Co-reporter:Qiong Wang, Fang Huang, Langhuan Jiang, Chuanzhi Sun, Jianbiao Liu, and Dezhan Chen
Inorganic Chemistry May 15, 2017 Volume 56(Issue 10) pp:5984-5984
Publication Date(Web):May 4, 2017
DOI:10.1021/acs.inorgchem.7b00739
The reaction mechanism and the origins of regio- and enantioselectivities for Pd-catalyzed asymmetric arylation of aliphatic α-amino anion equivalents were investigated computationally. The results indicate that the reaction proceeds via mainly six sequential steps: deprotonation at α′-site of imine, coordination of α-amino anion to Pd-catalyst, oxidative addition, transmetalation, reductive elimination, as well as the final dissociation to release the product and regenerate the catalyst. The transmetalation is a key step on which both enantioselectivity and regioselectivity depend. The charge inversions of α- and α′-C atoms and the orbital interaction between Pd center and α-C in transmetalation step are responsible for the regioselectivity. Additionally, the intermediates before the dissociation step are critical in controlling the enantioselectivity. Noncovalent interactions analyses indicate that the enantioselectivity primarily arises from the CH···π interactions of isopropyl (iPr) groups with the fluorene and the benzene rings for PdL1-catalyzed reaction.
Co-reporter:Xiehuang Sheng;Chao Shan;Jianbiao Liu;Jintong Yang;Bin Sun
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 20) pp:13153-13159
Publication Date(Web):2017/05/24
DOI:10.1039/C7CP00804J
Ferroptosis is a recently discovered iron-dependent form of non-apoptotic cell death caused by the accumulation of membrane lipid peroxidation products, which is involved in various pathological conditions of the brain, kidney, liver and heart. A potent spiroquinoxalinamine derivative named liproxstatin-1 is discovered by high-throughput screening, which is able to suppress ferroptosis via lipid peroxide scavenging in vivo. Thus, molecular simulations, density functional theory (DFT) and variational transition-state theory with a small-curvature tunneling (SCT) coefficient are utilized to elucidate the detailed mechanisms of inactivation of a lipid peroxide radical by liproxstatin-1. H-atom abstracted from liproxstatin-1 by a CH3OO˙ radical occurs preferentially at the aromatic amine site (1′-NH) under thermodynamic and frontier molecular orbital analysis. The value of a calculated rate constant at 300 K is up to 6.38 × 103 M−1 S−1, indicating that the quantum tunneling effect is responsible for making a free radical trapping reaction more efficient by liproxstatin-1. The production of a liproxstatin-1 radical is easily regenerated to the active reduced form by ubiquinol in the body to avoid secondary damage by free radicals. A benzene ring and the higher HOMO energy are beneficial to enhance the lipid radical scavenging activity based on the structure–activity relationship study. Overall, the present results provide theoretical insights into the exploration of novel ferroptosis inhibitors.
Co-reporter:Wenjuan Wang;Fang Huang;Chuanzhi Sun;Jianbiao Liu;Xiehuang Sheng
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 16) pp:10413-10426
Publication Date(Web):2017/04/19
DOI:10.1039/C6CP08068E
The detailed formation mechanisms of C-ribonucleoside and N-ribonucleoside via the reaction of 2,4,6-triaminopyrimidine (TAP) with (D)-ribose in aqueous solution were explored using density functional theory (DFT). The calculations indicate that five isomers (α,β-furanose, α,β-pyranose and open-chain aldehyde) of (D)-ribose can exist in equilibrium in aqueous solution. In contrast to cyclic isomers, an open-chain aldehyde is most feasible to react with TAP. In general, the formation pathways of C-nucleoside and N-nucleoside proceed in three steps including nucleophilic addition, dehydration and cyclization. The calculated apparent activation energies are 28.8 kcal mol−1 and 29.2 kcal mol−1, respectively. It suggests that both C- and N-nucleoside can be formed in aqueous solution, which is in good agreement with the experimental results. The water molecule plays an important “H-bridge” role by the hydrogen atom relay. Finally, a model structure of nucleobase, which will be beneficial for the C–C glycosidic bond formation, is proposed.
Co-reporter:Weixi Fan;Chuanzhi Sun;Fang Huang;Jianbiao Liu;Xue Zhao;Xiehuang Sheng
Dalton Transactions 2017 vol. 46(Issue 16) pp:5288-5296
Publication Date(Web):2017/04/19
DOI:10.1039/C7DT00547D
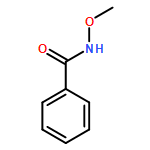
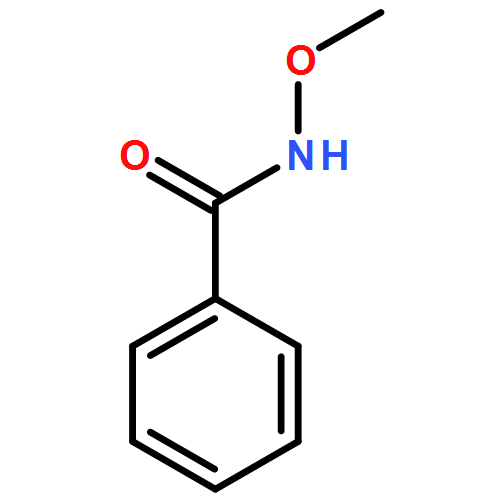
Computational studies have been applied to gain insight into the mechanism of Pd(II) catalyzed α-C–H functionalization of N-methoxy cinnamamide. The results show that the whole catalytic cycle proceeds via sequential six steps, including (i) catalyst Pd(t-BuNC)2 oxidation with O2, (ii) O–H deprotonation, (iii) t-BuNC migratory insertion to the Pd–C bond, (iv) acyl migration, (v) C–H activation and (vi) reductive elimination. The regioselectivity for different C–H activation sites depends on the coordination structures of α-C or β-C to the palladium(II) center. The coordination of α-C to the palladium(II) center shows a regular planar quadrilateral structure, which is stable. However, the β-C coordinating to the palladium(II) center mainly exhibits a distorted quadrilateral structure, which is relatively unstable. Thus, the barrier of α-C–H activation is much lower than that of β-C–H activation. The present results provide a deep understanding of the site-selectivity of C–H activation.
Co-reporter:Lihuan Xie, Fang Huang, Chuanzhi Sun, Jianbiao Liu, Dezhan Chen
Computational and Theoretical Chemistry 2016 Volume 1079() pp:11-22
Publication Date(Web):1 March 2016
DOI:10.1016/j.comptc.2016.01.006
•Mechanisms between 2-aminooxazole/2-aminothiazole and glyceraldehyde are studied.•CC formation determines the reaction diastereoselectivity.•Oxazole-hemiaminal is proposed can be formed based on the calculations.•Phosphate is very important in assisting proton transfers.Density functional theory is utilized to elucidate the detailed mechanisms of the reactions between 2-aminooxazole O2/2-aminothiazole S2 and glyceraldehyde 2. According to our calculations, in O2/2 system, aminooxazoline O3 is formed via two steps including CC formation and cyclization. CC formation determines the reaction diastereoselectivity and ribo-aminooxazoline is the most favorable product. Although oxazole-hemiaminal O6 is not detected in the experiment, it is deduced can be formed theoretically. In S2/2 system, aminothiazoline S3 is hard to be generated because of the less nucleophilic ability of S2. The formation of thiazole-hemiaminal S6 is more favorable than S3 formation in kinetics but somewhat unfavorable in thermodynamics. However, the transformation from S6 to thiazole-aminal S7 is favorable both in kinetics and thermodynamics, which provides a driving force for the formation and transformation of S6. Additionally, our calculations indicate that phosphate is very important in assisting proton transfer in all of the transformations.The synthesis of nucleotides, bypassing free ribose and nuclebase, has recently attracted great attention. It has been reported that pyrimidine nucleotide precursor can be generated from 2-aminooxazole and glyceraldehyde. However, attempt to generate deoxynucleotide from 2-aminothiazole and glyceraldehyde is failed experimentally. Density functional theory has been utilized to elucidate the similarities and distinctions of the two systems. From the energy profiles and optimized structures, conclusions are drawn about mechanisms and experimental phenomena are explained.
Co-reporter:Zhong Xing, Fang Huang, Chuanzhi Sun, Xue Zhao, Jianbiao Liu, and Dezhan Chen
Inorganic Chemistry 2015 Volume 54(Issue 8) pp:3958-3969
Publication Date(Web):April 9, 2015
DOI:10.1021/acs.inorgchem.5b00134
Density functional theory has been applied to gain insight into the Cp*Rh(OAc)2-catalyzed C–H activation and intermolecular annulation of benzamide derivatives with allenes. The study shows that the reactions proceed in three steps: (1) C–H activation induced by Rh catalyst reacting with benzamide derivatives, (2) carborhodation of allene, and (3) regeneration of Rh catalyst. The results indicate that the N–H deprotonation makes the following C–H activation much easier. The regio- and stereoselectivities of 1a (N-pivaloyloxy benzamide)/2a (cyclohexylallene) and 1b (N-pivaloyloxy-4-methyl-benzamide)/2b (1,1-dimethyl allene) depend on the allene carborhodation step. The steric hindrance effect is the dominant factor. We also discuss the reaction mechanism of 1c (N-methoxy benzamide)/2a. The chemoselectivity between 1c/2a is determined by the N–O cleavage step. Replacement of OPiv by OMe leads to loss of the stabilization effect provided by C═O in OPiv. Additionally, Cp*Rh(OAc)(OPiv) is produced in the Cp*Rh(OAc)2 regeneration step, which can work as catalyst as well.
Co-reporter:Chuanzhi Sun, Xiaodan He, Wenjuan Wang, Guohong Chu, Shuxiang Zhang, Dezhan Chen
Computational and Theoretical Chemistry 2014 Volume 1046() pp:49-56
Publication Date(Web):15 October 2014
DOI:10.1016/j.comptc.2014.07.014
•Uncover the mechanism for selenium-based catalyzed bromolactonization.•Explain the key role of the catalytic center selenium plays in the reaction.•The reaction sequence in one-pot reaction is well researched and explained.The mechanisms of the uncatalyzed and selenium-based catalyzed bromolactonization are studied using B3LYP and MP2 method. The pathways of reaction are investigated in detail. The results suggest that the uncatalyzed reaction proceeds in three major steps involving electrophilic addition, ring-closure and hydrogen transfer, while the selenium-based catalyzed process involves Cat-NBS interaction, hydrogen transfer, and ring-closure. The electrophilic addition is the rate-determining step of the uncatalyzed process, while Cat-NBS interaction is that of the catalyzed process. Selenium of Cat is the catalytic center which changes the charge density of Br on NBS, and it makes the electrophilic addition easier. Thus, the energy barrier is sharply reduced. There are four regioselective pathways for the formation of exo-five-lactone 3 and endo-six-lactone 4. The preferred pathway is confirmed relating to exo-five-lactone 3.The mechanism for bromolactonization reaction without and with catalyst was investigated, respectively. The comparison of uncatalyzed and catalyzed process reveals the key role what the catalyst plays.
Co-reporter:Nan Lu, Lin Meng, Dezhan Chen, and Guiqiu Zhang
The Journal of Physical Chemistry A 2012 Volume 116(Issue 1) pp:670-679
Publication Date(Web):October 10, 2011
DOI:10.1021/jp209308a
We have investigated important intermediates and key transition states of the organocatalyzed cascade double Michael addition using density functional theory. The calculated results suggest that the reaction contains intermolecular nucleophilic addition and intramolecular cyclization, both involving the formation of two stereocenters. The iminium–enamine catalysis of secondary amine unit enables the cascade addition to proceed consecutively. As an electron transport, the iminium attracts the electron stream to promote the nucleophilic addition. Then enamine causes the electron stream to catalyze the cyclization. As H bond donor, the catalyst forms three types of C–H···O H bond with substrates. The enantioselectivity and diastereoselectivity are dominated by the catalyst backbone. Two group links of pyrrole–phenyl and pyrrole–silyl ether orient the reaction in paths with smaller rotations of the linked single bonds. Our conclusion is supported by NBO analysis and the predicted ee, dr values according to the experiment.
Co-reporter:Nan Lu, Lin Meng, Dezhan Chen and Guiqiu Zhang
RSC Advances 2011 vol. 1(Issue 6) pp:1113-1118
Publication Date(Web):22 Sep 2011
DOI:10.1039/C1RA00015B
We, the named authors, hereby wholly retract this RSC Advances article.
Signed: Nan Lu, Lin Meng, Dezhan Chen and Guiqiu Zhang, China, February 2012.
Retraction endorsed by Sarah Ruthven, Managing Editor. Retraction published 1st February 2012.
Co-reporter:Nan Lu, Lin Meng, Dezhan Chen, Guiqiu Zhang
Journal of Molecular Catalysis A: Chemical 2011 Volume 339(1–2) pp:99-107
Publication Date(Web):1 April 2011
DOI:10.1016/j.molcata.2011.03.003
TangPhos-catalyzed asymmetric γ addition of thiols to allenoates has been investigated according to density functional theory. The uncatalyzed addition occurs at β-carbon via a process which involves C–S bond formation and proton transfer from S to γ-carbon. The β-thioester is generated. In TangPhos-catalyzed case, the nucleophilic thiol attacks γ-carbon after the addition of TangPhos to β-carbon. The proton transfers firstly from P of TangPhos to carbonyl O and then to β-carbon. The γ-thioester is obtained. Step1 is rate-limiting. As nucleophilic catalyst, P2 forms strong covalent bond with β-carbon which shifts the positive charge of C2, leaving C3 as the electrophilic center for γ addition. The regioselectivity is consequently altered. As Lewis base, P1 deprotonates thiol enhancing the nucleophility of S and facilitates the proton transfer to β-carbon as a medium. Among four competitive pathways, ER path is the most favorable one with smallest rotation of the single bond linking two chiral rings in TangPhos. The primary domination on enantioselectivity of chiral rings is assisted by t-butyl group, which also prefers ER path with the least steric hindrance. Our conclusion is supported by NBO analysis and the predicted ee values according to the experiment.Graphical abstractHighlights► This is the first theoretical study on TangPhos-catalyzed asymmetric γ addition. ► Reaction involves nucleophilic attack of S to γ-carbon and two times proton transfer. ► Bond β-carbon – P2 shifts positive charge of C2 leaving C3 as electrophilic center. ► P1 abstracts proton of thiol and facilitates proton transfer as a shuttle. ► Enantioselectivity is exerted by rigid chiral rings and bulky t-butyl of TangPhos.
Co-reporter:Dezhan Chen, Nan Lu, Guiqiu Zhang, Shizhen Mi
Tetrahedron: Asymmetry 2009 Volume 20(Issue 12) pp:1365-1368
Publication Date(Web):2 July 2009
DOI:10.1016/j.tetasy.2009.05.027
The mechanism of the enantioselective control of an organocatalyst with central and axial chiral elements in the Michael addition of 2,4-pentandione to a nitroalkene is investigated using density functional theory (DFT) calculations. Two enantioselective channels are characterized in detail. Enantioselectivity is determined in the C–C bond coupling and the proton transfer is identified as the energetic bottleneck. Generally, the level of enantioselectivity of the catalysts depends on the geometrical match or mismatch of two asymmetric elements. The ‘closed’ geometry of a catalyst makes the cooperation of two chiralities possible, so that the central and axial chiralities work together to enhance the enantioselective control. The ‘open’ structure of catalyst makes cooperation of the two asymmetric elements impossible, so that its enantioselectivity dominated only by one type of chirality is decreased.C37H31F6N3S[α]D20=-30.5 (c 0.8, CHCl3)
Co-reporter:Honghong Zhang, Dezhan Chen, Guiqiu Zhang, Shizhen Mi, Nan Lu
Journal of Molecular Structure: THEOCHEM 2009 Volume 908(1–3) pp:12-18
Publication Date(Web):30 August 2009
DOI:10.1016/j.theochem.2009.04.039
The mechanism of the aminolysis of dimethyl phenylphosphinate with ammonia was investigated by the density functional theory at the B3LYP level with basis set 6-31G(d,p) in the gas phase. Single point energy evaluations were also performed for more precise energy predictions at the B3LYP and the BB1K levels with the 6-311++G(d,p) basis set. Solvent effects on the uncatalyzed process were assessed by the polarized continuum model with B3LYP/6-311++G(d,p) and BB1K/6-311++G(d,p) based on the gas-phase optimized geometries. Transition state structures and energies were determined for concerted and neutral stepwise pathways. The theoretical results show that the stepwise pathway is more favorable than the concerted process. The general base catalysis of the process was also examined. The catalytic role of a second ammonia molecule is executed by facilitating the hydrogen transfer processes and by decreasing all energy barriers. The results show that the most favorable pathway of the reaction is through the general-base-catalyzed neutral stepwise mechanism.
Co-reporter:Dezhan Chen, Zhen Wang, Honghong Zhang
Journal of Molecular Structure: THEOCHEM 2008 Volume 859(1–3) pp:11-17
Publication Date(Web):30 June 2008
DOI:10.1016/j.theochem.2008.02.033
Quantum chemical investigations on diarylethene derivatives with various substituents linked to different positions of thienyl rings have been carried out. The calculations on molecular structures show that the distance between two reactive carbons, which has close relationship with the photochromic reactivity and cyclization quantum yields, is prominently affected by different alkyl and alkoxy groups at 2- and 2′-positions. The ground-state energy gaps (ΔE) between closed-ring and open-ring isomers change in parallel with the aromatic stabilization energy (ΔE′) for the same series of substituents at reactive carbons. Once ΔE or ΔE′ becomes smaller, the thermal cycloreversion energy barrier becomes larger and closed-ring isomers would exhibit excellently thermal stability. The absorption wavelengths of closed-ring isomers have also been investigated using time-dependent density functional theory (TD-B3LYP/6-31G(d) or TD-PBE0/6-31G(d)). Theoretical calculations can well reproduce experimental absorption spectrum data. Furthermore, this work rationally predicts absorption wavelengths of some unsynthesized diarylethenes with various groups at 2- and 2′- or 4- and 4′-positions. The theoretical study would provide some basic insight for the novel photochromic molecular design.
Co-reporter:De-Zhan Chen, Dao-Ping Wang, De-Xin Kong, Xiao Zhang
Journal of Photochemistry and Photobiology A: Chemistry 2005 Volume 170(Issue 1) pp:37-43
Publication Date(Web):28 February 2005
DOI:10.1016/j.jphotochem.2004.07.015
Ab initio and CIS theories were employed to study a new type of perylenequinonoid photosensitizer (PQP)—hypomycin B (HMB), which has only one pair of adjacent hydroxyl group and carbonyl group in its peri-region. Geometries, Mulliken charge population, energy, and photophysical and photochemical process in excited state were discussed in detail. The results indicate that HMB exhibits quite similar intramolecular proton transfer (IPT) to perylenequinone, which offers an indirect understanding that the IPT process in perylenequinonoid derivatives (PQD) is a single proton transfer process. There exists the crossing of the potential energy surface of the singlet and triplet so that the intersystem crossing (ISC) can occur. The ISC will increase triplet quantum efficiency, which is the basis for photosensitization of PQP.
Co-reporter:Dezhan Chen, Liang Shen
Journal of Photochemistry and Photobiology A: Chemistry 2005 Volume 171(Issue 3) pp:275-279
Publication Date(Web):5 May 2005
DOI:10.1016/j.jphotochem.2004.10.010
The photophysical and photochemical characteristics of the aluminum ion-complexed perylenequinone (Al3+-PQ) at different chain length have been explored employing the quantum chemical method in this paper. It is revealed firstly, the optimized structures of the complexes at different chain lengths are all one-dimension polymeric chains in the ground state. Secondly, the chelation reaction not only decreases the molecular EHOMO, ELUMO and ΔE, but also shifts the excitation spectra bathochromatically. In addition, based on the present calculation results, the first triplet excitation energy of the complexes at different chain length can directly be transferred to molecular oxygen to generate the singlet excited molecular oxygen (1O2) (Type II). On the contrary, the semiquinone anion radicals (Al3+-PQ−) cannot react with triplet molecular oxygen to form the superoxide anion radical (O2−), which indicates photosensitization mechanism Type I is not involved in the photodynamic action of Al3+-PQ.
Co-reporter:De-Zhan Chen, Dao-Ping Wang, Hong-Yu Zhang, Bo Tang
Chemical Physics Letters 2002 Volume 353(1–2) pp:119-126
Publication Date(Web):13 February 2002
DOI:10.1016/S0009-2614(01)01471-3
The intramolecular proton transfer of perylenequinone (PQ) in the ground state S0 and the excited states S1 and T1 has been studied at the HF/CIS/6-31g level of theory. It is found that the process can be viewed as a proton transfer in S0 and S1, while it is coupled with an electronic transfer through the conjugated system in T1. The barrier of proton transfer in the S1 is 10.38 kJ/mol, which agrees with the experimental result of hypocrellin A and offers good evidence that the experimental observed process of hypocrellin A is also single proton transfer.
Co-reporter:Weixi Fan, Chuanzhi Sun, Fang Huang, Jianbiao Liu, Xue Zhao, Xiehuang Sheng and Dezhan Chen
Dalton Transactions 2017 - vol. 46(Issue 16) pp:NaN5296-5296
Publication Date(Web):2017/03/23
DOI:10.1039/C7DT00547D
Computational studies have been applied to gain insight into the mechanism of Pd(II) catalyzed α-C–H functionalization of N-methoxy cinnamamide. The results show that the whole catalytic cycle proceeds via sequential six steps, including (i) catalyst Pd(t-BuNC)2 oxidation with O2, (ii) O–H deprotonation, (iii) t-BuNC migratory insertion to the Pd–C bond, (iv) acyl migration, (v) C–H activation and (vi) reductive elimination. The regioselectivity for different C–H activation sites depends on the coordination structures of α-C or β-C to the palladium(II) center. The coordination of α-C to the palladium(II) center shows a regular planar quadrilateral structure, which is stable. However, the β-C coordinating to the palladium(II) center mainly exhibits a distorted quadrilateral structure, which is relatively unstable. Thus, the barrier of α-C–H activation is much lower than that of β-C–H activation. The present results provide a deep understanding of the site-selectivity of C–H activation.
Co-reporter:Wenjuan Wang, Fang Huang, Chuanzhi Sun, Jianbiao Liu, Xiehuang Sheng and Dezhan Chen
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 16) pp:NaN10426-10426
Publication Date(Web):2017/03/24
DOI:10.1039/C6CP08068E
The detailed formation mechanisms of C-ribonucleoside and N-ribonucleoside via the reaction of 2,4,6-triaminopyrimidine (TAP) with (D)-ribose in aqueous solution were explored using density functional theory (DFT). The calculations indicate that five isomers (α,β-furanose, α,β-pyranose and open-chain aldehyde) of (D)-ribose can exist in equilibrium in aqueous solution. In contrast to cyclic isomers, an open-chain aldehyde is most feasible to react with TAP. In general, the formation pathways of C-nucleoside and N-nucleoside proceed in three steps including nucleophilic addition, dehydration and cyclization. The calculated apparent activation energies are 28.8 kcal mol−1 and 29.2 kcal mol−1, respectively. It suggests that both C- and N-nucleoside can be formed in aqueous solution, which is in good agreement with the experimental results. The water molecule plays an important “H-bridge” role by the hydrogen atom relay. Finally, a model structure of nucleobase, which will be beneficial for the C–C glycosidic bond formation, is proposed.
Co-reporter:Xiehuang Sheng, Chao Shan, Jianbiao Liu, Jintong Yang, Bin Sun and Dezhan Chen
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 20) pp:NaN13159-13159
Publication Date(Web):2017/05/08
DOI:10.1039/C7CP00804J
Ferroptosis is a recently discovered iron-dependent form of non-apoptotic cell death caused by the accumulation of membrane lipid peroxidation products, which is involved in various pathological conditions of the brain, kidney, liver and heart. A potent spiroquinoxalinamine derivative named liproxstatin-1 is discovered by high-throughput screening, which is able to suppress ferroptosis via lipid peroxide scavenging in vivo. Thus, molecular simulations, density functional theory (DFT) and variational transition-state theory with a small-curvature tunneling (SCT) coefficient are utilized to elucidate the detailed mechanisms of inactivation of a lipid peroxide radical by liproxstatin-1. H-atom abstracted from liproxstatin-1 by a CH3OO˙ radical occurs preferentially at the aromatic amine site (1′-NH) under thermodynamic and frontier molecular orbital analysis. The value of a calculated rate constant at 300 K is up to 6.38 × 103 M−1 S−1, indicating that the quantum tunneling effect is responsible for making a free radical trapping reaction more efficient by liproxstatin-1. The production of a liproxstatin-1 radical is easily regenerated to the active reduced form by ubiquinol in the body to avoid secondary damage by free radicals. A benzene ring and the higher HOMO energy are beneficial to enhance the lipid radical scavenging activity based on the structure–activity relationship study. Overall, the present results provide theoretical insights into the exploration of novel ferroptosis inhibitors.














![Phosphine, diphenyl[(1Z)-2-phenylethenyl]-](/data/chemimg/3747900/14090-07-4.png)
![Phosphine, diphenyl[(1Z)-2-phenylethenyl]-](/data/chemimg/3747900/14090-07-4_b.png)
![Phosphine, diphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14090-06-3.png)
![Phosphine, diphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14090-06-3_b.png)