Co-reporter:Zhengjun Wang;Qian Ge;Chen Chen;Xinxin Jin
Applied Biochemistry and Biotechnology 2017 Volume 181( Issue 2) pp:562-572
Publication Date(Web):2017 February
DOI:10.1007/s12010-016-2231-4
In this study, we cloned a full-length cDNA and the genomic DNA sequence of SmCCoAOMT (GenBank ID JQ007585) from Salvia miltiorrhiza. The 744-bp open-reading frame encodes a protein of 247 amino acids that shares 95 % similarity with one in Vitis vinifera. Real-time quantitative PCR analysis revealed that SmCCoAOMT is most highly expressed in the stems and can be induced by methyl jasmonate (MeJA) and XC-1 treatment. To evaluate its function in vivo, we generated RNA interference transgenic plants through Agrobacterium tumefaciens-mediated gene transfer. Compared with untransformed control plants, the transgenics had significantly less lignin and the expression of lignin-biosynthetic genes SmCCR and SmCOMT was depressed. In 90-day-old roots from plants of transgenic line M5, accumulations of rosmarinic acid and salvianolic acid B (Sal B) were greatly reduced by 0.89- and 0.69-fold, respectively. This low-Sal B phenotype was stable in the roots, with the level of accumulation being approximately 43.58 mg g−1 dry weight, which was 52 % of the amount measured in the untransformed control. Our results suggest that SmCCoAOMT is involved in lignin biosynthesis and affects the accumulation of phenolic acids. This study also provides potential guidance for using lignin-related genes to genetically engineer Salvia miltiorrhiza.
Co-reporter:Wenping Hua;Weiwei Kong;XiaoYan Cao;Chen Chen;Qian Liu;Xiangmin Li
Genes & Genomics 2017 Volume 39( Issue 5) pp:509-520
Publication Date(Web):2017 May
DOI:10.1007/s13258-017-0516-9
Dioscorea zingiberensis is the main plant source of diosgenin, a precursor for the production of steroid hormones used in the pharmaceutical industry. The extraction process of diosgenin from D. zingiberensis can generate high-acid and high-strength wastewater on a large scale and can threaten the environment. Bioengineering microorganisms to produce diosgenin is an effective way to avoid pollution. However, little is known about the genes that are involved in the biosynthesis of diosgenin. We obtained 85,010 unigenes (average length of 1142 bases) from the D. zingiberensis transcriptome through RNA-seq. A large number of unigenes (59,368; 69.83%) were annotated, and 2488 unigenes were assigned to 27 secondary-metabolite pathways. In our database, 66 unigenes encoding up to 40 key enzymes were found to be present in diosgenin biosynthesis pathways. In addition, we found 203 unigenes encoding CYP450 proteins and 47 unigenes encoding UGT proteins that may be involved in modifications of a downstream pathway. The expression patterns of key diosgenin biosynthesis genes were studied to identify the most important members of the enzyme family. These results add to the available genetic data of D. zingiberensis and lay the foundation for the further production of diosgenin using genetic engineering.
Co-reporter:Z. Wang;Q. Ge;Z. Wang
Russian Journal of Plant Physiology 2017 Volume 64( Issue 4) pp:553-559
Publication Date(Web):24 June 2017
DOI:10.1134/S1021443717040197
Salvia miltiorrhiza Bunge produces salvianolic acid, a water-soluble phenolic acid that is widely used in treating cardiovascular disease. The objective of this study is to employ AtPAP1 (Production of Anthocyanin Pigmentation) and SmCCR1 (cinnamoyl CoA reductase) for improving the phenolic acids of S. miltiorrhiza. First, results showed that AtPAP1 significantly increased the promoter activities of SmCCR1 by transient transformation of tobacco. To enrich the precursors available for phenolic acid biosynthesis, we present a strategy of combinational genetic manipulation by Agrobacterium tumefaciens-mediated gene transfer that simultaneously over-expressed AtPAP1 and down-regulated SmCCR1. Compared with the control, the amount of lignin was significantly reduced and its composition was changed in transgenic plants. While, the main phenolic acids, including salvianolic acid B and rosmarinic acid, were induced 2.0- and 1.8-fold compared with control lines in two-month-old plants. Meanwhile, the contents of total phenolics and total flavonoids were significantly improved in transformed plants. Expression of related genes throughout the phenylpropanoid pathways were altered, following the same trends as shown for their products, i.e., phenolic acids accumulation and lignin reduction. These results demonstrate that such methods for genetic modification, which increase the expression of transcription factors while suppressing that of branch genes, are of great value when genetically engineering plants for other secondary products.
Co-reporter:Chen Chen, Yuan Zhang, Kuliman Qiakefu, Xuan Zhang, Li-Min Han, Wen-Ping Hua, Ya-Ping Yan, and Zhe-Zhi Wang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 41) pp:7760-7769
Publication Date(Web):October 3, 2016
DOI:10.1021/acs.jafc.6b02844
Tanshinones are a group of active diterpenes with pharmacological properties that are widely used in the treatment of cardiovascular diseases. Jasmonate (JA) acts as an elicitor to enhance tanshinone biosynthesis in Salvia miltiorrhiza. However, because of high labor costs and undesirable chemical characteristics, the use of JA elicitation is still in the experimental stage. In our experiments, the overexpression of Lycopersicon esculentum (tomato) Prosystemin (LePS) in transgenic plants of S. miltiorrhiza increased their JA concentrations, significantly enhanced the production of tanshinone, and activated the expression of key genes in the tanshinone biosynthesis pathway. Meanwhile, the relative levels of metabolites related to defense such as sterols, terpenes, and phenolic acids were also increased in our OEP lines. In addition, when the larvae of cotton bollworms (Heliothis armigera) were fed with leaves from transgenic lines, their mortality rates rose by nearly 4-fold when compared to that of larvae exposed to leaves from the nontransformed wild type. Our study provides a new strategy for genetic engineering by which tanshinone production and pest resistance can be improved in S. miltiorrhiza. This is accomplished by simulating the wounding signal that increases the endogenous levels of JA.Keywords: insect resistance; JA; Salvia miltiorrhiza; tanshinone accumulation; wound signal;
Co-reporter:Yu-ping Liu, Xu Su, Yi-han He, Li-min Han, Ya-ya Huang, Zhe-zhi Wang
Biochemical Systematics and Ecology 2015 Volume 59() pp:159-167
Publication Date(Web):April 2015
DOI:10.1016/j.bse.2015.01.014
•We explored responses of arid-tolerant alpine plants to climatic oscillations.•We examined evolutionary history of endemic perennial species in western QTP.•The largest glaciation promoted the deep divergence by allopatric differentiation.•It experienced a recent regional expansion relevant to Pleistocene climate changes.•Multiple refugia were retained during the LGM in the high-altitude regions.To explore the responses of arid-tolerant alpine plants to Pleistocene climatic oscillations, we examined the evolutionary history of Orinus thoroldii, a grass species endemic to the western Qinghai-Tibetan Plateau (QTP) in China. Three maternally inherited chloroplast DNA (cpDNA) markers (matK, rbcL, and trnH-psbA) and the bi-parentally inherited nuclear ribosomal internal transcribed spacer (nrITS) were sequenced for 243 individuals from 28 populations. In all, seven chloroplast haplotypes (H1–H7) and seven nrITS types (S1–S7) were detected. Whereas two haplotypes were widespread, each of the remaining five was restricted to single populations or shared with only its adjacent population. Phylogenetic analyses of the cpDNA haplotypes revealed two distinct lineages distributed in the eastern and western regions of the western QTP. Our analysis of molecular variance (AMOVA) demonstrated the major partitioning (66.56%) of cpDNA variation between the western and eastern groups. A significant phylogeographical structure was detected (NST = 0.81, GST = 0.72; P < 0.05) based on the cpDNA dataset. The distribution of nrITS types also showed a certain geographical pattern. Whereas types S1 and S5 were widely distributed across the region, the others were exclusively restricted to specific populations. However, the two lineages discerned by the cpDNA sequence variations were not supported by phylogenetic and AMOVA analyses of nrITS variation. Based on documented mutation rates for grasses, we estimated that the two cpDNA lineages of this species diverged at 0.64 Ma during the Pleistocene. This divergence was very consistent with the largest glaciation recorded in the QTP. Long-distance pollen migrations probably blurred the divergence boundary at the nrITS sequence variations. These findings imply that O. thoroldii probably experienced a long-term allopatric divergence plus more recent regional expansions that corresponded to Pleistocene climate changes. Our data also suggest that this grass species survived throughout the Pleistocene glacial stage within the extremely adverse habitats of the QTP.
Co-reporter:Yucui Wu;Congling Liu;Jing Kuang;Qian Ge;Yuan Zhang;Zhezhi Wang
Protoplasma 2014 Volume 251( Issue 5) pp:1191-1199
Publication Date(Web):2014 September
DOI:10.1007/s00709-014-0626-z
Salinity and drought are important abiotic stresses limiting plant growth and development. Late embryogenesis abundant (LEA) proteins are a group of proteins associated with tolerance to water-related stress. We previously cloned an LEA gene, SmLEA, from Salvia miltiorrhiza Bunge. Phylogenetic analysis indicated that SmLEA belongs to Group LEA14, which is involved in the dehydration response. To determine its function in detail, we have now overexpressed SmLEA in Escherichia coli and S. miltiorrhiza. The logarithmic increase in accumulations of SmLEA proteins in E. coli occurred earlier under salinity than under standard conditions. SmLEA-transformed S. miltiorrhiza plants also showed faster root elongation and a lower malondialdehyde concentration than the empty vector control plants did when cultured on MS media supplemented with 60 mM NaCl or 150 mM mannitol. Moreover, SmLEA-overexpressing transgenics experienced a less rapid rate of water loss. Under either salinity or drought, overexpressing plants had greater superoxide dismutase activity and a higher glutathione concentration. These results suggest that SmLEA may be useful in efforts to improve drought and salinity tolerance in S. miltiorrhiza. Our data also provide a good foundation for further studies into the stress resistance mechanism and molecular breeding of this valuable medicinal plant.
Co-reporter:Q. Ge;Y. P. Xiao;Z. Z. Wang
Russian Journal of Plant Physiology 2014 Volume 61( Issue 6) pp:862-872
Publication Date(Web):2014 November
DOI:10.1134/S1021443714060065
Methyl jasmonate (MeJA) has an important role in modulating the accumulation of secondary metabolites in Salvia miltiorrhiza Bunge. However, little research has been reported on genes involved in the JA signaling pathway in this species. Jasmonate ZIM-domain (JAZ) transcriptional repressors are the key regulators of the hormonal response. We cloned a novel JAZ gene, SmJAZ1, from S. miltiorrhiza by screening its transcriptome database. Its full-length sequence is 2024 bp long, including a 1308-bp promoter, and comprises two introns and three exons that encode a polypeptide of 180 amino acid residues. The predicted SmJAZ1 contains the ZIM domain and Jas domain, which are highly conserved regions in the JAZ family. Most JAZ genes can be induced by treatment with MeJA or wounding, a phenomenon that is characteristic of JAZs. Similar to other JAZ genes, our real-time quantitative PCR analysis showed that SmJAZ1 expression increased rapidly, by 2.1-fold, within 1 h after MeJA treatment. It was also strongly induced, to 6.0-fold, within 0.5 h after wounding. Therefore, we believe that this gene is a member of the JAZ family. Although it was detected in four tissues examined here, expression was significantly elevated in the stems and leaves. This gene also responded to treatments with other phytohormones and plant growth regulators (ABA, gibberellin, naphthalene acetic acid, and salicylic acid) and to low-temperature stress. The results obtained provide general characteristics for SmJAZ1 and contribute to our understanding of the jasmonate signaling pathway in S. miltiorrhiza.
Co-reporter:Yuan Zhang;Xing Li;Zhezhi Wang
Biochemical Genetics 2013 Volume 51( Issue 9-10) pp:707-721
Publication Date(Web):2013 October
DOI:10.1007/s10528-013-9600-2
Salvia miltiorrhiza is a well-known medicinal plant and functional food in Asia. The range of quality among its germplasms is irregular, which inevitably affects its health and economic benefits. We established a system comprising phenotypic characteristics, ISSR markers, and active constituents to facilitate the comprehensive evaluation of this species. Concerning the phenotypic traits, 55 germplasms were characterized and divided into eight groups. ISSR molecular markers were employed to detect polymorphisms and distinguish germplasms by integrating with phenotypic characteristics. In terms of economic benefits, we were able to screen out the tall-stem and purple-stem types, both with good yields (root biomass) and high contents of active constituents, as members of a preferred-quality group in S. miltiorrhiza. Our study is the first to provide an overall assessment of different S. miltiorrhiza germplasms. The integrated investigation of the phenotypic, genetic, and phytochemical diversity of S. miltiorrhiza germplasms demonstrates the great potential of diverse genotypes to be exploited by plant breeders.
Co-reporter:Ying Wang;Wenping Hua;Jian Wang;Abdelali Hannoufa
Molecular Genetics and Genomics 2013 Volume 288( Issue 3-4) pp:131-139
Publication Date(Web):2013 April
DOI:10.1007/s00438-013-0736-x
Lotus corniculatus L. is used worldwide as a forage crop due to its abundance of secondary metabolites and its ability to grow in severe environments. Although the entire genome of L. corniculatus var. japonicus R. is being sequenced, the differences in morphology and production of secondary metabolites between these two related species have led us to investigate this variability at the genetic level, in particular the differences in flavonoid biosynthesis. Our goal is to use the resulting information to develop more valuable forage crops and medicinal materials. Here, we conducted Illumina/Solexa sequencing to profile the transcriptome of L. corniculatus. We produced 26,492,952 short reads that corresponded to 2.38 gigabytes of total nucleotides. These reads were then assembled into 45,698 unigenes, of which a large number associated with secondary metabolism were annotated. In addition, we identified 2,998 unigenes based on homology with L. japonicus transcription factors (TFs) and grouped them into 55 families. Meanwhile, a comparison of four tag-based digital gene expression libraries, built from the flowers, pods, leaves, and roots, revealed distinct patterns of spatial expression of candidate unigenes in flavonoid biosynthesis. Based on these results, we identified many key enzymes from L. corniculatus which were different from reference genes of L. japonicus, and five TFs that are potential enhancers in flavonoid biosynthesis. Our results provide initial genetics resources that will be valuable in efforts to manipulate the flavonoid metabolic pathway in plants.
Co-reporter:D. H. Wang;Y. Q. Chen;Y. Wang;Z. Z. Wang
Russian Journal of Plant Physiology 2013 Volume 60( Issue 1) pp:155-164
Publication Date(Web):2013 January
DOI:10.1134/S1021443712060179
To ensure survival, plants must adjust to various abiotic stresses in their environment. Identifying their stress-responsive genes is important for understanding the molecular mechanisms underlying the process of adaptation. From Salvia miltiorrhiza Bunge seedlings, we isolated and characterized two stress-related genes that are highly conserved. Both plasma membrane protein 3 genes (SmPMP3-1 and SmPMP3-2) encode two low-molecular-weight hydrophobic proteins. Cloning of their 5′-flanking regions revealed that they contain many stress-related cis-acting regulatory elements and putative protein-binding sites. Although expression of both was induced by salt and abscisic acid, treatment with salicylate, methyl jasmonate, cold, and dehydration led to down-regulation of SmPMP3-1, whereas SmPMP3-2 was up-regulation. Thus, our results demonstrated the important regulatory effects of SmPMP3 in response to various abiotic stresses.
Co-reporter:Zhengjun Wang;Langjun Cui;Chen Chen;Xiaojing Liu
Plant Molecular Biology Reporter 2012 Volume 30( Issue 5) pp:1229-1236
Publication Date(Web):2012/10/01
DOI:10.1007/s11105-012-0444-4
The biosynthesis of salvianolic acid B shares the phenylpropanoid pathway with lignin, and cinnamoyl CoA reductase (CCR; EC 1.2.1.44) is a specific enzyme in the lignin pathway. In this study, a CCR gene (SmCCR1) from Salvia miltiorrhiza Bunge was cloned using DNA walking technology (GenBank ID: JF798634). The full-length SmCCR1 is 2,489 bp long and consists of four introns and five exons encoding a polypeptide of 324 amino acid residues. Sequence alignment revealed that SmCCR1 shares 83 % identity with CCR sequences reported in Camellia oleifera and other plant species. Expression pattern analysis indicated that expression of SmCCR1 can be induced by exposure to Xanthomonas campestris pv. Campestris or methyl jasmonate. To demonstrate its functioning, we selected a 296-bp fragment and established an RNA interference construct that was introduced into S. miltiorrhiza by Agrobacterium tumefaciens-mediated gene transfer. Transgenic plants exhibited dwarfing phenotypes, and both syringyl and guaiacyl lignin monomers were decreased more than 60 %. In contrast, biosynthesis of phenolic acids—danshensu, rosmarinic acid, and salvianolic acid B—was strongly induced by 2.03-, 1.41-, and 1.45-fold, respectively, in the roots of transgenic plants from line CCR-10. Consistent with these phytochemical changes, downregulation of SmCCR1 also affected the expression of related genes in the phenolics and lignin biosynthetic pathways. Our results also provide potential opportunities for engineering danshensu and salvianolic acid B production in S. miltiorrhiza.
Co-reporter:Peng Zheng, Kejin Zhang, Zhezhi Wang
Biochemical Systematics and Ecology 2011 Volume 39(4–6) pp:704-710
Publication Date(Web):August–December 2011
DOI:10.1016/j.bse.2011.06.002
G. macrophylla pall, G. dahurica Fisch, G. straminea Maxim, and G. crassicaulis Duthie ex Burk are the major cultivated Gentiana species in Gansu Province, China. The present study has collected 107 wild samples from 19 natural populations and focused on the analysis of their medicinal ingredients, genetic diversity, and relationship, concerning four species’s worrying situation and solutions. High performance liquid chromatography (HPLC) results show that their gentiopicroside contents are 9.75 ± 2.0%, 9.29 ± 2.6%, 8.18 ± 3.2%, and 7.96 ± 3.2% respectively and higher than the criterion standard for Radix Gentianae in Chinese Pharmacopoeia. Low genetic polymorphism was found among these four species with HE (expected heterozygosity) = 0.147, GST (interspecies coefficient of gene differentiation) = 0.3207, and Nm (gene flow) = 1.0593. The unweighted pair group method with arithmetic mean (UPGMA) analysis was performed with POPGENE 1.31 software and Nei’s method but did not clearly differentiate these four species. Their medicinal ingredients did not correlate with the genetic variants (r = −0.179, P = 0.586). The results indicate that there is no significant difference among four species in regard to the traits of medicinal qualities. All four Gentiana species can be widely cultivated to not only meet the demand of domestic and international market for Chinese traditional medicine, but also help the local conservation of the wild Gentiana resources in China.Highlights► The gentiopicroside content of four gentiana were higher than Chinese medical criterion. ► The genetic variants among four species did not significantly relate to the gentiopicroside content. ► These species can be widely cultivated in China. ► This may contribute to meet medicinal market demand, and protect the local wild gentiana resources.
Co-reporter:Yuan Zhang, Ya-Ping Yan, and Zhe-Zhi Wang
Journal of Agricultural and Food Chemistry 2010 Volume 58(Issue 23) pp:12168-12175
Publication Date(Web):November 8, 2010
DOI:10.1021/jf103203e
Phenolic acids are health-promoting but low content secondary metabolites in Salvia miltiorrhiza. Here, the Arabidopsis transcription factor Production of Anthocyanin Pigment 1 (AtPAP1) was expressed in S. miltiorrhiza and improved the antioxidant capacity in transgenic plants up to 3-fold. Salvianolic acid B (Sal B) biosynthesis was strongly induced (10-fold higher) in 1 month old transgenic plantlets, a growth stage not normally characterized by significant levels of phenolic acids. This high-Sal B phenotype was stable in roots during vegetative growth, with tissues accumulating approximately 73.27 mg/g of dry weight. Total phenolics, total flavonoids, anthocyanin, and lignin were also significantly enhanced. Consistent with these biological and phytochemical changes, expression of phenolic acid biosynthetic genes was stimulated. Our results demonstrate that AtPAP1 has an additional, previously unknown, role as a transcriptional activator of phenolic acid biosynthesis in S. miltiorrhiza. The results provide a promising strategy for engineering phenolics production in economically significant medicinal plants.
Co-reporter:Xiao-Yan Cao
Phytochemical Analysis 2010 Volume 21( Issue 4) pp:348-354
Publication Date(Web):
DOI:10.1002/pca.1206
Abstract
Introduction – Radix Gentianae Macrophyllae, a traditional Chinese medicine, has been frequently used to dispel rheumatism and ease pain. There are four species of Gentiana (G. macrophylla, G. straminea, G. dahurica and G. crassicaulis) recorded as herbal drugs in the Chinese Pharmacopoeia and two other Gentiana species (G. officinalis and G. siphonantha) are often used as substitutes. Currently, the LC fingerprint comparison among different species and evidence for the equivalent application of these herbs are lacking.
Objective – To develop an HPLC method for the simultaneous determination of four iridoid and secoiridoid glycosides and a comparative study of six species of Gentiana.
Methodology – HPLC analysis was performed on a C18 column (Phenomenex, 150 × 4.6 mm, 5 µm particle size) with gradient elution using 0.4% aqueous phosphoric acid and methanol at 242 nm.
Results – The proposed method was precise, accurate and sensitive enough for simultaneous quantitative evaluation of four iridoid and secoiridoid glycosides (loganic acid, swertiamarin, gentiopicroside and sweroside) in the six species of Gentiana. Contents of the four marker compounds varied from each other even among the samples from the same species and the LC chromatograms of the six species of Gentiana showed high similarities.
Conslusion – The close similarity of LC chromatograms and chemical composition of the four genuine Gentiana species explain their popular usage as Radix Gentianae Macrophyllae in Chinese medicine. By comparing the four genuine Gentiana species, it is suggested that the two substitutes could be used as Radix Gentianae Macrophyllae to relieve the scarcity of resources. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:YaPing Yan;ZheZhi Wang;Wei Tian;ZhongMin Dong
Science China Life Sciences 2010 Volume 53( Issue 2) pp:273-285
Publication Date(Web):2010 February
DOI:10.1007/s11427-010-0005-8
Salvia miltiorrhiza Bge. is a well-known traditional Chinese herb. Its roots have been formulated and used clinically for the treatment of various diseases. However, little genetic information has so far been available and this fact has become a major obstacle for molecular studies. To address this lack of genetic information, an Expressed Sequence Tag (EST) library from whole plantlets of S. miltiorrhiza was generated. From the 12959 cDNA clones that were randomly selected and subjected to single-pass sequencing from their 5′ ends, 10288 ESTs (with sizes⩾100 bp) were selected and assembled into 1288 contigs, leaving 2937 singletons, for a total of 4225 unigenes. These were analyzed using BLASTX (against protein databases), RPS-BLAST (against a conserved domain database) as well as the web-based KEGG Automatic Annotation Server for metabolic enzyme assignment. Based on the metabolic enzyme assignment, expression patterns of 14 secondary metabolic enzyme genes in different organs and under different treatments were verified using real-time PCR analysis. Additionally, a total of 122 microsatellites were identified from the ESTs, with 89 having sufficient flanking sequences for primer design. This set of ESTs represents a significant proportion of the S. miltiorrhiza transcriptome, and gives preliminary insights into the gene complement of S. miltiorrhiza. They will prove useful for uncovering secondary metabolic pathways, analyzing cDNA-array based gene expression, genetic manipulation to improve yield of desirable secondary products, and molecular marker identification.
Co-reporter:Jie Song;Zhezhi Wang
Molecular Biology Reports 2009 Volume 36( Issue 5) pp:
Publication Date(Web):2009 May
DOI:10.1007/s11033-008-9266-8
Phenylalanine ammonia-lyase (PAL) is one of the branch point enzymes between primary and secondary metabolism. It plays an important role during plant development and defense. A PAL gene designated as SmPAL1 was cloned from Salvia miltiorrhiza using genome walking technology. The full-length SmPAL1 was 2,827 bp in size and consisted of an intron and two extrons encoding a 711-amino-acid polypeptide. Sequence alignment revealed that SmPAL1 shared more than 80% identity with the PAL sequences reported in Arabidopsis thaliana and other plants. The 5′ flanking sequence of SmPAL1 was also cloned, and a group of putative cis-acting elements such as TATA box, CAAT box, G box and TC-rich repeats were identified. Transcription pattern analysis indicated that SmPAL1 expressed in all tissues examined, but more highly in leaf. Besides, expression of SmPAL1 was found to be induced by various treatments including ABA, wounding, and dehydration. To further confirm its function, SmPAL1 was expressed in Escherichia coli strain M15 with pQE-30 vector. The recombinant protein exhibited high PAL activity and could catalyze the conversion of l-Phe to trans-cinnamic acid. This study will enable us to further understand the role SmPAL1 plays in the synthesis of active pharmaceutical compounds in S. miltiorrhiza at molecular level.
Co-reporter:Ya-ping Yan
Plant Cell, Tissue and Organ Culture (PCTOC) 2007 Volume 88( Issue 2) pp:175-184
Publication Date(Web):2007 February
DOI:10.1007/s11240-006-9187-y
A high-frequency and simple procedure for Agrobacterium tumefaciens-mediated genetic transformation of the medicinal plant Salvia miltiorrhiza was developed. Leaf discs were pre-cultured on MS medium supplemented with 6.6 μmol l−1 BAP and 0.5 μmol l−1 NAA for one day, then co-cultured with A. tumefaciens strain EHA105 harboring the plasmid pCAMBIA 2301 for three days on the same medium. Regenerated buds were obtained on selection medium (co-culture medium supplemented with 60 mg l−1 kanamycin and 200 mg l−1 cefotaxime) after two cycles’ culture of 10 days each and then transferred to fresh MS medium with 60 mg l−1 kanamycin for rooting. Fifteen days later, the rooted plantlets were obtained and then successfully transplanted to soil. The transgenic nature of the regenerated plants was confirmed by PCR, Southern hybridization analysis and GUS histochemical assay. Averagely, 1.1 independent verified transgenics per explant plated were obtained through this protocol. Adopting this procedure, positive transformed plants could be obtained within 2–3 months from mature seeds germination to transplant to soil, and more than 1,000 transgenic plants with several engineered constructs encoding different genes of interest were produced in our lab in the past two years.
Co-reporter:Yuan Zhang, Zhe-zhi Wang
Comptes Rendus Biologies (September 2009) Volume 332(Issue 9) pp:816-826
Publication Date(Web):1 September 2009
DOI:10.1016/j.crvi.2009.05.006
Two traditional Chinese medicines (Phlomis umbrosa Turcz. and Phlomis megalantha Diels), as well as five pure phenolic compounds (protocatechic, chlorogenic, benzoic, rosmarinic acid, and rutin) have been studied for antioxidant activities in acetone and methanol extracts from leaves. An HPLC method was developed to quantify the amounts of 14 phenolic compounds in the leaf extracts. The antioxidant capacities of the studied species are high. Almost all samples were capable of directly scavenging DPPH and superoxide free radicals, inhibiting linoleic acid oxidation, acting as reducing agents, and reducing plasmid DNA damage induced by hydroxyl radicals. Among different extracts, the acetone extract of P. megalantha exhibited the highest antioxidant activity. The major phenolic compounds identified were protocatechic, chlorogenic, caffeic, rosmarinic acid, and (−)-epicatechin. Antioxidant activities of pure compounds and correlation analysis indicated that protocatechic and rosmarinic acids were the major contributors to the observed antioxidant activities of the investigated Phlomis extracts. To cite this article: Y. Zhang, Z.-z. Wang, C. R. Biologies 332 (2009).
Co-reporter:Hua Wenping, Zhang Yuan, Song Jie, Zhao Lijun, Wang Zhezhi
Genomics (October 2011) Volume 98(Issue 4) pp:272-279
Publication Date(Web):1 October 2011
DOI:10.1016/j.ygeno.2011.03.012
Medicinal Salvia miltiorrhiza is a Chinese herb commonly used for treating cardiovascular diseases and neuroasthenic insomnia. However, little is known at the genetics level about how its compounds are synthesized in that plant. Here, we obtained 56,774 unigenes (average length = 467 bases) in its transcriptome by performing Solexa deep sequencing over the entire growing cycle. Unigenes (34,340; 60.49%) were annotated and 2545 unigenes were assigned to specific pathways. Unigenes (1539) were identified as part of five major, secondary-metabolite pathways, covering almost all nodes in the phenylpropanoid and terpenoid pathways. Using Blast search against AGRIS, 1341 unigenes were found homologous to 686 Arabidopsis transcription factor genes. Real-time PCR was also used to verify the spatio-temporal expression patterns of several novel transcripts related to biosynthesis of active ingredients in that species. These results not only enrich the gene resource but also benefit research into its molecular genetics and functional genomics.
Co-reporter:Donghao Wang, Wei Yao, Yin Song, Wenchao Liu, Zhezhi Wang
Journal of Plant Physiology (15 December 2012) Volume 169(Issue 18) pp:1838-1848
Publication Date(Web):15 December 2012
DOI:10.1016/j.jplph.2012.07.015
To adapt to changes in their growing environment, plants express several stress-responsive genes. For example, the products of galactinol synthase (Gols) genes play a key role in regulating the levels of raffinose family oligosaccharides and conferring resistance to stress. We cloned and characterized three Gols genes in Salvia miltiorrhiza. Their expression followed three distinct patterns. Compared with the control, SmGols1 was up-regulated by temperature changes but was suppressed by exposure to methyl jasmonate or short-term drought. This gene had the greatest abundance of transcripts and was assigned a general function of carbon storage. SmGols2 responded to all stress and hormone treatments, and transcripts were maintained at a high level. Finally, expression of SmGols3 was weaker than the other two genes, but was increased significantly under different treatments. Over the experimental period, its expression declined to normal levels in response to all treatments except exposure to 100 μM ABA, long-term drought, heat (42 °C), or chilling (8 °C). Based on our finding of cis-elements in the 5′ flanking regions, we concluded that these genes seem to be regulated by several HSF transcription factors. We also targeted their 90-bp conserved sequences and used them for RNA interference analysis. Some were knocked down to various extents in our transgenic lines. Fluctuations in their malondialdehyde contents under different stress treatments, as well as the rate of water loss in transformed plants, suggested that lipid peroxidation was more likely to occur in the transgenics than in the control. These results indicate that SmGols genes could have a main function in responding to cold or heat. Therefore, we believe that it is important to investigate this mechanism for tolerance in S. miltiorrhiza and to examine how expression of these SmGols and other homologs are influenced by abiotic stresses.
Co-reporter:Zhang-Min Yang, Yu-E. Yang, Yu Chen, Jing Cao, Cui Zhang, Ling-Ling Liu, Zhe-Zhi Wang, Xu-Min Wang, Ying-Ming Wang, Inn-Ho Tsai
Toxicon (1 December 2015) Volume 107(Part B) pp:175-186
Publication Date(Web):1 December 2015
DOI:10.1016/j.toxicon.2015.08.010
•Gloydius intermedius venom contains abundant crotoxin-like neurotoxins, kallikrein-like proteases and BPP.•The venom gland ESTs reveal the most abundant venom families: SP > BPP > LAAO > PLA2.•Similarity between Gintexin and Crotoxin subunits supports G. intermedius being the Asian sister of neurotoxic rattlers.•Co-evolution of neurotoxic and hypotensive toxins occurs in viperid venoms.The venomics of Gloydius intermedius were investigated using expressed sequence tags (ESTs) analyses, 2D gel-electrophoresis combined with MALDI-TOF/TOF, and LC-MS/MS. A total of 1920 ESTs from the venom gland cDNA library were sequenced; 74% of them belonged to toxin-families. The four most abundant families among the toxin transcripts were: serine protease (SP, 36.2%), bradykinin potentiating peptide (25.3%), l-amino acid oxidase (LAAO, 13.1%), and phospholipase A2 (PLA2, 9.9%). Moreover, the full sequences of four PLA2s, eight SPs, cysteine-rich secretory protein (CRISP), C-type-lectin-like-protein (CTLP), hyaluronidase, metalloproteinase, and nerve growth factor were deduced from the cDNA sequences. Excluding the CRISP and hyaluronidase, most of the G. intermedius venom proteins bear 92–99% sequence identities to those of other pitviper venoms. The most abundant components are PLA2s (37%), SPs (20%) and LAAO (6%), while metalloproteinase, CTLP, and other components each account for <3% of the total venom proteins. The abundance of Gintexin (a crotoxin-like neurotoxin) and low levels of hemorrhagic metalloproteases, disintegrins and CTLPs highlight the great venom differences between G. intermedius and other hemorrhagic pitvipers. The bimorphism of hemorrhagic and neurotoxic venoms among Gloydius is confirmed; our results shed more lights on the co-evolution of both neurotoxicity and hypotension in some viperid venoms.Download full-size image

![1,3-Naphthalenedicarboxylic acid, 2-(3,4-dihydroxyphenyl)-1,2-dihydro-7,8-dihydroxy-, 1,3-bis[1-carboxy-2-(3,4-dihydroxyphenyl)ethyl] ester](/data/chemimg/103800/389065-74-1.png)
![1,3-Naphthalenedicarboxylic acid, 2-(3,4-dihydroxyphenyl)-1,2-dihydro-7,8-dihydroxy-, 1,3-bis[1-carboxy-2-(3,4-dihydroxyphenyl)ethyl] ester](/data/chemimg/103800/389065-74-1_b.png)
![2-Propenoic acid, 3-[2-[(1E)-2-(3,4-dihydroxyphenyl)ethenyl]-3,4-dihydroxyphenyl]-, (2E)-](/data/chemimg/103800/158732-59-3.png)
![2-Propenoic acid, 3-[2-[(1E)-2-(3,4-dihydroxyphenyl)ethenyl]-3,4-dihydroxyphenyl]-, (2E)-](/data/chemimg/103800/158732-59-3_b.png)
![Benzenepropanoic acid, a-[[(2E)-3-[2-(carboxymethyl)-3,4-dihydroxyphenyl]-1-oxo-2-propen-1-yl]oxy]-3,4-dihydroxy-,(aR)-](http://img.cochemist.com/ccimg/143000/142998-47-8.png)
![Benzenepropanoic acid, a-[[(2E)-3-[2-(carboxymethyl)-3,4-dihydroxyphenyl]-1-oxo-2-propen-1-yl]oxy]-3,4-dihydroxy-,(aR)-](http://img.cochemist.com/ccimg/143000/142998-47-8_b.png)







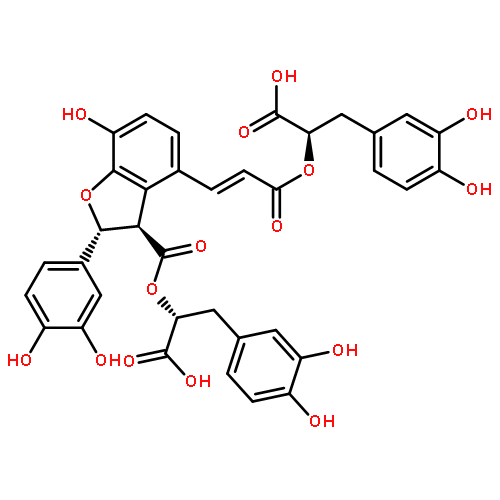


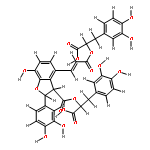
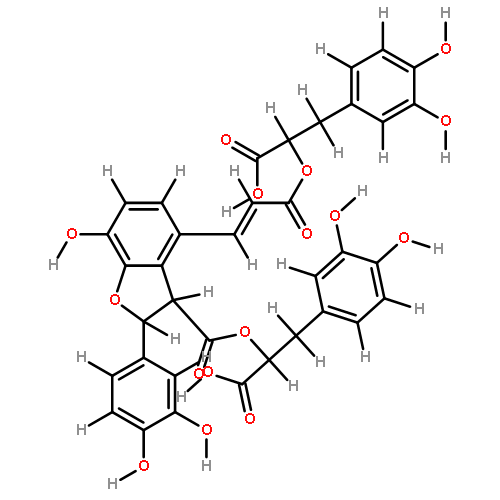
![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4.png)
![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4_b.png)
![Phenanthro[3,2-b]furan-7,11-dione,1,2,3,4-tetrahydro-4,4,8-trimethyl-](http://img.cochemist.com/ccimg/21000/20958-15-0.png)
![Phenanthro[3,2-b]furan-7,11-dione,1,2,3,4-tetrahydro-4,4,8-trimethyl-](http://img.cochemist.com/ccimg/21000/20958-15-0_b.png)
![methyl 1-(beta-D-glucopyranosyloxy)-6-hydroxy-7-methyl-1,4a,5,6,7,7a-hexahydrocyclopenta[c]pyran-4-c](http://img.cochemist.com/ccimg/18600/18524-94-2.png)
![methyl 1-(beta-D-glucopyranosyloxy)-6-hydroxy-7-methyl-1,4a,5,6,7,7a-hexahydrocyclopenta[c]pyran-4-c](http://img.cochemist.com/ccimg/18600/18524-94-2_b.png)
![(S)-6-(Hydroxymethyl)-1,6-dimethyl-6,7,8,9-tetrahydrophenanthro[1,2-b]furan-10,11-dione](http://img.cochemist.com/ccimg/17400/17397-93-2.png)
![(S)-6-(Hydroxymethyl)-1,6-dimethyl-6,7,8,9-tetrahydrophenanthro[1,2-b]furan-10,11-dione](http://img.cochemist.com/ccimg/17400/17397-93-2_b.png)