Co-reporter:Jing Yuwen, Sumit Chakraborty, William W. Brennessel, and William D. Jones
ACS Catalysis May 5, 2017 Volume 7(Issue 5) pp:3735-3735
Publication Date(Web):April 20, 2017
DOI:10.1021/acscatal.7b00623
Here, we report an additive-free catalytic system for hydrogenation of carboxylic acid esters to alcohols with a well-defined cobalt pincer catalyst precursor. Various substrates, including methyl, ethyl, and benzyl esters, have been evaluated under hydrogenation conditions; however, methyl esters have low reactivity compared to those of the corresponding ethyl and benzyl esters. The biomass-derived γ-valerolactone successfully formed 1,4-pentanediol with a turnover number of 3890 with this system. Metal–ligand cooperativity is probed with the related [PN(Me)P] derivative of the cobalt catalyst, and the results suggest a nonbifunctional hydrogenation mechanism.Keywords: alcohols; catalysis; cobalt; esters; hydrogenation;
Co-reporter:Sumit Chakraborty, William W. Brennessel, and William D. Jones
Journal of the American Chemical Society June 18, 2014 Volume 136(Issue 24) pp:8564-8567
Publication Date(Web):May 30, 2014
DOI:10.1021/ja504523b
A well-defined iron complex (3) supported by a bis(phosphino)amine pincer ligand efficiently catalyzes both acceptorless dehydrogenation and hydrogenation of N-heterocycles. The products from these reactions are isolated in good yields. Complex 3, the active catalytic species in the dehydrogenation reaction, is independently synthesized and characterized, and its structure is confirmed by X-ray crystallography. A trans-dihydride intermediate (4) is proposed to be involved in the hydrogenation reaction, and its existence is verified by NMR and trapping experiments.
Co-reporter:Miles Wilklow-Marnell, William W. Brennessel, and William D. Jones
Organometallics August 28, 2017 Volume 36(Issue 16) pp:3125-3125
Publication Date(Web):August 16, 2017
DOI:10.1021/acs.organomet.7b00466
The reaction of iPrPCPIrH4 (iPrPCP = κ3-2,6-C6H3(CH2P(iPr)2)2) with ≥2 equiv of 2,3,4,5,6-pentafluoroacetophenone (AP-F5) in aromatic solvents at room temperature leads to the hydrogenation of AP-F5 and formation of the corresponding 5-coordinate alkoxy hydride species (I) within several minutes. Heating at ≥120 °C quickly provides the cyclometalated trans C-H product iPrPCPIr(κ-O,C-OC8H3F4)H (III), which slowly isomerizes to the cis C-H product iPrPCPIr(κ-C,O-OC8H3F4)H (V). The fate of the fluoride during formation of III and V is not entirely clear, though the production of HF is implicated by significant glass etching and several crystal structures. When iPrPCPIrH4 is allowed to react overnight with ≥2 equiv of AP-F5 at room temperature, formation of C–F oxidative addition product iPrPCPIr(κ-O,C-OC8H3F4)F (II) is observed by 31P and 19F NMR spectroscopy. When iPrPCPIrH4 activated with tert-butylethylene is employed, formation of II occurs within 10 min. Analogous reactivity is observed with several other fluorinated aryl ketones, and the crystal structure of the C–F oxidative addition product of 2,6-difluoroacetophenone (IIAF2) has been determined. This represents the first well-defined examples of stable iridium fluorides formed by C(sp2)–F oxidative addition.
Co-reporter:Lloyd Munjanja, Hongmei Yuan, William W. Brennessel, William D. Jones
Journal of Organometallic Chemistry 2017 Volume 847(Volume 847) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jorganchem.2017.02.015
•Cp*Rh(III) complexes having functional N,O chelate ligands have been synthesized.•These complexes act as catalysts for the dehydrogenation of alcohols.•These complexes act as catalysts for the hydrogenation of ketones.•These compounds form nanoparticles which serve as the active catalysts.Cp*Rh(III) complexes 1a and 1b (Cp* = 1, 2, 3, 4, 5-pentamethylcyclopentadienyl) having functional N,O chelate ligands have been synthesized and characterized by 1H, 13C{1H} NMR spectroscopy, elemental analysis, IR spectroscopy and X-ray diffraction. Reactivity of these complexes has been investigated towards dehydrogenation of alcohols and hydrogenation of ketones. It was found that these compounds are precursors to the formation of Rh nanoparticles which serve as catalysts, as evidenced by mercury poisoning of the catalysis and direct observation of the particles by TEM, EDX, and XRD.Synopsis: Cp*Rh(III) complexes with functional N,O chelate ligands have been synthesized and examined for catalytic activity as alcohol dehydrogenation catalyst precursors.Download high-res image (121KB)Download full-size image.
Co-reporter:Srinivasan Ramakrishnan, Sumit Chakraborty, William W. Brennessel, Christopher E. D. Chidsey and William D. Jones
Chemical Science 2016 vol. 7(Issue 1) pp:117-127
Publication Date(Web):29 Oct 2015
DOI:10.1039/C5SC03189C
A series of square-planar nickel hydride complexes supported by bis(phosphinite) pincer ligands with varying substituents (–OMe, –Me, and –But) on the pincer backbone have been synthesized and completely characterized by NMR spectroscopy, IR spectroscopy, elemental analysis, and X-ray crystallography. Their cyclic voltammograms show irreversible oxidation peaks (peak potentials from 101 to 316 mV vs. Fc+/Fc) with peak currents consistent with overall one-electron oxidations. Chemical oxidation by the one-electron oxidant Ce(NBu4)2(NO3)6 was studied by NMR spectroscopy, which provided quantitative evidence for post-oxidative H2 evolution leading to a solvent-coordinated nickel(II) species with the pincer backbone intact. Bulk electrolysis of the unsubstituted nickel hydride (3a) showed an overall one-electron stoichiometry and gas chromatographic analysis of the headspace gas after electrolysis further confirmed stoichiometric production of dihydrogen. Due to the extremely high rate of the post-oxidative chemical process, electrochemical simulations have been used to establish a lower limit of the bimolecular rate constant (kf > 107 M−1 s−1) for the H2 evolution step. To the best of our knowledge, this is the fastest known oxidative H2 evolution process observed in transition metal hydrides. Quantum chemical calculations based on DFT indicate that the one-electron oxidation of the nickel hydride complex provides a strong chemical driving force (−90.3 kcal mol−1) for the production of H2 at highly oxidizing potentials.
Co-reporter:Ruibo Xu, Sumit Chakraborty, Sarina M. Bellows, Hongmei Yuan, Thomas R. Cundari, and William D. Jones
ACS Catalysis 2016 Volume 6(Issue 3) pp:2127
Publication Date(Web):February 11, 2016
DOI:10.1021/acscatal.5b02674
Hydrogenation of alkenes containing polarized C═C double bonds has been achieved with iron-based homogeneous catalysts bearing a bis(phosphino)amine pincer ligand. Under standard catalytic conditions (5 mol % of (PNHPiPr)Fe(H)2(CO) (PNHPiPr = NH(CH2CH2PiPr2)2), 23 °C, 1 atm of H2), styrene derivatives containing electron-withdrawing para substituents reacted much more quickly than both the parent styrene and substituted styrenes with an electron-donating group. Selective hydrogenation of C═C double bonds occurs in the presence of other reducible functionalities such as −CO2Me, −CN, and N-heterocycles. For the α,β-unsaturated ketone benzalacetone, both C═C and C═O bonds have been reduced in the final product, but NMR analysis at the initial stage of catalysis demonstrates that the C═O bond is reduced much more rapidly than the C═C bond. Although Hanson and co-workers have proposed a nonbifunctional alkene hydrogenation mechanism for related nickel and cobalt catalysts, the iron system described here operates via a stepwise metal–ligand cooperative pathway of Fe–H hydride transfer, resulting in an ionic intermediate, followed by N–H proton transfer from the pincer ligand to form the hydrogenated product. Experimental and computational studies indicate that the polarization of the C═C bond is imperative for hydrogenation with this iron catalyst.Keywords: bifunctional mechanism; DFT calculations; iron catalysis; metal−ligand cooperativity; olefin/alkene hydrogenation
Co-reporter:Jing Yuwen, Yunzhe Jiao, William W. Brennessel, and William D. Jones
Inorganic Chemistry 2016 Volume 55(Issue 18) pp:9482-9491
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.inorgchem.6b01992
The active fragment [Tp′Rh(PMe3)], generated from a thermal precursor Tp′Rh(PMe3)(CH3)H, underwent oxidative addition of water and alcohols to give O–H adducts of the type Tp′Rh(PMe3)(OR)H (R = H, Me, Et, nPr, nBu, CH2Ph, iPr, c-pentyl, CH2CF3, CH2CH2OH) at ambient temperature. These activation products eliminate water or alcohols in benzene, which allows determination of the relative metal–oxygen bond energies by using previously established kinetics techniques. Analysis of the relationship between the relative M–O bond strengths and O–H bond strengths showed a linear correlation with RM–O/O–H of 0.97 (3) for aliphatic alcohols. The two extraordinary substrates (R = CH2CF3, CH2CH2OH) both have stronger M–O bonds than would be predicted from this trend, suggesting the stabilization of the M–O bond when an electron-withdrawing substituent is present as previously seen in M–C bond strengths. In addition, the O–H activation products of aliphatic alcohols are thermally unstable at 80 °C, as rearrangement to form Tp′Rh(PMe3)H2 from β-elimination is observed after 1 or 2 d. Benzyl alcohol and 2,2,2-trifluoroethanol activation products were stable. For benzyl alcohol, although the O–H activation product was kinetically favored, the C–H activation products of the benzene ring were thermodynamically preferred.
Co-reporter:Lloyd Munjanja, Coralys Torres-López, William W. Brennessel, and William D. Jones
Organometallics 2016 Volume 35(Issue 11) pp:2010-2013
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.organomet.6b00304
The (dippe)palladium(0) fragment generated from [(dippe)Pd(μ-H)]2 (1) has been shown to form an intermediate η2-nitrile complex with acetonitrile (dippe)Pd(η2-C,N-CH3CN-BEt3) (2a) in the presence of BEt3 [(dippe = bis-diisopropylphosphino)ethane)]. On introducing a solution of 2a to 1 equiv of BPh3, rapid formation of (dippe)Pd(η2-C,N-CH3CNBPh3) (2a′) is observed. Heating 2a′ at 100 °C in THF-d8 results in the C–CN activation product 3a′, (dippe)Pd(CH3)(CNBPh3). Reaction of 1 with benzonitrile in the presence of BEt3 gives the C–CN activation product (dippe)Pd(Ph)(CN-BEt3) (3b) exclusively. The complexes 2a, 2a′, 3a′, and 3b were characterized by 1H, 31P{1H}, and 13C{1H} NMR spectroscopy, elemental analysis, IR spectroscopy, and X-ray diffraction.
Co-reporter:Kelsey R. BreretonSarina M. Bellows, Hengameh Fallah, Antonio A. Lopez, Robert M. Adams, Alexander J. M. Miller, William D. JonesThomas R. Cundari
The Journal of Physical Chemistry B 2016 Volume 120(Issue 50) pp:12911-12919
Publication Date(Web):December 8, 2016
DOI:10.1021/acs.jpcb.6b09864
Hydricity, or hydride donating ability, is a thermodynamic value that helps define the reactivity of transition metal hydrides. To avoid some of the challenges of experimental hydricity measurements in water, a computational method for the determination of aqueous hydricity values has been developed. With a thermochemical cycle involving deprotonation of the metal hydride (pKa), 2e– oxidation of the metal (E°), and 2e– reduction of the proton, hydricity values are provided along with other valuable thermodynamic information. The impact of empirical corrections (for example, calibrating reduction potentials with 2e– organic versus 1e– inorganic potentials) was assessed in the calculation of the reduction potentials, acidities, and hydricities of a series of iridium hydride complexes. Calculated hydricities are consistent with electronic trends and agree well with experimental values.
Co-reporter:Sumit Chakraborty; Paige E. Piszel; Cassandra E. Hayes; R. Tom Baker
Journal of the American Chemical Society 2015 Volume 137(Issue 45) pp:14264-14267
Publication Date(Web):November 2, 2015
DOI:10.1021/jacs.5b10257
A highly selective (>99%) tandem catalytic system for the conversion of ethanol (up to 37%) to n-butanol, through the Guerbet process, has been developed using a bifunctional iridium catalyst coupled with bulky nickel or copper hydroxides. These sterically crowded nickel and copper hydroxides catalyze the key aldol coupling reaction of acetaldehyde to exclusively yield the C4 coupling product, crotonaldehyde. Iridium-mediated dehydrogenation of ethanol to acetaldehyde has led to the development of an ethanol-to-butanol process operated at a lower temperature.
Co-reporter:Ruibo Xu, Sumit Chakraborty, Hongmei Yuan, and William D. Jones
ACS Catalysis 2015 Volume 5(Issue 11) pp:6350
Publication Date(Web):September 15, 2015
DOI:10.1021/acscatal.5b02002
Acceptorless, reversible dehydrogenation and hydrogenation reactions involving N-heterocycles are reported with a well-defined cobalt complex supported by an aminobis(phosphine) [PN(H)P] pincer ligand. Several N-heterocycle substrates have been evaluated under dehydrogenation and hydrogenation conditions. The cobalt-catalyzed amine dehydrogenation step, a key step in the dehydrogenation process, has been independently verified. Control studies with related cycloalkanes suggest that a direct acceptorless alkane dehydrogenation pathway is unlikely. The metal–ligand cooperativity is probed with the related [PN(Me)P] derivative of the cobalt catalyst. These results suggest a bifunctional dehydrogenation pathway and a nonbifunctional hydrogenation mechanism.Keywords: acceptorless dehydrogenation; cobalt catalysis; H2 storage materials; hydrogenation; N-heterocycles; pincer ligands
Co-reporter:Natalie H. Chan, James H. Roache, William D. Jones
Inorganica Chimica Acta 2015 Volume 437() pp:36-40
Publication Date(Web):1 October 2015
DOI:10.1016/j.ica.2015.07.040
•Carbon–sulfur bond cleavage is achieved in benzothiophene.•Assignment of NMR resonances of κ2-benzothiophene.•X-ray structure of a dinuclear C–S activation product.Cp∗Co(C2H4)2 was reacted with benzothiophene and C–S activation of the vinyl-sulfur bond was observed. The resulting product, [Cp∗Co]2(μ-κ2-C,S-C8H6S) was characterized by 1H NMR, 13C{1H} NMR, 13C DEPT-135 NMR, and 1H–13C 2D HSQC spectroscopies. The broad Cp∗ methyl peaks in these spectra suggested a fluxional structure.The reactive fragment [Cp∗CoI] reacts with benzothiophene to give a C–S inserted dinuclear product.
Co-reporter:Yunzhe Jiao, William W. Brennessel, and William D. Jones
Organometallics 2015 Volume 34(Issue 8) pp:1552-1566
Publication Date(Web):April 15, 2015
DOI:10.1021/acs.organomet.5b00131
The reactive fragment [Tp′Rh(PMe3)], generated from the thermal precursor Tp′Rh(PMe3)(Me)H, is found to cleave the C–Cl bonds of chlorohydrocarbons under mild conditions. Reaction with chloromethane gives clean formation of an initial C–H activation product, which rearranges to form the C–Cl activation product at 30 °C. Reaction with dichloromethane or benzyl chloride gives a mixture of C–Cl activation products as well as products from chlorination. Reaction with chlorocyclohexane gives a mixture of intermediates from C–H activation, which react further upon heating to give a C–Cl cleavage product as well as the β-chloride elimination product Tp′Rh(PMe3)(Cl)H plus cyclohexene. Complete conversion from a C–H activation product to a C–Cl activation product was observed in the reaction with 1,2-dichloroethylene, where β-elimination is circumvented. Activation of 1-chlorobutane, 1,2-dichloroethane, or 1,4-dichlorobutane gives a mixture of C–Cl activation products as well as Tp′Rh(PMe3)(Cl)H plus olefin. Similar to the case for activation of methylene chloride, C–Cl activation and hydride/chloride exchange was observed in the reaction with benzyl chloride, where C–H activation was not seen. The reaction with chlorobenzene gives isomeric species resulting from C–H activation, which react further to give the corresponding chloride derivatives upon heating. Reaction with pentachlorobenzene gives a cyclometalated product from C–H bond cleavage in the phosphine ligand. These reactions are compared and contrasted with related photoreactions with the [Tp′Rh(CNneopentyl)] analogue, where C–H activation is solely observed in most cases. Mechanistic studies suggest the spectator ligand dependent reactivity relies greatly on the dissociation energy of the Tp′Rh–L bond.
Co-reporter:Ahmet Gunay, William W. Brennessel, and William D. Jones
Organometallics 2015 Volume 34(Issue 11) pp:2233-2239
Publication Date(Web):December 1, 2014
DOI:10.1021/om500999u
Carbon–carbon bond activation reactions of acetylene derivatives featuring sp–sp3 C–C bonds or both sp–sp2 and sp–sp single C–C bonds were studied via photolysis of platinum compounds. Novel Pt0–acetylene complexes with η2 coordination of the alkynes were synthesized and characterized. Irradiation of (dtbpe)Pt(η2-H3CC≡CCH3) (1), (dtbpe)Pt[η2-(H3C)3CC≡CC(CH3)3] (3), and [(dtbpe)Pt]2(μ2-η2:η2-H3CC≡CC≡CCH3) (6) with UV light (λ >300 nm) produced the activation product (dtbpe)Pt(D)(C6D5) (2) as a result of C–D bond activation of the solvent (C6D6), whereas (dtbpe)Pt(η2-F3CC≡CCF3) (4) and (dippe)Pt(η2-F3CC≡CCF3) (5) remained unchanged upon irradiation for 22 h. Photolysis of [(dtbpe)Pt]2(μ2-η2:η2-PhC≡CC≡CPh) (7) and (dippe)Pt(η2-PhC≡CC≡CPh) (9) resulted in [(dtbpe)(Ph)Pt]2(μ-C≡CC≡C−) (8) and (dippe)Pt(Ph)(C≡CC≡CPh) (10), respectively, showing exclusive C–C bond activation through sp–sp2 type C–C bonds. Both of the products stayed unchanged upon heating to 150 °C overnight.
Co-reporter:Aaron P. Walsh and William D. Jones
Organometallics 2015 Volume 34(Issue 13) pp:3400-3407
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.organomet.5b00369
The effect of the carboxylate used in a concerted metalation–deprotonation reaction is probed and shows a direct correlation of pKa to observed rate up to a pKa of 4.3, where the rate drops off at higher pKa. The rate of the C–H activation of 2-(4-methoxyphenyl)pyridine with [Cp*RhCl2]2 and carboxylate follows first-order kinetics in the active metal species, Cp*RhCl(κ2-OAc), and zero-order kinetics in substrate when in a 1:1 ratio. There is a first-order dependence on substrate observed when excess substrate is present. The evaluation of the mechanism using kinetic studies allowed for a mechanistic proposal in which a second Ph′Py coordinates prior to the rate-determining C–H activation.
Co-reporter:Sarina M. Bellows, Thomas R. Cundari, and William D. Jones
Organometallics 2015 Volume 34(Issue 16) pp:4032-4038
Publication Date(Web):August 6, 2015
DOI:10.1021/acs.organomet.5b00452
The chemical inertness of C(sp3)–H bonds has made their functionalization difficult, especially with earth-abundant 3d metals. The mechanism of C–H activation with the Co(III) complex Cp*(PMe3)Co(CH3)(OTf) was studied by DFT at the M06/6-31+G(d) level of theory and was determined to be in very good agreement with experimental data. The Co(III) complex can activate the C–H bond of methane through an oxidative hydrogen migration mechanism with an activation free energy of 32.4 kcal/mol in CH2Cl2. Although C2H6 and c-C6H12 have weaker C–H bonds than CH4, their C–H activation barriers were greater at 34.9 and 45.9 kcal/mol, respectively. The C(sp3)–H and C(sp2)–H activation of this Co(III) complex is primarily driven by the sterics (−E′s) of the substrates and supporting ligands and not the strength of the C–H bond.
Co-reporter:Sumit Chakraborty, Paige E. Piszel, William W. Brennessel, and William D. Jones
Organometallics 2015 Volume 34(Issue 21) pp:5203-5206
Publication Date(Web):October 22, 2015
DOI:10.1021/acs.organomet.5b00824
A single homogeneous nickel(II) complex, supported by the tris(3,5-dimethylpyrazolyl)borate ligand and 2-hydroxyquinoline ancillary ligand, is shown to catalyze both acceptorless dehydrogenation of alcohols and hydrogenation of carbonyl compounds under mild conditions. Products from the catalytic reactions were isolated with good yields. A mechanistic investigation highlights the critical role of the 2-hydroxyquinoline ligand in the catalysis and argues against a stepwise dehydrogenation pathway.
Co-reporter:Barbara Procacci; Yunzhe Jiao; Meagan E. Evans; William D. Jones; Robin N. Perutz;Adrian C. Whitwood
Journal of the American Chemical Society 2014 Volume 137(Issue 3) pp:1258-1272
Publication Date(Web):December 29, 2014
DOI:10.1021/ja5113172
The photochemical reactions of Tp′Rh(PMe3)H2 (1) and thermal reactions of Tp′Rh(PMe3)(CH3)H (1a, Tp′ = tris(3,5-dimethylpyrazolyl)borate) with substrates containing B–H, Si–H, C–F, and C–H bonds are reported. Complexes 1 and 1a are known activators of C–H bonds, including those of alkanes. Kinetic studies of reactions with HBpin and PhSiH3 show that photodissociation of H2 from 1 occurs prior to substrate attack, whereas thermal reaction of 1a proceeds by bimolecular reaction with the substrate. Complete intramolecular selectivity for B–H over C–H activation of HBpin (pin = pinacolate) leading to Tp′Rh(PMe3)(Bpin)H is observed. Similarly, the reaction with Et2SiH2 shows a strong preference for Si–H over C–H activation, generating Tp′Rh(PMe3)(SiEt2H)H. The Rh(Bpin)H and Rh(SiEt2H)H products were stable to heating in benzene in accord with DFT calculations that showed that reaction with benzene is endoergic. The intramolecular competition with PhSiH3 yields a ∼1:4 mixture of Tp′Rh(PMe3)(C6H4SiH3)H and Tp′Rh(PMe3)(SiPhH2)H, respectively. Reaction with pentafluoropyridine generates Tp′Rh(PMe3)(C5NF4)F, while reaction with 2,3,5,6-tetrafluoropyridine yields a mixture of C–H and C–F activated products. Hexafluorobenzene proves unreactive. Crystal structures are reported for B–H, Si–H, and C–F activated products, but in the latter case a bifluoride complex Tp′Rh(PMe3)(C5NF4)(FHF) was crystallized. Intermolecular competition reactions were studied by photoreaction of 1 in C6F6 with benzene and another substrate (HBpin, PhSiH3, or pentafluoropyridine) employing in situ laser photolysis in the NMR probe, resulting in a wide-ranging map of kinetic selectivities. The mechanisms of intramolecular and intermolecular selection are analyzed.
Co-reporter:Yunzhe Jiao, William W. Brennessel and William D. Jones
Chemical Science 2014 vol. 5(Issue 2) pp:804-812
Publication Date(Web):06 Dec 2013
DOI:10.1039/C3SC52748D
The thermal precursor Tp′Rh[P(OMe)3](Me)H was used to generate the active [Tp′Rh[P(OMe)3]] fragment, which activates C–H bonds of various hydrocarbons to form products of the type Tp′Rh[P(OMe)3](R)H (Tp′ = tris-(3,5-dimethylpyrazolyl)borate). Only one single activation product was observed in each case. The structures of Tp′Rh[P(OMe)3](R)X (X = H, Br, Cl) have been characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. The kinetics of reductive elimination of RH from Tp′Rh[P(OMe)3](R)H as well as competition experiments between substrates allow measurement of the Rh–C bond strengths relative to the Rh–Ph bond strength. Two separate linear correlations of the Rh–C bond energies versus H–C bond energies were found based on whether the alkyl group is α-substituted or not. While the correlation for α-substituted substrates gives a slope of 1.45, smaller than the slope (1.55) for unsubstituted hydrocarbons, the Rh–C bond energies of the former group are higher by ∼7 kcal mol−1. In comparison with the analogous [Tp′Rh(PMe3)] and [Tp′Rh(CNneopentyl)] systems, replacing the spectator ligand with a more electronic donating group slightly increases metal–carbon bond strengths as the trend in slopes of the correlations follows an order of CNneopentyl < P(OMe)3 ≤ PMe3.
Co-reporter:Sumit Chakraborty, Paraskevi O. Lagaditis, Moritz Förster, Elizabeth A. Bielinski, Nilay Hazari, Max C. Holthausen, William D. Jones, and Sven Schneider
ACS Catalysis 2014 Volume 4(Issue 11) pp:3994
Publication Date(Web):September 25, 2014
DOI:10.1021/cs5009656
Acceptorless dehydrogenation of alcohols, an important organic transformation, was accomplished with well-defined and inexpensive iron-based catalysts supported by a cooperating PNP pincer ligand. Benzylic and aliphatic secondary alcohols were dehydrogenated to the corresponding ketones in good isolated yields upon release of dihydrogen. Primary alcohols were dehydrogenated to esters and lactones, respectively. Mixed primary/secondary diols were oxidized at the secondary alcohol moiety with good chemoselectivity. The mechanism of the reaction was investigated using both experiment and DFT calculations, and the crucial role of metal–ligand cooperativity in the reaction was elucidated. The iron complexes are also excellent catalysts for the hydrogenation of challenging ketone substrates at ambient temperature under mild H2 pressure, the reverse of secondary alcohol dehydrogenation.Keywords: acceptorless dehydrogenation; catalysis; hydrogenation; iron; metal−ligand cooperativity
Co-reporter:Juanjuan Li;James Morris;William W. Brennessel
Journal of Chemical Crystallography 2014 Volume 44( Issue 1) pp:15-19
Publication Date(Web):2014 January
DOI:10.1007/s10870-013-0476-0
Ni(IMes)2 (IMes = N,N′-(2,4,6-trimethylphenyl)imidazole) was prepared and reacted with dimethyldisulfide. A novel trinuclear product (IMes)(SMe)Ni(μ-SMe)2Ni(μ-SMe)2Ni(SMe)(IMes) (1) was obtained containing both bridging and non-bridging methylsulfido groups. The crystal of 1 belongs to triclinic space group P-1 with unit cell dimensions a = 9.6664(5) Å, b = 12.8304(6) Å, c = 16.1842(8) Å, α = 70.0370(10)°, β = 73.0050(10)°, γ = 86.7240(10)°, V = 1802.20(15) Å3, Z = 1, Dcalcd = 1.294 Mg m−3, and F(000) = 730.
Co-reporter:Christopher R. Turlington, James Morris, Peter S. White, William W. Brennessel, William D. Jones, Maurice Brookhart, and Joseph L. Templeton
Organometallics 2014 Volume 33(Issue 17) pp:4442-4448
Publication Date(Web):August 27, 2014
DOI:10.1021/om500660n
The reactions of oxygen atom transfer reagents with Rh(Cp*) complexes, each with a bidentate ligand and an accessible coordination site, are described (Cp* = η5-pentamethylcyclopentadienyl). When [Rh(Cp*)(phpy)(NCArF)][B(ArF)4] (1, phpy = 2-phenylene-κC1′-pyridine-κN, NCArF = 3,5-bis(trifluoromethyl)benzonitrile, B(ArF)4 = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) was treated with the soluble oxygen atom transfer reagent 2-tert-butylsulfonyliodosylbenzene (sPhIO), oxygen atom insertion into the rhodium–carbon bond of coordinated phpy was observed. This resulted in the formation of a κ2 2-(2-pyridyl)phenoxide ligand. Following insertion to form a new bidentate ligand, a second equivalent of sPhIO, acting as a neutral, two-electron donor ligand, coordinated to the rhodium center through the iodosyl oxygen. Over time, the sPhIO ligand dissociates and dimerization occurs to generate a phenoxide-bridged dinuclear species. The 2-(2-pyridyl)phenoxide ligand could be protonated and cleaved from the mononuclear rhodium-(sPhIO) adduct by treating with a carboxylic acid (pivalic acid) at room temperature. In addition, when rhodium complex 1 was treated with excess phpy (14 equiv), hydrogen peroxide, and acetic acid, 5 equiv of 2-(2-pyridyl)phenol formed. Deactivation of the organometallic species, probably due to oxidative degradation of Cp*, severely limited this catalysis.
Co-reporter:Yunzhe Jiao ; Meagan E. Evans ; James Morris ; William W. Brennessel
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6994-7004
Publication Date(Web):April 23, 2013
DOI:10.1021/ja400966y
A series of substituted methyl derivatives of the type Tp′Rh(CNneopentyl)(CH2X)H (CH2X = CH2C(═O)CH3, CH2C≡CCH3, CH2O-t-Bu, CH2CF3, CH2F, CHF2) was synthesized either by photolysis of Tp′Rh(CNneopentyl)(PhNCNneopentyl) in neat CH3X or by exchange with the labile hydrocarbon in Tp′Rh(CNneopentyl)(n-pentyl)H or Tp′Rh(CNneopentyl)(CH3)H. Only a single product was observed in each case. Clean reductive elimination was observed for all compounds in C6D6. Structures of these complexes and their corresponding chlorinated derivatives have been characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. Relative Rh–C bond energies are calculated using previously established kinetic techniques, and two separate linear correlations are observed versus known C–H bond strengths, one for the parent hydrocarbons, and one for the substituted hydrocarbons. Both correlations have slopes of 1.4, and are separated vertically by 7.5 kcal mol–1 (−CH2X above −CxHy). In addition, it is now clear that preferences for linear vs branched olefin insertion products in substituted derivatives can be predicted on the basis of the strengths of the β-C–H bonds. The DFT calculations of the metal–carbon bond strengths in these Rh–CH2X derivatives with α-substitution show a trend that is in good agreement with the experimental results.
Co-reporter:Yunzhe Jiao ; James Morris ; William W. Brennessel
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16198-16212
Publication Date(Web):October 15, 2013
DOI:10.1021/ja4080985
Tp′Rh(PMe3)(CH3)H was synthesized as a precursor to produce the coordinatively unsaturated fragment [Tp′Rh(PMe3)], which reacts with benzene, mesitylene, 3,3-dimethyl-1-butene, 2-methoxy-2-methylpropane, 2-butyne, acetone, pentane, cyclopentane, trifluoroethane, fluoromethane, dimethyl ether, and difluoromethane at ambient temperature to give only one product in almost quantitative yield in each case. All of the complexes Tp′Rh(PMe3)(R)H were characterized by NMR spectroscopy, and their halogenated derivatives were fully characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. The active species [Tp′Rh(PMe3)] was also able to activate the alkynyl C–H bond of terminal alkynes to give activation products of the type Tp′Rh(PMe3)(C≡CR)H (R = t-Bu, SiMe3, hexyl, CF3, Ph, p-MeOC6H4, and p-CF3C6H4). The measured relative rhodium–carbon bond strengths display two linear correlations with the corresponding carbon–hydrogen bond strengths, giving a slope of 1.54 for α-unsubstituted hydrocarbons and a slope of 1.71 for substrates with α-substitution. Similar trends of energy correlations were established by DFT calculated metal–carbon bond strengths for the same groups of substrates.
Co-reporter:Aaron P. Walsh, William W. Brennessel, William D. Jones
Inorganica Chimica Acta 2013 Volume 407() pp:131-138
Publication Date(Web):1 October 2013
DOI:10.1016/j.ica.2013.07.039
•New metal isocyanide complexes are prepared and structurally characterized.•Derivative include Cp∗ and p-cymene complexes of rhodium, iridium, and ruthenium complexes with chloride and iodide ligands.•Formation of electrophilic intermediates is examined.Several new electrophilic metal isocyanide complexes have been fully characterized and reported herein. Isocyanide induced cleavage of the dimer, [LMCl2]2 {LM = Cp∗Ir, Cp∗Rh, or (p-cymene)Ru}, with 2,6-xylylisocyanide or 2,6-diethylphenylisocyanide produces complexes of the general formula LM(CNAr)Cl2. Halide metathesis of the dichloro complexes with sodium iodide produces the corresponding complexes with the general formula LM(CNAr)I2. For the analogous ruthenium complexes better results were achieved via isocyanide induced cleavage of [(p-cymene)RuI2]2 and was synthesized differently from previous reports. Several neutral complexes in reaction with AgPF6 in acetonitrile form cationic, solvent-coordinated complexes have been fully characterized. Most reactions with rhodium decomposed to either [Cp∗RhCl(MeCN)]2[(PF6)2] starting from the dichloro complexes, or [Cp∗Rh(MeCN)3,(PF6)2] and Cp∗Rh(CNAr)I2 starting from the diiodo complexes. Several bases were probed to see if cyclization could be induced, but were not successful in any case. Many of these complexes have been characterized by single crystal X-ray crystallography.Several new electrophilic metal isocyanide complexes of the type Cp∗M(CNaryl)X2 and (cymene)Ru(CNaryl)X2 (aryl = 2,6-xylyl, 2,6-Et2C6H3; X = Cl, I) have been fully characterized and reported herein. Many are characterized by single crystal X-ray diffraction, and the structures are compared. Several cationic solvated derivatives are also described.
Co-reporter:Kimberly A. Manbeck, William W. Brennessel, William D. Jones
Inorganica Chimica Acta 2013 Volume 397() pp:140-143
Publication Date(Web):1 March 2013
DOI:10.1016/j.ica.2012.12.002
A cationic rhodium aquo methyl complex, [Rh(bpy)(CH3)(H2O)3]2+·2[BF4]−, was prepared and examined for activity related to the Shilov PtII system, specifically the formation of methanol. Instead, the complex exhibited interesting behavior under basic conditions, with rare double “rollover” metalation of the coordinated bipyridine ligand and deuterium incorporation into the rhodium methyl group.Graphical abstractA cationic rhodium aquo methyl complex, [Rh(bpy)(CH3)(H2O)3]2+·2[BF4]−, was prepared and examined for activity related to the Shilov PtII system, specifically the formation of methanol.Highlights► Base catalyzed H/D exchange into methyl group from water. ► Double rollover metalation of bipyridine is observed. ► Hydroxide does not attack methylrhodium dication to give methanol.
Co-reporter:Sabuj Kundu, Benjamin E.R. Snyder, Aaron P. Walsh, William W. Brennessel, William D. Jones
Polyhedron 2013 Volume 58() pp:99-105
Publication Date(Web):13 July 2013
DOI:10.1016/j.poly.2012.07.071
The complex (dippe)Pt(NBE)2 (NBE = norbornene) reacts with thioethers RSR′ upon heating to give C–S oxidative addition products (RSR′ = Ph2S, PhSMe, PhSallyl, MeSallyl, PhSvinyl, PhCH2SMe, PhSCF3, and dithiane). Continued heating leads to disproportionation and formation of R′2 and (dippe)Pt(SR)2 in several cases.The complex (dippe)Pt(NBE)2 (NBE = norbornene) reacts with thioethers RSR′ upon heating to give C–S oxidative addition products (dippe)Pt(SR)(R′).
Co-reporter:Gyeongshin Choi ; James Morris ; William W. Brennessel
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9276-9284
Publication Date(Web):May 23, 2012
DOI:10.1021/ja301095r
C–H bond activation of terminal alkynes by [Tp′Rh(CNneopentyl)] (Tp′ = hydridotris-(3,5-dimethylpyrazolyl)borate) resulted in the formation of terminal C–H bond activation products Tp′Rh(CNneopentyl)(C≡CR)H (R = t-Bu, SiMe3, hexyl, CF3, p-MeOC6H4, Ph, and p-CF3C6H4). A combination of kinetic selectivity determined in competition reactions and activation energy for reductive elimination has allowed for the calculation of relative Rh–Calkynyl bond strengths. The bond strengths of Rh–Calkynyl products are noticeably higher than those of Rh–Caryl and Rh–Calkyl analogues. The relationship between M–C and C–H bond strengths showed a linear correlation (slope RM–C/H–C = 1.32), and follows energy correlations previously established for unsubstituted sp2 and sp3 C–H bonds in aliphatic and aromatic hydrocarbons.
Co-reporter:Sabuj Kundu, William W. Brennessel, and William D. Jones
Inorganic Chemistry 2011 Volume 50(Issue 19) pp:9443-9453
Publication Date(Web):September 7, 2011
DOI:10.1021/ic201102v
Synthesis and characterization of new (PONOP) [2,6-bis(di-tert-butylphosphinito)pyridine] metal (Ni, Pd, Pt) complexes are reported. Surprisingly, these compounds [(PONOP)MCl]Cl in the presence of 1 equiv of superhydride (LiEt3BH) formed a new class of complexes (H-PONOP)MCl, in which the pyridine ring in the PONOP ligand lost its aromaticity as a result of hydride attack at the para position of the ring. The new Ni–H compound [(H-PONOP)NiH] was synthesized by reacting (H-PONOP)NiCl with 1 equiv of superhydride. Analogous Pd and Pt compounds were prepared. Reactivity of these new pincer complexes toward MeLi and PhLi also has been studied. These Ni complexes catalyzed the hydrosilylation of aldehyde. In some cases characterization of new (PONOP)M complexes was difficult because of high instability due to degradation of the P–O bond.
Co-reporter:James Kovach, Maria Peralta, William W. Brennessel, William D. Jones
Journal of Molecular Structure 2011 Volume 992(1–3) pp:33-38
Publication Date(Web):19 April 2011
DOI:10.1016/j.molstruc.2011.02.027
We report the synthesis of α-diimine 1,4-bis(2,5-di-tert-butylphenyl)-2,3-dimethyl-1,4-diaza-1,3-butadiene, 1, and α-iminoketones 2-[(3,5-xylyl)imino]acenaphthylen-1-one, 4, and 2-[(4-chlorophenyl)imino]acenaphthylen-1-one, 5, all of which have been characterized by 1H NMR, 13C NMR, IR, and X-ray crystallography. Also, we report the previously unknown X-ray crystal structures of α-diimines Ar-BIAN (Ar-BIAN = bis(arylimino)acenaphthene; Ar = 3,5-xylyl, 2; Ar = 4-chlorophenyl, 3) and α-iminoketone 2-[(2,6-xylyl)imino]acenaphthylen-1-one, 6. In solution, 4 and 5 show fluxional behavior observed with variable temperature 1H NMR in DMSO-d6 which is attributed to isomerization between the E and Z form of the imine.
Co-reporter:Sabuj Kundu, William W. Brennessel, William D. Jones
Inorganica Chimica Acta 2011 Volume 379(Issue 1) pp:109-114
Publication Date(Web):15 December 2011
DOI:10.1016/j.ica.2011.09.048
C–N bond activation of tert-butyl isocyanide in methanol using 2,6-bis(di-tert-butylphosphinito)pyridine (PONOP) metal (Ni, Pd, Pt) complexes and (dippe)NiCl2 are reported. t-BuOMe and t-BuCl were detected as organic products by GC–MS. Substitution of the metal-chloride by one molecule of tert-butyl isocyanide followed by carbonium ion loss/nucleophilic attack by chloride anion or methanol led to formation of a metal-cyanide bond.Graphical abstractThe pincer PONOP complexes of Ni, Pd, and Pt have been found to react with tert-butyl isocyanide to give an adduct. This adduct is unstable, and loses tert-butyl carbonium ion to produce the cyanide product.Highlights► We have observed C–N cleavage in nitriles. ► We have investigated reactions of Ni, Pd, and Pt pincer complexes. ► We report the synthesis of Ni, Pd, and Pt pincer cyanide complexes.
Co-reporter:James Kovach, William W. Brennessel, William D. Jones
Inorganica Chimica Acta 2011 Volume 367(Issue 1) pp:108-113
Publication Date(Web):28 February 2011
DOI:10.1016/j.ica.2010.12.005
Treatment of Rh(acac)(CO)2 (acac = acetoacetonate) with perchloric acid followed by addition of an α-diimine (α-diimine = 1,4-bis(Ar)-2,3-dimethyl-1,4-diaza-1,3-butadiene, Ar = 3,5-dimethylphenyl, 1; 3,5-di-tert-butylphenyl, 2; and 3,4,5-trimethoxyphenyl, 3; phenyl, 4; and 4-chlorophenyl, 5) generates a series of complexes of the type [Rh(α-diimine)(CO)2][ClO4] 6–10 with varying electronic properties of the supporting diimine ligand. X-ray crystal structures have been determined for the α-diimine ligands 1–5, and complexes 6, 8, and 10.Graphical abstractRh(acac)(CO)2 (acac = acetoacetonate) is used for the preparation of several new rhodium-diiminedicarbonyl cations of the type [Rh(α-diimine)(CO)2][ClO4] (α-diimine = 1,4-bis(Ar)-2,3-dimethyl-1,4-diaza-1,3-butadiene, Ar = 3,5-dimethylphenyl, 1; 3,5-di-tert-butylphenyl, 2; and 3,4,5-trimethoxyphenyl, 3; phenyl, 4; and 4-chlorophenyl, 5). X-ray crystal structures have been determined for the α-diimine ligands 1–5, and complexes 6, 8, and 10.Research highlights► Rhodium dicarbonyl diimine complexes. ► Structures of aryldiimines. ► Metal carbonyls.
Co-reporter:Matthew R. Grochowski;William W. Brennessel
Journal of Chemical Crystallography 2011 Volume 41( Issue 6) pp:829-833
Publication Date(Web):2011 June
DOI:10.1007/s10870-011-0006-x
The unique dinuclear Ir(III) complex (Cp*IrCl)(μ-H)[μ-(η1:η3-C6H6S)](IrCp*) (1) has been synthesized and characterized by NMR spectroscopy (1H and 13C), elemental analysis, and single crystal X-ray diffraction. It is the first structurally determined complex in which an activated thiophene ligand displays an η3-allylic interaction. 1 appears to form from successive C-H bond activations of 2,5-dimethylthiophene, resulting in its bridging the two iridium centers. The η3-allylic interaction occurs with one of the Ir centers and has Ir–Cthio bond lengths ranging from 2.133(5)-2.207(5) Å; the C–C double bond involved in the interaction has a bond length of 1.438(7) Å compared to 1.348(8) Å for the uncoordinated C–C double bond. The 3-carbon of the thiophene ring bridges both iridium centers with bond lengths of 2.036(5) Å and 2.208(5) Å. 1 crystallizes in space group P−1 with cell constants a = 8.6303(6) Å, b = 9.0153(6) Å, c = 18.1089(12) Å, α = 84.728(1)°, β = 87.534(1)°, γ = 64.373(1)°, and Z = 2. The structure was solved by direct methods and refined to R = 0.0363 (F2 > 2σ(F2)) and wR = 0.0851 (F2). The NMR data indicate the solution state structure is consistent with the solid state structure.
Co-reporter:Sabuj Kundu, William W. Brennessel, and William D. Jones
Organometallics 2011 Volume 30(Issue 19) pp:5147-5154
Publication Date(Web):September 9, 2011
DOI:10.1021/om200452z
C–S bond activation of both cyclic and acyclic thioesters using (dippe)Pt(NBE)2 (NBE = norbornene) are reported. In the cases of S-methyl thioacetate and S-ethyl thioacetate, (dippe)Pt(C(O)Me)(SR) (R = Me, Et) formed initially. Further heating in the presence of excess thioester led to formation of the final complex (dippe)Pt(SR)2 along with acetone and RSMe as organic side products. PtIV intermediates are proposed to be involved in the reaction mechanism.
Co-reporter:Ting Li and William D. Jones
Organometallics 2011 Volume 30(Issue 3) pp:547-555
Publication Date(Web):December 30, 2010
DOI:10.1021/om100907y
The isomerization of 2-methyl-3-butenenitrile (2M3BN) carried out by [Ni(dippe)H]2 can follow either a C−CN activation pathway to form the linear product 3-pentenenitrile (3PN) or a C−H activation pathway to give the branched olefin product 2-methyl-2-butenenitrile (2M2BN). Both pathways have been studied by DFT calculation methods, and the results match well with those observed in stoichiometric experiments. A detailed mechanism has been proposed and tested on several other model bisphosphine ligands to investigate the bite angle and electronic effects on the selectivity of nickel bisphosphine catalysts.
Co-reporter:Taro Tanabe, Meagan E. Evans, William W. Brennessel, and William D. Jones
Organometallics 2011 Volume 30(Issue 4) pp:834-843
Publication Date(Web):January 6, 2011
DOI:10.1021/om101002m
The [Tp′Rh(PR3)] fragments, where Tp′ = tris(3,5-dimethylpyrazolyl)borate, R3 = Me3 (a) or PhMe2 (b), were shown to be applicable for the activation of the C−H and C−CN bonds of acetonitrile. The photoirradiation of Tp′Rh(PR3)H2 (1a,b) in acetonitrile at room temperature afforded the complexes Tp′Rh(PR3)(CH2CN)H (3a,b), by the selective oxidative addition of the primary C−H bond of acetonitrile to the [Tp′Rh(PR3)] fragment generated by the dehydrogenation of 1. The thermal reactions of Tp′Rh(PMe3)(Ph)H (2a) or Tp′Rh(κ2-C6H4-2-PMe2)H (2b) in acetonitrile at 80 °C also resulted in the formation of 3a and 3b, respectively, by the oxidative addition of the C−H bond to the [Tp′Rh(PR3)] fragment, generated by the reductive elimination of benzene (from 2a) or intramolecular reductive elimination of the PPhMe2 ligand (from 2b). Heating the isolated C−H bond activated complexes 3a,b in acetonitrile at higher temperature (100 °C) for longer times (≈5 days) resulted in the formation of the corresponding C−CN bond activated complexes, Tp′Rh(PR3)(CH3)(CN) (4a,b), respectively. The molecular structures of the C−H activated product 3a,b (kinetic product) and C−CN activated product 4a,b (thermodynamic product) were fully characterized by NMR spectroscopy, elemental analysis, and X-ray crystallographic analysis. Furthermore, the secondary C−H bond activated product of succinonitrile, Tp′Rh(PMe3)[CH(CN)CH2CN]H (5), was isolated by the thermal reaction of 2a with succinonitrile (20 equiv) at 100 °C, and the molecular structure was characterized by NMR spectroscopy and X-ray crystallographic analysis. The cleavage of CH3CN was also investigated using DFT models to compare thermodynamics and identify transition-state geometries.
Co-reporter:Brett D. Swartz, William W. Brennessel, and William D. Jones
Organometallics 2011 Volume 30(Issue 6) pp:1523-1529
Publication Date(Web):March 3, 2011
DOI:10.1021/om101069j
The reaction of [(dippe)PtH]2 with benzonitrile at 140 °C produced two products, the C−H-activated adduct (dippe)Pt(H)(2-C6H4CN) (2) and the C−CN-activated adduct (dippe)Pt(Ph)(CN) (5), with the kinetically favored C−H activation product forming in a significant majority (∼18:1). Further reaction showed a formal β-cyano elimination in 2, forming (dippe)Pt(H)(CN) (3) and benzene. Following the elimination, C−H activation of a second benzonitrile by 3 led to three regioisomers of (dippe)Pt(CN)(C6H4CN) (4).
Co-reporter:Meagan E. Evans and William D. Jones
Organometallics 2011 Volume 30(Issue 12) pp:3371-3377
Publication Date(Web):May 20, 2011
DOI:10.1021/om2002602
Density functional theory has been used to evaluate the energetics of C–H and C–CN bond activation of acetonitrile by two rhodium fragments, [(C5Me5)Rh(CNMe)] and [TpRh(CNMe)], each containing a π-acceptor ligand (isocyanide) (Tp = tris(pyrazolyl)borate). These new results are evaluated against previous calculations on similar rhodium systems, [(C5Me5)Rh(PMe3)] and [TpRh(PMe3)], which contain a σ-donating ligand (phosphine). Our new DFT results show the barrier to C–H bond activation to be lower than the barrier to C–CN activation by 6.7 kcal mol–1 (for [(C5Me5)Rh(CNMe)]) and 9.3 kcal mol–1 (for [TpRh(CNMe)]). The addition of a PCM solvation correction did not change the relative energies of these two barriers for either complex, which is in contrast to previous results observed for the two analogous rhodium–phosphine systems. These new calculations along with experimental results demonstrate the importance of an electron-rich metal center for the stabilization of the transition state for C–CN cleavage and ultimately on the metal’s ability to cleave the C–CN bond.
Co-reporter:Tülay A. Ateşin, Sabuj Kundu, Karlyn Skugrud, Katherine A. Lai, Brett D. Swartz, Ting Li, William W. Brennessel, and William D. Jones
Organometallics 2011 Volume 30(Issue 17) pp:4578-4588
Publication Date(Web):August 15, 2011
DOI:10.1021/om200352d
Exchange reactions of 2- and 3-cyanothiophene, 2- and 3-methylthiophene, and 2- and 3-methoxythiophene, either with thiophene in the thiaplatinacycle Pt(dippe)(κ2-C,S-C4H4S) or with norbornene in Pt(dippe)(nor)2, were performed to probe the kinetic and thermodynamic selectivity of the C–S bond activation reactions. Kinetic data were collected by following these reactions by 31P{1H} NMR spectroscopy. The ground-state energies of the two possible products and the transition-state energies leading to the formation of these products were calculated using density functional theory. The comparison of the predicted selectivities from calculations with the experimentally observed selectivities showed good agreement for thermodynamic selectivity, but only moderate agreement for kinetic selectivity. The reactions with 2-cyanothiophene, 3-cyanothiophene, and 3-methoxythiophene gave kinetic products that were less favored thermodynamically. All of the other substituted thiophenes gave kinetic products that were also preferred thermodynamically. These results indicate that the selectivities seen in the C–S bond activation reactions of substituted thiophenes with the [Pt(dippe)] fragment are initially under kinetic control.
Co-reporter:Matthew R. Grochowski, James Morris, William W. Brennessel, and William D. Jones
Organometallics 2011 Volume 30(Issue 21) pp:5604-5610
Publication Date(Web):October 13, 2011
DOI:10.1021/om200342f
The complex [Rh(dippe)(μ-Cl)]2 (1) was reduced with potassium metal to produce the highly reactive Rh–I species [Rh(dippe)(μ-K·THF)]2 (2). 2 was characterized by NMR spectroscopy (31P, 1H) and IR spectroscopy after forming the derivative K[Rh(dippe)(CO)2] (2a). The C–CN bond of benzonitrile was cleaved by oxidative addition when it was reacted stoichiometrically with 2 to form the anionic Rh(I) complex K[Rh(dippe)(CN)(Ph)] (3). 3 has been characterized by NMR spectroscopy (31P, 1H, 13C), and by the use of 13C-labeled benzonitrile. C–CN bond cleavage was also attempted by reacting benzonitrile with the zwitterionic complex [Rh(dippe)(η6-Ph-BPh3)] (4). A product 5 was formed from reaction with 2 equiv of benzonitrile. Reaction with 13C-labeled benzonitrile has ruled out the possibility of C–CN cleavage in this species, and a C–H bond activation pathway is operative instead.
Co-reporter:Matthew R. Grochowski ; Ting Li ; William W. Brennessel
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12412-12421
Publication Date(Web):August 12, 2010
DOI:10.1021/ja104158h
The processes of C−C and C−S bond cleavage have been studied with the homogeneous organometallic compound [Ni(dippe)H]2 (1). When 1 is reacted with 2-cyanothiophene at room temperature, cleavage of the nitrile-substituted C−S bond occurs, forming the Ni−metallacycle complex (dippe)Ni(κ2-S,C-SCH═CHCH═C(CN)) (2a), which has been fully characterized by NMR spectroscopy and X-ray diffraction. 2a was converted to the C−CN cleavage product (dippe)Ni(CN)(2-thiophenyl) (3) when heated in solution. On closer inspection, four other intermediates were observed by 31P NMR spectroscopy at low temperature. Structures for the intermediates were elucidated through a combination of independent synthesis, theoretical calculations, chemical characterization, and experimental precedent. A kinetic product (dippe)Ni(κ2-S,C-SC(CN)=CHCH═CH) (2b) was formed from cleavage of the nonsubstituted C−S bond, as well as a Ni(0) η2-nitrile intermediate, (dippe)Ni(η2-C,N-2-cyanothiophene) (4), and a dinuclear mixed Ni(0)−Ni(II) product (6b). A complete DFT analysis of this system has been carried out to reveal comparative details about the two bond cleavage transition states.
Co-reporter:Meagan E. Evans ; Ting Li
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:16278-16284
Publication Date(Web):October 22, 2010
DOI:10.1021/ja107927b
The photochemical reaction of (C5Me5)Rh(PMe3)H2 (1) in neat acetonitrile leads to formation of the C−H activation product, (C5Me5)Rh(PMe3)(CH2CN)H (2). Thermolysis of this product in acetonitrile or benzene leads to thermal rearrangement to the C−C activation product, (C5Me5)Rh(PMe3)(CH3)(CN) (4). Similar results were observed for the reaction of 1 with benzonitrile. The photolysis of 1 in neat benzonitrile results in C−H activation at the ortho, meta, and para positions. Thermolysis of the mixture in neat benzonitrile results in clean conversion to the C−C activation product, (C5Me5)Rh(PMe3)(C6H5)(CN) (5). DFT calculations on the acetonitrile system show the barrier to C−H activation to be 4.3 kcal mol−1 lower than the barrier to C−C activation. A high-energy intermediate was also located and found to connect the transition states leading to C−H and C−C activation. This intermediate has an agostic hydrogen interaction with the rhodium center. Reactions of acetonitrile and benzonitrile with the fragment [Tp′Rh(CNneopentyl)] show only C−H and no C−C activation. These reactions with rhodium are compared and contrasted to related reactions with [Ni(dippe)H]2, which show only C−CN bond cleavage.
Co-reporter:Andreas Steffen, Richard M. Ward, William D. Jones, Todd B. Marder
Coordination Chemistry Reviews 2010 Volume 254(17–18) pp:1950-1976
Publication Date(Web):September 2010
DOI:10.1016/j.ccr.2010.03.002
The review discusses the synthesis, structures and properties of transition metal and main-group containing heterocyclopentadienes, with particular focus on dibenzometallacyclopentadienes (2,2′-biphenyl complexes), diethynylmetallacyclopentadienes, boroles and related main-group systems, and their catalytic and optoelectronic properties.
Co-reporter:Taro Tanabe, William W. Brennessel, Eric Clot, Odile Eisenstein and William D. Jones
Dalton Transactions 2010 vol. 39(Issue 43) pp:10495-10509
Publication Date(Web):05 Oct 2010
DOI:10.1039/C0DT00157K
A series of complexes of the type Tp′Rh(PR3)(ArF)H, where PR3 = PMe3 (3) and PMe2Ph (9), ArF = C6F5 (a), 2,3,4,5-C6F4H (b), 2,3,5,6-C6F4H (c), 2,4,6-C6F3H2 (d), 2,3-C6F2H3 (e), 2,5-C6F2H3 (g), and 2-C6FH4 (h) and Tp′ = tris(3,5-dimethylpyrazolyl)borate, has been synthesized as stable crystalline compounds by the reactions of the [Tp′Rh(PR3)] fragment with the corresponding fluorinated aromatic hydrocarbons, and their structures were characterized by NMR spectroscopy and elemental analysis together with X-ray crystallography. The kinetics of the reductive eliminations of fluoroarenes from complexes 3a–h in benzene-d6 solutions at 140 °C were investigated, but were complicated by the formation of the rhodium(I) bisphosphine complex, Tp′Rh(PMe3)2 (4). On the other hand, thermal reactions of (9) in THF-d8 solutions at 120 °C resulted in the formation of an intramolecular C–H bond activated complex of the phenyl group on the phosphorus atom, Tp′Rh(κ2-C6H4-2-PMe2)H (7), which prevents the formation of the corresponding bisphosphine complex. The experimentally determined rates of the reductive eliminations of fluoroarenes from the complexes 9a–h and their kinetic selectivities for formation in competition with the metallacycle have been used to determine relative Rh–CArF bond energies. The Rh–CArF bond energy is found to be dependent on the number of ortho fluorines. A plot of Rh–CArFvs. C–H bond strengths resulted in a line with a slope RM–C/C–H of 2.15 that closely matches the DFT calculated value (slope = 2.05).
Co-reporter:Brett D. Swartz, Tülay A. Ateşin, Matthew R. Grochowski, Stephen S. Oster, William W. Brennessel, William D. Jones
Inorganica Chimica Acta 2010 Volume 363(Issue 3) pp:517-522
Publication Date(Web):15 February 2010
DOI:10.1016/j.ica.2009.05.013
Two unusual lithium coordinated binuclear platinum- and rhodium-hydride complexes [M(dippe)(H)]2·LiHBEt3 were synthesized and characterized by NMR spectroscopy and X-ray crystallography. Not only does the lithium ion interact with the metal hydrides, but also with the B–H bond of the borohydride.An unusual binuclear platinum-hydride complex [Pt(dippe)(H)]2.LiHBEt3 shows a lithium ion bridging the two platinum hydrides and coordinating to the hydride of the HBEt3 unit. A related compound, [Rh(dippe)]2(μ-H)2·LiHBEt3], has also been prepared and structurally characterized, but now shows bridging hydrides coordinated to the LiHBEt3 moiety.
Co-reporter:Bradley M. Kraft, Eric Clot, Odile Eisenstein, William W. Brennessel, William D. Jones
Journal of Fluorine Chemistry 2010 Volume 131(Issue 11) pp:1122-1132
Publication Date(Web):November 2010
DOI:10.1016/j.jfluchem.2010.05.003
Cp*2ZrH2 (1) (Cp*: pentamethylcyclopentadienyl) reacts with cyclic perfluorinated olefins to give Cp*2ZrHF (2) and hydrodefluorinated products under very mild conditions. Initial C–F bond activation occurs selectively at the vinylic positions of the cycloolefin to exchange fluorine for hydrogen. Several mechanisms are discussed for this H/F exchange: (a) olefin insertion/β-fluoride elimination, (b) olefin insertion/α-fluoride elimination, and (c) hydride/fluoride σ-bond metathesis. Following H/F σ-bond metathesis exchange of both vinylic C–F bonds of perfluorocyclobutene, 1 then reacts with allylic C–F bonds by insertion/β-fluoride elimination. A similar sequence is observed with perfluorocyclopentene. Cp*2ZrHF reacts selectively with vinylic C–F bonds of perfluorocyclobutene to give 3,3,4,4-tetrafluorocyclobutene and Cp*2ZrF2 without further hydrodefluorination occurring. In the presence of excess 1 and H2, perfluorocyclobutene and perfluorocyclopentene are reduced to cyclobutane and cyclopentane in 46% and 16% yield, respectively. DFT calculations exclude the pathway by way of the olefin insertion/α-fluoride elimination and suggest that the pathway by way of hydride/fluoride σ-bond metathesis is preferred.Cp*2ZrH2 reduces perfluorocycloolefins by a sigma-bond metathesis pathway.
Co-reporter:Ting Li, Juventino J. García, William W. Brennessel and William D. Jones
Organometallics 2010 Volume 29(Issue 11) pp:2430-2445
Publication Date(Web):May 12, 2010
DOI:10.1021/om100001m
[Ni(dippe)H]2 has been reacted with a variety of aromatic nitriles. Both experimental and DFT calculation results have shown that an η2-arene complex with nickel coordinated to the C═C double bond next to the cyano substituent is the crucial intermediate leading to C−CN bond activation. Furthermore, the fluxional processes of the η2-arene species were investigated by low-temperature experiments as well as computational methods. In the case of dicyanobenzenes, a mechanism similar to that found for PhCN was found with the Ni(dippe) fragment rotating as it migrated around the phenyl ring through a series of η3-allyl-like transition states. For polycyclic aromatic nitriles, only certain η2-arenes were stable enough to contribute to the fluxional process, and nickel migrates via an η4-coordinated transition state. The transition states connecting the η2-nitrile complex to the η2-arene intermediate and the η2-arene intermediate to the C−CN bond activation products are at much higher energies compared to those for migration around the ring. In the reaction of 9-cyanoanthracene, the instability of the η2-arene precursor and the high-energy activation barrier resulted in the absence of the C−CN oxidative addition product. The complex with 9-cyanophenanthrene undergoes only C−CN cleavage upon photolysis.
Co-reporter:Ling Li, Yunzhe Jiao, William W. Brennessel, and William D. Jones
Organometallics 2010 Volume 29(Issue 20) pp:4593-4605
Publication Date(Web):September 21, 2010
DOI:10.1021/om100796q
The reactivity of four different cyclometalated iridium and rhodium complexes (1, Ir−N−Me; 2, Rh−N−Me; 3, Ir−N−Py; 4, Rh−N−Py) with ancillary ligands with different electronic and steric properties has been investigated by reactions of ethylene (a), propylene (b), carbon monoxide (c), tert-butylisocyanide (d), acetylene (e), and phenylacetylene (f). Only coordination products were obtained for the reactions of ethylene and propylene with 1 and 3, while inserted and rearranged products were achieved for the reactions with 2 and 4. Insertion of a single equivalent of acetylene was observed for the reactions with 2, 3, and 4, whereas reaction with 1 produces a product in which 4 equiv of acetylene has undergone insertion. The reactions with carbon monoxide showed clean M−C bond insertion products, while tert-butylisocyanide formed only terminal adducts. Two equivalents of phenylacetylene were observed to insert for all of the cyclometalated complexes. The regioselectivity was also investigated for each cyclometalated complex by using a series of internal unsymmetrical alkynes, and the results revealed that the regioselectivity was controlled by both steric and electronic factors. The insertion compounds were fully characterized by 1H NMR spectroscopy, 13C NMR spectroscopy, elemental analysis, and X-ray determinations for selected cases.
Co-reporter:Meagan E. Evans ; Catherine L. Burke ; Sornanong Yaibuathes ; Eric Clot ; Odile Eisenstein
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13464-13473
Publication Date(Web):August 26, 2009
DOI:10.1021/ja905057w
C−H bond activation of fluorinated aromatic hydrocarbons by [Tp′Rh(CNneopentyl)] resulted in the formation of products of the type Tp′Rh(CNneopentyl)(arylF)H. The stability of the Rh−Caryl product is shown to be strongly dependent on the number of ortho fluorines and only mildly dependent on the total number of fluorine substituents. Complexes with aryl groups containing two ortho fluorines have barriers to reductive elimination that are ∼5 kcal mol−1 higher than for those with a single ortho fluorine. Competition experiments along with ΔGre⧧ values allow for the determination of relative Rh−Caryl bond strengths and illustrate the large ortho fluorine effect on the strength of the Rh−Caryl bond. A large change in Rh−Caryl bond strength was measured for small changes in the respective calculated C−H bond strengths. Relating M−C to C−H bond strengths resulted in a line (slope = 2.14) that closely matches the theoretically calculated value (slope = 1.96). This is the first experimental quantization of an ortho fluorine effect as predicted by theory.
Co-reporter:Douglas D. Wick, William D. Jones
Inorganica Chimica Acta 2009 Volume 362(Issue 12) pp:4416-4421
Publication Date(Web):15 September 2009
DOI:10.1016/j.ica.2009.03.022
Treatment of Tp′Rh(PMe3)Cl2 and Tp′Rh(CNCH2CMe3)Cl2 with Cp2ZrH2 produces Tp′Rh(PMe3)H2 and Tp′Rh(CNCH2CMe3)H2, respectively, in excellent yield. Photolysis of benzene solutions of each dihydride complex generates hydrogen and the fragment [Tp′Rh(L)] which inserts into the solvent C–H bond. The phosphine dihydride has also been shown to be a catalyst for the hydrogenation of biphenylene, showing a capability to cleave C–C bonds. Reductive elimination of benzene from Tp′Rh(PMe3)PhH is nearly 250 times slower than from Cp*Rh(PMe3)PhH.Dihydride complexes of both Cp*RhLH2 and Tp′RhLH2 where L = PMe3 or CNCH2CMe3 are synthesized and their reactivity with C–H and C–C bonds compared.
Co-reporter:Meagan E. Evans, Ting Li, Andrew J. Vetter, Ryan D. Rieth and William D. Jones
The Journal of Organic Chemistry 2009 Volume 74(Issue 18) pp:6907-6914
Publication Date(Web):August 11, 2009
DOI:10.1021/jo9012223
Several transition-metal systems have been used to establish correlations between metal−carbon and carbon−hydrogen bonds. Here, the [Tp′RhL] fragment, where Tp′ = tris(3,5-dimethylpyrazolyl)borate and L = neopentyl isocyanide, is used to investigate C−H bond activation in a series of linear alkylnitriles and chloroalkanes. Using a combination of kinetic techniques, relative free energies can be found for the compounds TpRhL(CH3)H, Tp′RhL[(CH2)nCN]H (n = 1−5), and Tp′RhL[(CH2)mCl]H (m = 1, 3, 4, 5). It is found that the CN and Cl substituents dramatically strengthen the M−C bond more than anticipated if in the α-position, with the effect on bond strength diminishing substantially as the X group moves further from the metal (i.e, β, γ, δ). Examination of M−C vs C−H bond strengths shows that the Tp′RhL(CH2X)H compounds (X = phenyl, vinyl, CN, Cl) all show a good correlation, as do the alkyl, aryl, and vinyl derivatives. The compounds in the former group, however, have stronger M−C bonds than expected based on the C−H bond strengths and consequently, their correlation is separate from the other unsubstituted compounds.
Co-reporter:Matthew R. Grochowski, William W. Brennessel and William D. Jones
Organometallics 2009 Volume 28(Issue 9) pp:2661-2667
Publication Date(Web):April 17, 2009
DOI:10.1021/om900114m
Reaction of [Cp*IrHCl]2 (Cp* = η5-C5Me5) with 2-methylthiophene and 2,5-dimethylthiophene at 120 °C in the presence of H2 results in the cleavage of the thiophene carbon−sulfur bond(s). In both cases the thiophenes are ring-opened and hydrogenated, resulting in dinuclear Ir complexes with bridging thiolates. The primary product in the reaction involving 2,5-dimethylthiophene is [Cp*IrCl]2(μ-H)(μ-S-2-hexyl). This product has been characterized and is present in diastereomeric pairs. In the reaction with 2-methylthiophene a complex mixture consisting of five products is produced. The product distribution consists of mono- and disubstituted bridging thiolate complexes, three of which have been structurally characterized by single-crystal X-ray diffraction. Independent synthesis of each of these products has been performed, and characterization of the reaction mixture has been accomplished by 1H and 13C NMR spectroscopies, as well as by ESI-MS and elemental analysis. Reaction with 2-acetylthiophene showed very similar reactivity; an X-ray structure confirmed the nature of the diastereomeric pairs present.
Co-reporter:Ling Li, William W. Brennessel and William D. Jones
Organometallics 2009 Volume 28(Issue 12) pp:3492-3500
Publication Date(Web):May 4, 2009
DOI:10.1021/om9000742
Sodium acetate promoted C−H activation in a series of para-substituted phenyl imines has been examined using [Cp*MCl2]2 (M = Ir, Rh). The regioselectivity was investigated using a series of meta-substituted phenyl imines (−OMe, −CH3, −F, −COOMe, −CF3, and −CN) and 2-phenylpyridines (−OMe, −CH3, and −CF3). It was found that substrates with electron-donating substituents react significantly faster than substrates with electron-withdrawing substituents, which is consistent with an electrophilic C−H activation mechanism. It was also found that the regioselectivity of the C−H activation was extremely sensitive to steric effects, with a meta methyl group leading to only one regioisomer. Solvent and temperature studies showed that the reaction rate can be increased both by increasing temperature and by using polar solvents such as methanol. The regioselectivity was solvent dependent for the reaction with [Cp*IrCl2]2 but independent of solvent for the reaction with [Cp*RhCl2]2. The regioselectivity was temperature independent for both metals. With added acid, the aromatic C−H activation was shown to be reversible. Kinetic studies were performed, leading to the conclusion that [Cp*M(OAc)]+ is the key catalytic species responsible for the electrophilic C−H activation.
Co-reporter:Ahmet Gunay, Christian Müller, Rene J. Lachicotte, William W. Brennessel and William D. Jones
Organometallics 2009 Volume 28(Issue 22) pp:6524-6530
Publication Date(Web):October 30, 2009
DOI:10.1021/om900413a
Carbon−carbon bond activation reactions of asymmetric acetylene derivatives of the type L2Pt(PhC≡CR) were studied with 1,2-bis(diisopropylphosphino)ethane (dippe), 1,2-bis(di-tert-butylphosphino)ethane (dtbpe), and 1-diisopropylphosphino-2-dimethylaminoethane (dippdmae) chelates. (dippe)Pt(η2-PhC≡CCH3) (1a), (dippe)Pt(η2-PhC≡CCF3) (1b), and (dippe)Pt(η2-PhC≡CC(CH3)3) (1c) showed no thermal reactivity at 160 °C, but 1b showed evidence for C−C cleavage to form (dippe)Pt(Ph)(C≡CCF3) upon irradiation with UV light (>300 nm). In comparison, dtbpe analogues of these metal complexes, (dtbpe)Pt(η2-PhC≡CCH3) (2a), (dtbpe)Pt(η2-PhC≡CCF3) (2b), and (dtbpe)Pt(η2-PhC≡CC(CH3)3) (2c), showed either C−H or C−C activation products upon photolysis. 2b produced (dtbpe)Pt(Ph)(C≡CCF3), but 2a or 2c showed the formation of (dtbpe)Pt(D)(C6D5) (2D) by activation of the C6D6 solvent. Compounds 2a−c showed no thermal reactivity at 160 °C. Two complexes with the hemilabile chelate dippdmae were synthesized and fully characterized, (dippdmae)Pt(η2-PhC≡CCF3) (3b) and (dippdmae)Pt(η2-PhC≡CC(CH3)3) (3c). C−C cleavage products of the type (L2)Pt(Ph)(C≡CCF3) were observed only upon photolysis of compounds 1b, 2b, and 3b.
Co-reporter:Tülay A. Ateşin ; Abdurrahman Ç. Ateşin ; Karlyn Skugrud
Inorganic Chemistry 2008 Volume 47(Issue 11) pp:4596-4604
Publication Date(Web):May 1, 2008
DOI:10.1021/ic702273a
The reaction of 2-cyanothiophene with a zerovalent platinum bisalkylphosphine fragment yields two thiaplatinacycles derived from the cleavage of the substituted and unsubstituted C—S bonds. While cleavage away from the cyano group is preferred kinetically, cleavage adjacent to the cyano group is preferred thermodynamically. Density functional theory using B3LYP level of theory on a model of this system is consistent with experimental results in that the transition state energy leading to the formation of the kinetically favored C—S bond cleavage product is lower by 5.3 kcal mol−1 than the barrier leading to the thermodynamically favored product. There is a 6.7 kcal mol−1 difference between these two products. The cyano substituent at the 2- position of thiophene did not substantially change the mechanism involved in the C—S bond cleavage of thiophene previously reported.
Co-reporter:Tülay A. Ateşin
Inorganic Chemistry 2008 Volume 47(Issue 23) pp:10889-10894
Publication Date(Web):October 28, 2008
DOI:10.1021/ic8009892
Theoretical studies were performed on the C−S bond activation reactions of 2-/3-cyanothiophene, 2-/3-methoxythiophene, and 2-/3-methylthiophene with the [Rh(PMe3)(C5Me5)] fragment to compare with the selectivity of these reactions observed in the experimental study, with the goal of determining whether the latter represent kinetic or thermodynamic products. Density functional theory (DFT) calculations have been used to optimize the ground-state structures of the two possible insertion products and the transition state structures leading to the formation of the products arising from the above cleavage reactions to address this question. With the 2-cyano and 2-methoxy substituents, the observed formation of one product resulting from the exclusive insertion of the rhodium into the more hindered substituted C−S bond was found to be consistent with the calculated energy differences between the ground states of the two possible products (7.6 and 2.6 kcal mol−1). With 2-methylthiophene, the product resulting from the activation of the unsubstituted C−S bond is calculated to be favored by 5.8 kcal mol−1, in agreement with observed results. The ∼1:1 ratio of products with 3-cyano and 3-methyl substituted thiophenes are also found to be consistent with the small calculated energy differences (0.4 and 0.8 kcal mol−1) between the ground states of the two insertion products. Although the observed high selectivity in the formation of a single C−S bond activation product with 3-methoxythiophene appears to be underestimated in the calculations, the observed products for all substituted thiophenes correlate with the calculated thermodynamic products. In addition, the kinetic selectivities predicted based on the calculated C−S bond activation barriers are different from those observed experimentally. Consequently, these investigations demonstrate that DFT calculations can be used reliably to differentiate if an experimentally observed C−S bond activation reaction proceeds under thermodynamic or kinetic control.
Co-reporter:Andrew W. Myers, Lingzhen Dong, Tülay A. Ateşin, Roger Skugrud, Christine Flaschenriem, William D. Jones
Inorganica Chimica Acta 2008 Volume 361(Issue 11) pp:3263-3270
Publication Date(Web):27 July 2008
DOI:10.1016/j.ica.2007.09.023
The reactions of (C5Me5)Rh(PMe3)(Ph)H with 2-methoxythiophene, 3-methoxythiophene, 2-cyanothiophene, 3-cyanothiophene, 2-trimethylsilylthiophene, ethylenesulfide, trimethylenesulfide, and several polymethylthiophenes have been investigated. These thiophene derivatives give C–S and in one case C–H insertion products. Ethylene sulfide and trimethylene sulfide undergo ring opening or desulfurization.The reactions of the reactive species [(C5Me5)Rh(PMe3)] with 2- and 3-substituted thiophenes (R = OMe, CN, SiMe3) have been investigated. Insertion reactions of ethylenesulfide, trimethylenesulfide, and several polymethylthiophenes have also been examined.
Co-reporter:Tülay A. Ateşin and William D. Jones
Organometallics 2008 Volume 27(Issue 1) pp:53-60
Publication Date(Web):December 20, 2007
DOI:10.1021/om700679j
The reaction of thiophene with a zerovalent platinum bisalkylphosphine fragment yields a highly stable thiaplatinacycle derived from cleavage of the C−S bond. Calculations on the [Pt(dmpe)] model system using Density Functional Theory are consistent with experimental results obtained with [Pt(dippe)] in that the reaction is exothermic overall and furthermore predict that the initial η2-coordination of thiophene through the C═C double bond is energetically more favorable than coordination through the sulfur atom (ΔG = 9.3 kcal/mol). There are three well-defined transition states along the pathway to the oxidative addition product from both of these coordination modes. Two of these lead to a higher energy η2-C,S-coordinated intermediate, while the third one leads to cleavage of the C−S bond from the η2-C,S complex. As the reaction was carried out in a polar solvent (THF), the effect of solvation was taken into account by using the polarizable continuum model. The thermodynamic preference for the initial coordination of thiophene through the C═C bond is found to be greater in THF (ΔG = 11.4 kcal/mol). More importantly, the total free energy of the transition state from the C═C coordinated complex is now lower than that of the S-coordinated complex in solution. Therefore, the initial η2-coordination of thiophene through the C═C double bond results in the kinetically preferred pathway. Molecular orbital analyses were carried out to rationalize the results.
Co-reporter:Tülay A. Ateşin ; Ting Li ; Sébastien Lachaize ; Juventino J. García
Organometallics 2008 Volume 27(Issue 15) pp:3811-3817
Publication Date(Web):June 28, 2008
DOI:10.1021/om800424s
The nickel(0) fragment [Ni(dippe)] was reacted with benzonitrile and initially formed both η2-nitrile and η2-arene complexes at −60 °C. When the sample was warmed to room temperature, the latter completely converted to the η2-nitrile product, which is known to give an equilibrium mixture with the Ni(II) oxidative addition product [(dippe)Ni(Ph)(CN)]. Thermodynamic parameters for this equilibrium have been obtained in both polar and nonpolar solvents (THF vs toluene). Use of density functional theory showed three relatively stable η2-arene intermediates, as well as six well-defined transition states located on the potential energy surface between the η2-nitrile complex and the C−CN bond activation product. Among these transition states, those for the migration of the nickel metal between the carbon−carbon bonds of the phenyl ring are at lower energies than those connecting the η2-nitrile complex to the η2-arene intermediate and the η2-arene intermediate to the C−CN bond activation product. Calculations were carried out both in the gas phase and in solution using the PCM model, which was critical for simulation of the different polar solvent environments in these experiments.
Co-reporter:Tülay A. Ateşin
Organometallics 2008 Volume 27(Issue 15) pp:3666-3670
Publication Date(Web):July 17, 2008
DOI:10.1021/om700899n
Density functional theory (DFT) calculations on the C−S bond activation reaction of thiophene with the [(C5Me5)Rh(PMe3)] fragment have been reinvestigated, giving two new isomeric C−S bond activation transition states, in which the coordinated thiophene molecule tilts toward either the C5Me5 ligand or the PMe3 ligand. Through intrinsic reaction coordinate (IRC) calculations, these transition states were found to connect the oxidative addition product with two isomeric η2-C,S coordinated intermediates. These latter intermediates in turn connected to two isomeric η1-S and η2-C,C coordinated species. The energetics and mechanistic details are described.
Co-reporter:Ryan D. Rieth;William W. Brennessel
European Journal of Inorganic Chemistry 2007 Volume 2007(Issue 18) pp:
Publication Date(Web):4 JAN 2007
DOI:10.1002/ejic.200600802
The hafnium hydride Cp*2HfH2 is reacted with a series of fluorocarbons to examine the scope of C–F bond activation. Aromatic, vinylic, and aliphatic C–F bonds all show some degree of reactivity, and possible mechanisms are discussed. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Andrew J. Vetter;Ryan D. Rieth
PNAS 2007 Volume 104 (Issue 17 ) pp:6957-6962
Publication Date(Web):2007-04-24
DOI:10.1073/pnas.0609726104
The photochemical reaction of Tp′Rh(L)(η2-PhN=L) [Tp′ = tris-(3,5-dimethyl pyrazolyl)borate, L = CNCH2CMe3] to form the coordinatively unsaturated reactive fragment, [Tp′Rh(L)], in the presence of alkylnitriles has been studied.
The [Tp′Rh(L)] complex has been shown to selectively activate the primary CH terminus of acetonitrile, propionitrile, butyronitrile, and valeronitrile. The resulting hydrides showed uncharacteristic
stability in the presence of C6D6 and their rates of reductive elimination were monitored by 1H NMR spectroscopy. Competition reactions permit the establishment of the relative stabilities of the activation products.
Co-reporter:Tülay A. Ateşin, Stephen S. Oster, Karlyn Skugrud, William D. Jones
Inorganica Chimica Acta 2006 Volume 359(Issue 9) pp:2798-2805
Publication Date(Web):1 June 2006
DOI:10.1016/j.ica.2005.10.052
X-ray structural and NMR spectroscopic data for the ring-opened thiophene complexes [Pd(dippe)(T)] (2), and [Pt(dippe)(T)] (3) are now presented. The complex [Ni(dippe)(T)] (1), where T = (η2-C,S-C4H4S), was reported by our group, previously.The structural and bonding properties of complexes 2 and 3 were compared with those of complex 1. DFT calculations were carried out to rationalize their relative stabilities and structural properties. Compound 1 loses thiophene at ambient temperature in solution, while compound 2 decomposes rapidly in both acetone-d6 and THF-d8 with kobs = 7.15(9) × 10−5 and 7.7(3) × 10−5 s−1, respectively, to give products that varied by solvent. Complex 3 does not lose thiophene at temperatures below 100 °C. The ΔG0 values determined from DFT calculations are consistent with the observed stabilities of the complexes. The single crystal X-ray structures of all three complexes contain a disordered thienyl fragment in the asymmetric unit due to the interchange of the position of sulfur in the metal-inserted thiophenic ring. The thiophenic moiety is relatively flat in 1, 2 and 3, which is attributed to the open ligand environment at the M(dippe) fragment. All three complexes possess square-planar geometry around the metal center and have bond-length alternation among the thiophenic carbons, which indicates double bond localization. The calculated bond lengths are in good agreement with experimental data. Molecular orbital (MO) and natural bonding orbital (NBO) analyses were carried out to rationalize the results.The reactions of the unsaturated metal fragments [M(dippe)] where M = Ni, Pd, or Pt with thiophene are described (dippe = bis-(diisopropylphosphino)ethane). DFT calculations are used to support the electronic and geometric features of the compounds.
Co-reporter:Nicole M. Brunkan, William D. Jones
Journal of Organometallic Chemistry 2003 Volume 683(Issue 1) pp:77-82
Publication Date(Web):7 October 2003
DOI:10.1016/S0022-328X(03)00431-5
The complex (dippe)Ni(η3-allyl)(CN) has been prepared and fully characterized (dippe=bis-(diisopropylphosphino)ethane), including X-ray diffraction studies, as a square pyramidal structure. The complex shows dynamic 1H-NMR behavior consistent with substantial structural rearrangements upon π to σ allyl interconversion. A comparison is made with (dippe)Ni(η3-allyl)Br, which also displays a square pyramidal structure.The complex (dippe)Ni(η3-allyl)(CN) has been prepared and fully characterized (dippe=bis-(diisopropylphosphino)ethane), including X-ray diffraction studies, as a square pyramidal structure. The complex shows dynamic 1H-NMR behavior consistent with substantial structural rearrangements upon π to σ allyl interconversion. A comparison is made with (dippe)Ni(η3-allyl)Br, which also displays a square pyramidal structure.
Co-reporter:Josemon Jacob, Colleen M. Cavalier, William D. Jones, Stephen A. Godleski, Ronald R. Valente
Journal of Molecular Catalysis A: Chemical 2002 Volumes 182–183() pp:565-570
Publication Date(Web):31 May 2002
DOI:10.1016/S1381-1169(01)00493-9
Dicobaltoctacarbonyl has been found to be an effective catalyst for the conversion of diallylanilines to quinolines. Arylimines are also found to undergo heteroannulation in the presence of diallylaniline as allyl fragment donor to give quinolines. Imines can also be allylated to give quinoline derivatives. Initial studies of the scope and mechanism of the reaction are presented.
Co-reporter:Sumit Chakraborty ; William W. Brennessel
Journal of the American Chemical Society () pp:
Publication Date(Web):May 30, 2014
DOI:10.1021/ja504523b
A well-defined iron complex (3) supported by a bis(phosphino)amine pincer ligand efficiently catalyzes both acceptorless dehydrogenation and hydrogenation of N-heterocycles. The products from these reactions are isolated in good yields. Complex 3, the active catalytic species in the dehydrogenation reaction, is independently synthesized and characterized, and its structure is confirmed by X-ray crystallography. A trans-dihydride intermediate (4) is proposed to be involved in the hydrogenation reaction, and its existence is verified by NMR and trapping experiments.
Co-reporter:Yunzhe Jiao, William W. Brennessel and William D. Jones
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN812-812
Publication Date(Web):2013/12/06
DOI:10.1039/C3SC52748D
The thermal precursor Tp′Rh[P(OMe)3](Me)H was used to generate the active [Tp′Rh[P(OMe)3]] fragment, which activates C–H bonds of various hydrocarbons to form products of the type Tp′Rh[P(OMe)3](R)H (Tp′ = tris-(3,5-dimethylpyrazolyl)borate). Only one single activation product was observed in each case. The structures of Tp′Rh[P(OMe)3](R)X (X = H, Br, Cl) have been characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. The kinetics of reductive elimination of RH from Tp′Rh[P(OMe)3](R)H as well as competition experiments between substrates allow measurement of the Rh–C bond strengths relative to the Rh–Ph bond strength. Two separate linear correlations of the Rh–C bond energies versus H–C bond energies were found based on whether the alkyl group is α-substituted or not. While the correlation for α-substituted substrates gives a slope of 1.45, smaller than the slope (1.55) for unsubstituted hydrocarbons, the Rh–C bond energies of the former group are higher by ∼7 kcal mol−1. In comparison with the analogous [Tp′Rh(PMe3)] and [Tp′Rh(CNneopentyl)] systems, replacing the spectator ligand with a more electronic donating group slightly increases metal–carbon bond strengths as the trend in slopes of the correlations follows an order of CNneopentyl < P(OMe)3 ≤ PMe3.
Co-reporter:Srinivasan Ramakrishnan, Sumit Chakraborty, William W. Brennessel, Christopher E. D. Chidsey and William D. Jones
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN127-127
Publication Date(Web):2015/10/29
DOI:10.1039/C5SC03189C
A series of square-planar nickel hydride complexes supported by bis(phosphinite) pincer ligands with varying substituents (–OMe, –Me, and –But) on the pincer backbone have been synthesized and completely characterized by NMR spectroscopy, IR spectroscopy, elemental analysis, and X-ray crystallography. Their cyclic voltammograms show irreversible oxidation peaks (peak potentials from 101 to 316 mV vs. Fc+/Fc) with peak currents consistent with overall one-electron oxidations. Chemical oxidation by the one-electron oxidant Ce(NBu4)2(NO3)6 was studied by NMR spectroscopy, which provided quantitative evidence for post-oxidative H2 evolution leading to a solvent-coordinated nickel(II) species with the pincer backbone intact. Bulk electrolysis of the unsubstituted nickel hydride (3a) showed an overall one-electron stoichiometry and gas chromatographic analysis of the headspace gas after electrolysis further confirmed stoichiometric production of dihydrogen. Due to the extremely high rate of the post-oxidative chemical process, electrochemical simulations have been used to establish a lower limit of the bimolecular rate constant (kf > 107 M−1 s−1) for the H2 evolution step. To the best of our knowledge, this is the fastest known oxidative H2 evolution process observed in transition metal hydrides. Quantum chemical calculations based on DFT indicate that the one-electron oxidation of the nickel hydride complex provides a strong chemical driving force (−90.3 kcal mol−1) for the production of H2 at highly oxidizing potentials.
Co-reporter:Taro Tanabe, William W. Brennessel, Eric Clot, Odile Eisenstein and William D. Jones
Dalton Transactions 2010 - vol. 39(Issue 43) pp:NaN10509-10509
Publication Date(Web):2010/10/05
DOI:10.1039/C0DT00157K
A series of complexes of the type Tp′Rh(PR3)(ArF)H, where PR3 = PMe3 (3) and PMe2Ph (9), ArF = C6F5 (a), 2,3,4,5-C6F4H (b), 2,3,5,6-C6F4H (c), 2,4,6-C6F3H2 (d), 2,3-C6F2H3 (e), 2,5-C6F2H3 (g), and 2-C6FH4 (h) and Tp′ = tris(3,5-dimethylpyrazolyl)borate, has been synthesized as stable crystalline compounds by the reactions of the [Tp′Rh(PR3)] fragment with the corresponding fluorinated aromatic hydrocarbons, and their structures were characterized by NMR spectroscopy and elemental analysis together with X-ray crystallography. The kinetics of the reductive eliminations of fluoroarenes from complexes 3a–h in benzene-d6 solutions at 140 °C were investigated, but were complicated by the formation of the rhodium(I) bisphosphine complex, Tp′Rh(PMe3)2 (4). On the other hand, thermal reactions of (9) in THF-d8 solutions at 120 °C resulted in the formation of an intramolecular C–H bond activated complex of the phenyl group on the phosphorus atom, Tp′Rh(κ2-C6H4-2-PMe2)H (7), which prevents the formation of the corresponding bisphosphine complex. The experimentally determined rates of the reductive eliminations of fluoroarenes from the complexes 9a–h and their kinetic selectivities for formation in competition with the metallacycle have been used to determine relative Rh–CArF bond energies. The Rh–CArF bond energy is found to be dependent on the number of ortho fluorines. A plot of Rh–CArFvs. C–H bond strengths resulted in a line with a slope RM–C/C–H of 2.15 that closely matches the DFT calculated value (slope = 2.05).
.jpg)



















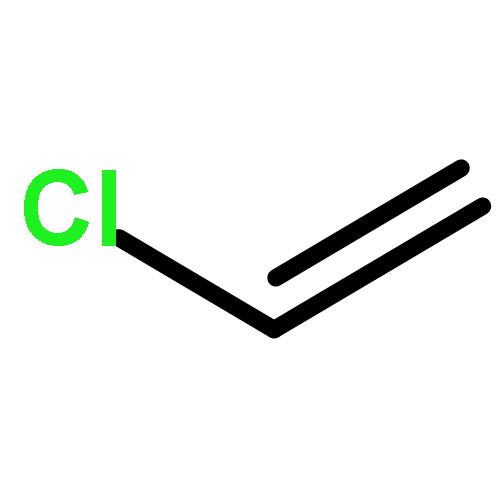









![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0.png)
![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0_b.png)































































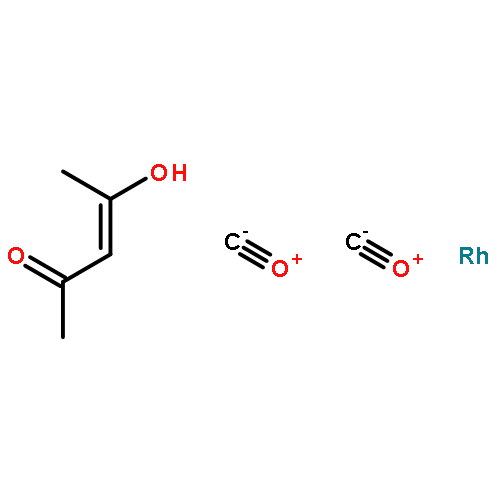
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)





























![[Pt(bicyclo[2.2.1]hept-2-en)3]](http://img.cochemist.com/ccimg/57200/57158-98-2.png)
![[Pt(bicyclo[2.2.1]hept-2-en)3]](http://img.cochemist.com/ccimg/57200/57158-98-2_b.png)

