. To our knowledge, this is the first time stainless steel has been used to moderate the redox chemistry of iron in organic synthesis.






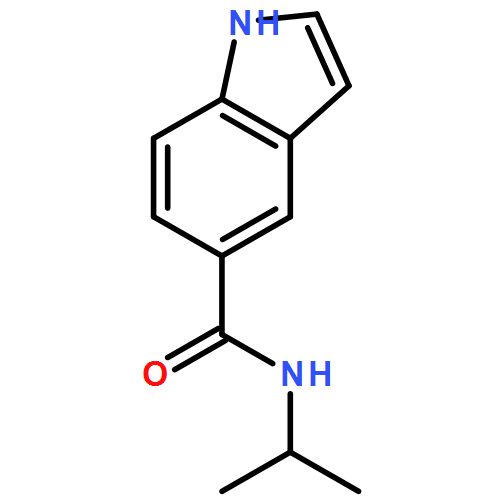
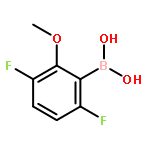
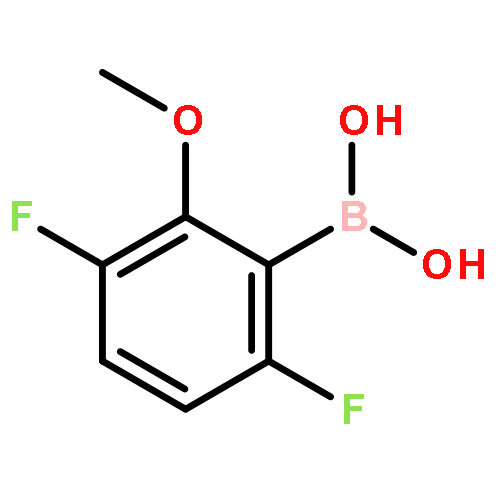
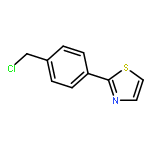
![4-chloro-7-ethyl-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/866700/866648-70-6.png)
![4-chloro-7-ethyl-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/866700/866648-70-6_b.png)
![Oxazole, 2-[4-(bromomethyl)phenyl]-](http://img.cochemist.com/ccimg/866300/866261-56-5.png)
![Oxazole, 2-[4-(bromomethyl)phenyl]-](http://img.cochemist.com/ccimg/866300/866261-56-5_b.png)
![3-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/839800/839712-12-8.png)
![3-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-1,1-dimethylurea](http://img.cochemist.com/ccimg/839800/839712-12-8_b.png)
![4-Aza-1-azoniabicyclo[2.2.2]octane, 1-hexyl-, bromide](http://img.cochemist.com/ccimg/783400/783354-56-3.png)
![4-Aza-1-azoniabicyclo[2.2.2]octane, 1-hexyl-, bromide](http://img.cochemist.com/ccimg/783400/783354-56-3_b.png)


![<br>1-(1'-(2-Methylbenzyl)-[1,4'-bipiperidin]-4-yl)-1H-benzo[d]imidazol-2(3H)-o ne](http://img.cochemist.com/ccimg/634700/634616-95-8.png)
![<br>1-(1'-(2-Methylbenzyl)-[1,4'-bipiperidin]-4-yl)-1H-benzo[d]imidazol-2(3H)-o ne](http://img.cochemist.com/ccimg/634700/634616-95-8_b.png)


![1-[3-(4-BUTYLPIPERIDIN-1-YL)PROPYL]-3,4-DIHYDROQUINOLIN-2-ONE](http://img.cochemist.com/ccimg/560100/560085-11-2.png)
![1-[3-(4-BUTYLPIPERIDIN-1-YL)PROPYL]-3,4-DIHYDROQUINOLIN-2-ONE](http://img.cochemist.com/ccimg/560100/560085-11-2_b.png)






![4-[2-(4-FLUOROPHENYL)THIOPHEN-3-YL]PYRIDINE](http://img.cochemist.com/ccimg/443700/443685-67-4.png)
![4-[2-(4-FLUOROPHENYL)THIOPHEN-3-YL]PYRIDINE](http://img.cochemist.com/ccimg/443700/443685-67-4_b.png)






![6,7-DIHYDRO-[1,4]DIOXINO[2,3-F][1,3]BENZOTHIAZOL-2-AMINE](http://img.cochemist.com/ccimg/313300/313223-82-4.png)
![6,7-DIHYDRO-[1,4]DIOXINO[2,3-F][1,3]BENZOTHIAZOL-2-AMINE](http://img.cochemist.com/ccimg/313300/313223-82-4_b.png)

![Boronic acid, [3-(benzoylamino)phenyl]-](http://img.cochemist.com/ccimg/252700/252663-49-3.png)
![Boronic acid, [3-(benzoylamino)phenyl]-](http://img.cochemist.com/ccimg/252700/252663-49-3_b.png)
![7-Hydroxy-5-oxo-5H-thiazolo[3,2-a]pyrimidine-6-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/224400/224313-88-6.png)
![7-Hydroxy-5-oxo-5H-thiazolo[3,2-a]pyrimidine-6-carboxylic acid methyl ester](http://img.cochemist.com/ccimg/224400/224313-88-6_b.png)








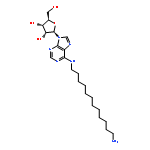
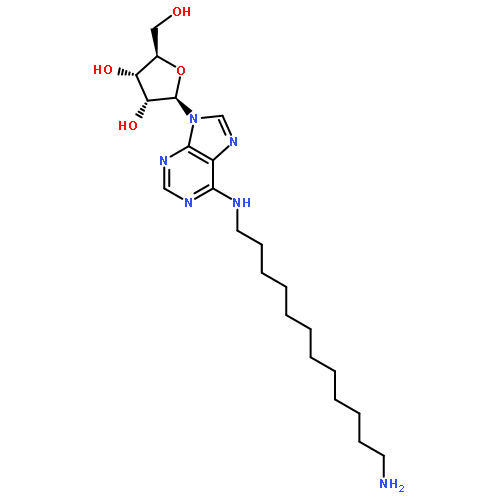
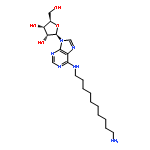
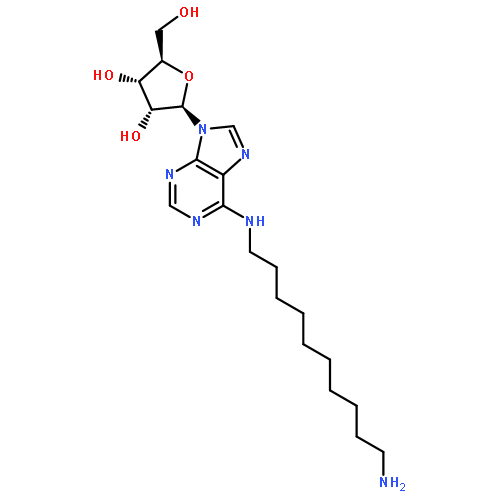











![Phenol,3-[(3S)-1-propyl-3-piperidinyl]-](/data/chemimg/984600/85966-89-8.png)
![Phenol,3-[(3S)-1-propyl-3-piperidinyl]-](/data/chemimg/984600/85966-89-8_b.png)


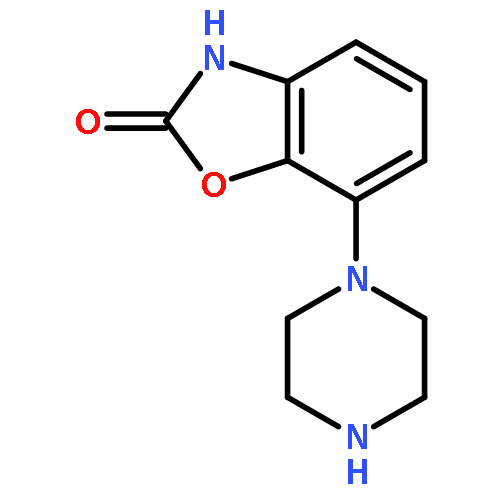
![7-Aminobenzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/81300/81282-60-2.png)
![7-Aminobenzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/81300/81282-60-2_b.png)
![2-Propenoic acid, 3-[1,1'-biphenyl]-4-yl-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/81100/81069-41-2.png)
![2-Propenoic acid, 3-[1,1'-biphenyl]-4-yl-, ethyl ester, (2E)-](http://img.cochemist.com/ccimg/81100/81069-41-2_b.png)
![N'-[(1E)-1-(2-THIENYL)PROPYLIDENE]NICOTINOHYDRAZIDE](http://img.cochemist.com/ccimg/75300/75240-91-4.png)
![N'-[(1E)-1-(2-THIENYL)PROPYLIDENE]NICOTINOHYDRAZIDE](http://img.cochemist.com/ccimg/75300/75240-91-4_b.png)
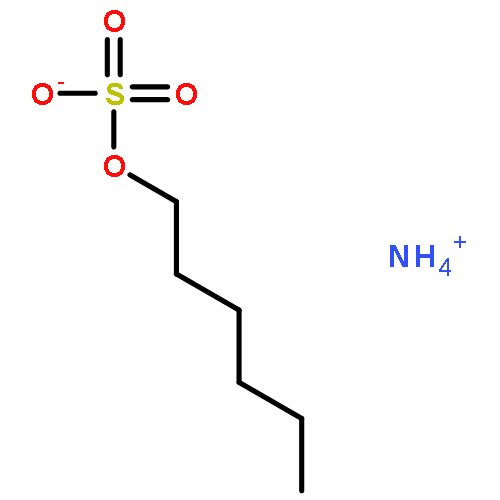
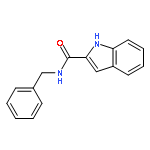
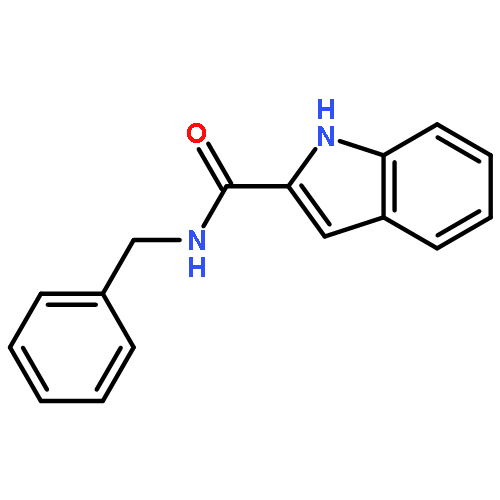
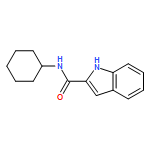

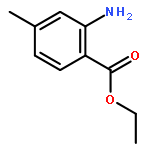
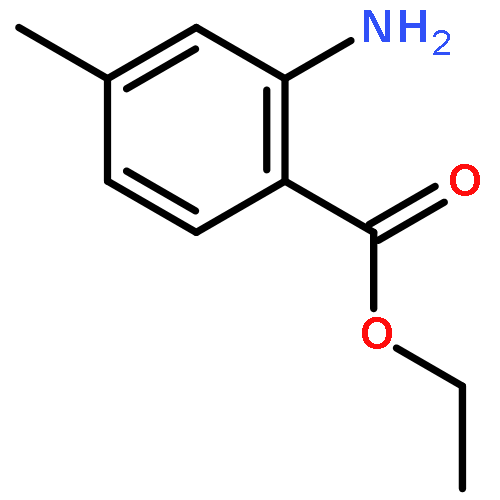
![N6,N6-Dimethylbenzo[d]thiazole-2,6-diamine](http://img.cochemist.com/ccimg/64400/64334-41-4.png)
![N6,N6-Dimethylbenzo[d]thiazole-2,6-diamine](http://img.cochemist.com/ccimg/64400/64334-41-4_b.png)

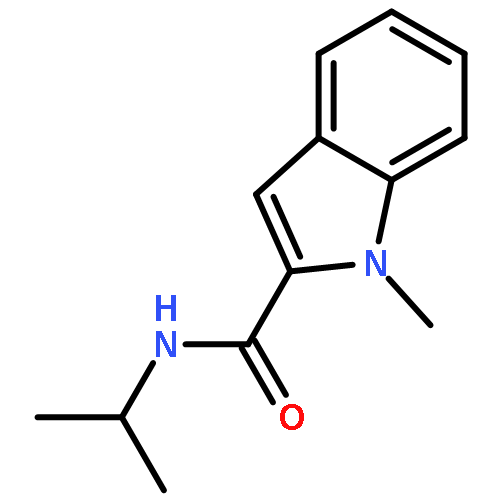




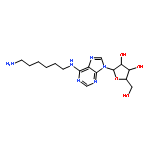
![6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-6-oxido-7,8-dihydro-5H-[1,3]dioxolo[4,5-g]isoquinolin-6-ium-5-yl]-3H-isobenzofuran-1-one](http://img.cochemist.com/ccimg/54400/54383-36-7.png)
![6,7-dimethoxy-3-[(5S)-4-methoxy-6-methyl-6-oxido-7,8-dihydro-5H-[1,3]dioxolo[4,5-g]isoquinolin-6-ium-5-yl]-3H-isobenzofuran-1-one](http://img.cochemist.com/ccimg/54400/54383-36-7_b.png)



![2-Propenoic acid, 3-[1,1'-biphenyl]-4-yl-, ethyl ester](http://img.cochemist.com/ccimg/40800/40795-99-1.png)
![2-Propenoic acid, 3-[1,1'-biphenyl]-4-yl-, ethyl ester](http://img.cochemist.com/ccimg/40800/40795-99-1_b.png)
![4-AZA-1-AZONIABICYCLO[2.2.2]OCTANE, 1-METHYL-](http://img.cochemist.com/ccimg/40500/40473-48-1.png)
![4-AZA-1-AZONIABICYCLO[2.2.2]OCTANE, 1-METHYL-](http://img.cochemist.com/ccimg/40500/40473-48-1_b.png)


![2-[6-(2-aminoethylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/35700/35662-04-5.png)
![2-[6-(2-aminoethylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/35700/35662-04-5_b.png)




![2-Butenoic acid,4-[(4-methylphenyl)amino]-4-oxo-, (2Z)-](http://img.cochemist.com/ccimg/24900/24870-11-9.png)
![2-Butenoic acid,4-[(4-methylphenyl)amino]-4-oxo-, (2Z)-](http://img.cochemist.com/ccimg/24900/24870-11-9_b.png)
![2-Aminobenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18200/18101-58-1.png)
![2-Aminobenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18200/18101-58-1_b.png)


![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8.png)
![5-Methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-amine](http://img.cochemist.com/ccimg/17900/17899-48-8_b.png)





























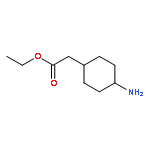
![Ethyl trans-2-[4-(Boc-amino)cyclohexyl]acetate](http://img.cochemist.com/ccimg/946600/946598-34-1.png)
![Ethyl trans-2-[4-(Boc-amino)cyclohexyl]acetate](http://img.cochemist.com/ccimg/946600/946598-34-1_b.png)
![trans-N-[4-[2-(7-Cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-1H-indole-2-carboxamide](http://img.cochemist.com/ccimg/215900/215802-15-6.png)
![trans-N-[4-[2-(7-Cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-1H-indole-2-carboxamide](http://img.cochemist.com/ccimg/215900/215802-15-6_b.png)
![Carbamic acid, [4-[(methylsulfonyl)oxy]butyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174700/174626-25-6.png)
![Carbamic acid, [4-[(methylsulfonyl)oxy]butyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174700/174626-25-6_b.png)