Co-reporter:Dachang Bai, Qingqian Jia, Teng Xu, Qiuqiu Zhang, Fen Wu, Chaorui Ma, Bingxian Liu, Junbiao Chang, and Xingwei Li
The Journal of Organic Chemistry September 15, 2017 Volume 82(Issue 18) pp:9877-9877
Publication Date(Web):August 17, 2017
DOI:10.1021/acs.joc.7b01574
Rh(III)-catalyzed synthesis of nitro-functionalized indenes has been realized via C–H activation of arylnitrones and annulation with nitroolefins. The reaction proceeded in moderate to high yields with good functional group tolerance under ambient atmosphere.
Co-reporter:Xukai Zhou, Yixin Luo, Lingheng Kong, Youwei Xu, Guangfan Zheng, Yu Lan, and Xingwei Li
ACS Catalysis October 6, 2017 Volume 7(Issue 10) pp:7296-7296
Publication Date(Web):September 12, 2017
DOI:10.1021/acscatal.7b02248
An efficient, atom-economical, and regioselective insertion of indoles into terminal alkynes has been realized via cobalt(III)-catalyzed C–H activation under mild conditions, leading to efficient synthesis of α-gem-vinylindoles. The insertion of the alkynes follows a rare 1,2-selectivity, and silyl alkynes, alkyl alkynes, propargyl alcohols, and protected propargyl amines are all applicable. The mechanism of this hydroarylation system has been studied in detail by a combination of experimental and computational approaches. In the reaction of silyl terminal alkynes, the regioselectivity is dictated by the steric effects of the alkyne substituent, especially in the protonolysis stage. However, for protected propargyl amines, the selectivity results from electronic effects during the insertion step, with protonolysis being insignificant in the determination of selectivity. An internal alkyne also coupled in high efficiency but with low regioselectivity. Comparisons of cobalt, rhodium, and iridium catalysts have also been made in terms of regioselectivity and reactivity, and both are high for cobalt catalysts.Keywords: alkyne insertion; cobalt; C−H activation; hydroarylation; indole;
Co-reporter:Bingxian Liu, Panjie Hu, Ying Zhang, Yunyun Li, Dachang Bai, and Xingwei Li
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.orglett.7b02678
Rh(III)-catalyzed [3 + 2] annulation of cyclic N-sulfonyl or N-acyl ketimines with activated alkenes has been realized, leading to the synthesis of spirocycles with three continuous stereogenic centers. This atom-economic reaction proceeded efficiently under mild and redox-neutral conditions via a C–H activation pathway, and the coupling is diastereodivergent, with the diastereoselectivity being controlled by silver additives.
Co-reporter:Jintao Xia, Lingheng Kong, Xukai Zhou, Guangfan Zheng, and Xingwei Li
Organic Letters November 3, 2017 Volume 19(Issue 21) pp:5972-5972
Publication Date(Web):October 25, 2017
DOI:10.1021/acs.orglett.7b02983
An efficient synthesis of disubstituted acrylic acids has been realized via Rh(III)-catalyzed C–H activation of (hetero)arenes and coupling with four-membered methyleneoxetanones under redox-neutral conditions. In most cases, the reactions are silver-free, and the products are exclusively E-selective with a broad substrate scope. The transformation proceeds via ortho C–H activation followed by selective olefin insertion and β-oxygen elimination.
Co-reporter:Xiaohong Chen, Guangfan Zheng, Yunyun Li, Guoyong Song, and Xingwei Li
Organic Letters November 17, 2017 Volume 19(Issue 22) pp:6184-6184
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.orglett.7b03099
A Rh(III)-catalyzed site-selective C–H activation of C(3)-functionalized indoles in a coupling with diazo esters has been realized with carbonyl as a weakly coordinating group. The coupling selectivity is dictated by the temperature and additives, affording either C4-alkylated indoles or C2-annulated lactones in moderate to excellent efficiency.
Co-reporter:Jia-Qiang Wu, Shang-Shi Zhang, Hui Gao, Zisong Qi, Chu-Jun Zhou, Wei-Wei Ji, Yao Liu, Yunyun Chen, Qingjiang Li, Xingwei Li, and Honggen Wang
Journal of the American Chemical Society March 8, 2017 Volume 139(Issue 9) pp:3537-3537
Publication Date(Web):February 8, 2017
DOI:10.1021/jacs.7b00118
Fluorinated heterocycles play an important role in pharmaceutical and agrochemical industries. Herein, we report on the synthesis of four types of fluorinated heterocycles via rhodium(III)-catalyzed C—H activation of arenes/alkenes and versatile coupling with 2,2-difluorovinyl tosylate. With N-OMe benzamide being a directing group (DG), the reaction delivered a monofluorinated alkene with the retention of the tosylate functionality. Subsequent one-pot acid treatment allowed the efficient synthesis of 4-fluoroisoquinolin-1(2H)-ones and 5-fluoropyridin-2(1H)-ones. When N—OPiv benzamides were used, however, [4 + 2] cyclization occurred to provide gem-difluorinated dihydroisoquinolin-1(2H)-ones. Synthetic applications have been demonstrated and the ready availability of both the arene and the coupling partner highlighted the synthetic potentials of these protocols. Mechanistically, these two processes share a common process involving N—H deprotonation, C—H activation, and olefin insertion to form a 7-membered rhodacycle. Thereafter, different reaction pathways featuring β-F elimination and C—N bond formation are followed on the basis of density functional theory (DFT) studies. These two pathways are DG-dependent and led to the open chain and cyclization products, respectively. The mechanistic rationale was supported by detailed DFT studies. In particular, the origins of the intriguing selectivity in the competing β-F elimination versus C—N bond formation were elucidated. It was found that β-F elimination is a facile event and proceeds via a syn-coplanar transition state with a low energy barrier. The C—N bond formation proceeds via a facile migratory insertion of the Rh—C(alkyl) into the Rh(V) amido species. In both reactions, the migratory insertion of the alkene is turnover-limiting, which stays in good agreement with the experimental studies.
Co-reporter:Jintao Xia, Xifa Yang, Yunyun Li, and Xingwei Li
Organic Letters June 16, 2017 Volume 19(Issue 12) pp:
Publication Date(Web):June 1, 2017
DOI:10.1021/acs.orglett.7b01356
Ir(III)-catalyzed synthesis of benzimidazoles has been realized under redox-neutral conditions by annulation of aniline derivatives with dioxazolones. The reaction proceeded via a C–H activation–amidation–cyclization pathway with a decent substrate scope.
Co-reporter:Yunyun Li, Qiang Wang, Xifa Yang, Fang Xie, and Xingwei Li
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 12, 2017
DOI:10.1021/acs.orglett.7b01365
Rh-catalyzed C–H activation of phenacyl phosphoniums in coupling with α-diazocarbonyl compounds has been realized with the assistance of a mutifunctional phosphonium ylidic directing group, providing expedient accesses to 1-naphthols and isocoumarins. Switchable synthesis of 1-naphthols and isocoumarins was realized by substrate control, where these transformations were enabled by initial C–H activation and subsequent intramolecular Wittig reaction or nucleophilic C–O formation.
Co-reporter:Lingheng Kong, Xukai Zhou, Youwei Xu, and Xingwei Li
Organic Letters July 7, 2017 Volume 19(Issue 13) pp:
Publication Date(Web):June 23, 2017
DOI:10.1021/acs.orglett.7b01650
Rh(III)-catalyzed activation and acylation of sp3 C–H bonds has been realized with diarylcyclopropenone as an acylating reagent. Both benzylic C–H in 8-methylquinolines and unactivated C–H in 2-alkylpyridines are applicable in this C–H acylation reaction, providing enones in good yields under redox-neutral conditions.
Co-reporter:Youwei Xu, Xifa Yang, Xukai Zhou, Lingheng Kong, and Xingwei Li
Organic Letters August 18, 2017 Volume 19(Issue 16) pp:
Publication Date(Web):August 7, 2017
DOI:10.1021/acs.orglett.7b01974
Direct and efficient synthesis of 1-naphthols has been realized via Rh(III)-catalyzed C–H activation of sulfoxonium ylides and subsequent annulation with alkynes, where the sulfoxonium ylide functioned as a new traceless bifunctional directing group. This reaction occurred under redox-neutral conditions with a broad substrate scope.
Co-reporter:Bingxian Liu, Panjie Hu, Xukai Zhou, Dachang Bai, Junbiao Chang, and Xingwei Li
Organic Letters April 21, 2017 Volume 19(Issue 8) pp:
Publication Date(Web):April 4, 2017
DOI:10.1021/acs.orglett.7b00690
A Rh(III)-catalyzed addition of benzylic C(sp3)–H bond to α,β-unsaturated ketones/aldehydes has been realized, leading to efficient synthesis of γ-aryl ketones/aldehydes. This atom-economic reaction proceeded under mild and redox-neutral conditions with a broad substrate scope. Besides benzylic C–H, allylic C–H bonds are also applicable when assisted by O-methyl ketoxime directing groups.
Co-reporter:Lingheng Kong;Bingxian Liu;Xukai Zhou;Fen Wang
Chemical Communications 2017 vol. 53(Issue 74) pp:10326-10329
Publication Date(Web):2017/09/14
DOI:10.1039/C7CC06048C
Rhodium(III)-catalyzed mild benzylic α-fluoroalkenylation of 8-methylquinolines with gem-difluorostyrenes has been developed. This reaction occurred via C–H activation and C–F cleavage and is applicable to a wide range of substrates, leading to the synthesis of Z-alkenyl fluorides under mild and redox-neutral conditions with high regio- and stereoselectivity.
Co-reporter:Xukai Zhou;Yupeng Pan; Xingwei Li
Angewandte Chemie 2017 Volume 129(Issue 28) pp:8275-8279
Publication Date(Web):2017/07/03
DOI:10.1002/ange.201704036
AbstractRhodium(III)- and cobalt(III)-catalyzed C−H activation of indoles and coupling with 1,6-enynes is discussed. Under rhodium(III) catalysis, the alkyne insertion follows 2,1-regioselectivity with a subsequent type-I intramolecular Diels–Alder reaction (IMDA) to afford [6,5]-fused cycles. When catalyzed by the cobalt(III) congener, 1,2-insertion of the alkyne is preferred, and followed by a rare type-II IMDA, thus leading to bridged [3,3,1]-cycles. This selectivity of the alkyne insertion was mainly tuned by the steric sensitivity of the catalyst.
Co-reporter:Xukai Zhou;Zisong Qi;Songjie Yu;Lingheng Kong;Yang Li;Wan-Fa Tian
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 10) pp:1620-1625
Publication Date(Web):2017/05/17
DOI:10.1002/adsc.201601278
AbstractAn efficient synthesis of 2-substituted quinolines from readily available cyclopropanols and imidamides has been developed, where the cyclopropanol acts as a C3 synthon. With the assistance of a bifunctional imidamide directing group, the reaction occurred via sequential C–H/C–C cleavage and C–C/C–N bond formation.
Co-reporter:Xukai Zhou;Zisong Qi;Songjie Yu;Lingheng Kong;Yang Li;Wan-Fa Tian
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 10) pp:1599-1599
Publication Date(Web):2017/05/17
DOI:10.1002/adsc.201700240
The front cover picture, provided by Xingwei Li and co-workers, illustrates an efficient synthesis of 2-substituted quinolines from readily available cyclopropanols and imidamides, which has been developed with the cyclopropanol acting as a C3 synthon. With the assistance of a bifunctional imidamide directing group, the reaction occurred via sequential C−H/C−C cleavage and C−C/C−N bond formation. Details can be found in the communication on pages 1620–1625 (X. Zhou, Z. Qi, S. Yu, L. Kong, Y. Li, W.-F. Tian, X. Li, Adv. Synth. Catal. 2017, 359, 1620–1625; DOI: 10.1002/adsc.201601278).
Co-reporter:Manman Wang;Lingheng Kong;Fen Wang
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 24) pp:4411-4416
Publication Date(Web):2017/12/19
DOI:10.1002/adsc.201700899
AbstractA rhodium(III)-catalyzed C−H amination of benzamides and isoquinolones with anthranils has been realized under assistance of weakly coordinating amide, leading to a bifunctionalized amination product which can further cyclize to acridine under in situ or ex situ conditions.
Co-reporter:Xukai Zhou;Yupeng Pan; Xingwei Li
Angewandte Chemie International Edition 2017 Volume 56(Issue 28) pp:8163-8167
Publication Date(Web):2017/07/03
DOI:10.1002/anie.201704036
AbstractRhodium(III)- and cobalt(III)-catalyzed C−H activation of indoles and coupling with 1,6-enynes is discussed. Under rhodium(III) catalysis, the alkyne insertion follows 2,1-regioselectivity with a subsequent type-I intramolecular Diels–Alder reaction (IMDA) to afford [6,5]-fused cycles. When catalyzed by the cobalt(III) congener, 1,2-insertion of the alkyne is preferred, and followed by a rare type-II IMDA, thus leading to bridged [3,3,1]-cycles. This selectivity of the alkyne insertion was mainly tuned by the steric sensitivity of the catalyst.
Co-reporter:Yunyun Li;Xifa Yang;Lingheng Kong
Organic Chemistry Frontiers 2017 vol. 4(Issue 11) pp:2114-2118
Publication Date(Web):2017/10/24
DOI:10.1039/C7QO00510E
Phosphonium ylide acts as an efficient bifunctional directing group in Rh(III)-catalyzed C–H activation of arenes and oxidative coupling with activated olefins, leading to facile construction of indanones via a sequence of oxidative olefination and carboannulation. The phosphonium moiety functions as an oxophilic group, and dephosphination triggers a nucleophilic cyclization.
Co-reporter:Qiang Wang;Youwei Xu;Xifa Yang;Yunyun Li
Chemical Communications 2017 vol. 53(Issue 69) pp:9640-9643
Publication Date(Web):2017/08/24
DOI:10.1039/C7CC05000C
An efficient and redox-neutral naphthol synthesis has been realized via rhodium(III) catalyzed C–H activation of α-carbonyl nitrones and annulation with alkynes, where the nitrone group functioned as a traceless directing group.
Co-reporter:Qiang Wang, Yingzi Li, Zisong Qi, Fang Xie, Yu Lan, and Xingwei Li
ACS Catalysis 2016 Volume 6(Issue 3) pp:1971
Publication Date(Web):February 9, 2016
DOI:10.1021/acscatal.5b02297
C–H activation under redox-neutral conditions, especially by Rh(III) catalysis, has offered attractive synthetic strategies. Previous work in redox-neutral C–H activation relied heavily on the cleavage of oxidizing N–O and N–N directing groups, and cleavable N–S bonds have been rarely used, although they may offer complementary coupling patterns. In this work, N-sulfinyl ketoimines were designed as a novel substrate for the redox-neutral coupling with different activated olefins via a Rh-catalyzed C–H activation pathway. The coupling with acrylate esters afforded 1H-isoindoles with the formation of three chemical bonds around a quaternary carbon. Furthermore, the coupling with maleimides furnished pyrrolidone-fused isoquinolines. A broad scope of substrates has been established. The mechanism of the coupling with acrylates has been studied in detail by a combination of experimental and computational methods. This coupling proceeds via imine-assisted C–H activation of the arene followed by ortho C–H olefination to afford a Rh(III) olefin hydride intermediate which, upon deprotonation, may exist in equilibrium with a Rh(I) olefin species. Cleavage of the N–S bond occurs only after C–H olefination to generate a Rh(III) imide species. DFT studies indicated that the imide group can undergo migratory insertion to produce a Rh(III) secondary alkyl which isomerizes under the assistance of acetic acid to a Rh(III) tertiary alkyl that is prone to insertion of the second acrylate.Keywords: C−H bond activation; isoindole; N-sulfinyl imine; N−S bond cleavage; olefin; Rh(III) catalyst
Co-reporter:Xukai Zhou, Songjie Yu, Lingheng Kong, and Xingwei Li
ACS Catalysis 2016 Volume 6(Issue 2) pp:647
Publication Date(Web):December 21, 2015
DOI:10.1021/acscatal.5b02414
Rhodium-catalyzed C–H activation of arenes has been established as an important strategy for the rapid construction of new bonds. On the other hand, ring-opening of readily available cyclopropanols has served as a driving force for the coupling with various nucleophiles and electrophiles. Nevertheless, these two important areas evolved separately, and coupling of arenes with cyclopropanols via C–H activation has been rarely explored. In this work, the oxidative coupling between arenes and cyclopropanols has been realized with high efficiency and selectivity under Rh(III)-catalysis, providing an efficient route to access β-aryl ketones. Moreover, the C–H bond has been extended to benzylic C–H bonds.Keywords: cyclopropanols; C−H activation; rhodium(III) catalysis; ring opening; strained ring
Co-reporter:Songjie Yu, Yingzi Li, Lingheng Kong, Xukai Zhou, Guodong Tang, Yu Lan, and Xingwei Li
ACS Catalysis 2016 Volume 6(Issue 11) pp:7744
Publication Date(Web):October 13, 2016
DOI:10.1021/acscatal.6b02668
Carbonyl groups are ubiquitous in functional molecules. Although C–H bond acylation has been well-studied via different mechanisms, transition-metal-catalyzed redox-neutral C(sp3)–H acylation under mild conditions is unprecedented. In this work, ketene is designed as a acylating reagent for both C(sp3)–H and C(sp2)–H bonds under Rh(III) catalysis, affording a diverse array of carbonyl compounds in high yields and high atom economy under mild conditions.Keywords: acylation; arene; C−H activation; ketene; rhodium
Co-reporter:Xingwei Li, Xifa Yang, and Zisong Qi
ACS Catalysis 2016 Volume 6(Issue 10) pp:6372
Publication Date(Web):August 23, 2016
DOI:10.1021/acscatal.6b01936
An efficient Rh(III)-catalyzed redox-neutral [2+2+1] coupling between 8-formylquinolines and alkynes has been realized for the synthesis of cyclopentadienols with broad substrate scope and functional group tolerance. The reaction occurs via C (acyl)–H activation with double insertion of the alkyne in high atom-economy. Instead of simply undergoing a [2+2+1] cyclization, a subsequent formal intermolecular 1,5-shift of the hydroxyl group is involved, which affords a thermodynamically more stable, conjugated cyclopentadienol.Keywords: aldehyde; alkyne; cyclopentadienol; C−H activation; rhodium
Co-reporter:Zisong Qi, Lingheng Kong, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 17) pp:4392-4395
Publication Date(Web):August 24, 2016
DOI:10.1021/acs.orglett.6b02146
Vinyl benzoxazinanone was applied as an electrophilic allylating reagent for a series of arenes under redox-neutral Rh(III) catalysis. This reaction occurs in high efficiency under mild conditions to afford allylarenes bearing a sulfonamide functionality in exclusively E-selectivity. This allylation system combines C–H activation of arenes and scission of an unstrained six-membered ring.
Co-reporter:Lei Li, He Wang, Songjie Yu, Xifa Yang, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 15) pp:3662-3665
Publication Date(Web):July 14, 2016
DOI:10.1021/acs.orglett.6b01716
Cooperative cobalt- and copper-catalyzed C–H activation of imidate esters and oxidative coupling with anthranils allowed efficient synthesis of 1H-indazoles in the absence of metal oxidants. The anthranil acts as a convenient aminating reagent as well as an organic oxidant in this transformation. The copper catalyst likely functions at the stage of N–N formation.
Co-reporter:Qiang Wang, Fen Wang, Xifa Yang, Xukai Zhou, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 23) pp:6144-6147
Publication Date(Web):November 14, 2016
DOI:10.1021/acs.orglett.6b03155
Quinazoline N-oxides have been prepared from simple ketoximes and 1,4,2-dioxazol-5-ones via Rh(III)-catalyzed C–H activation–amidation of the ketoximes and subsequent Zn(II)-catalyzed cyclization. The substrate scope and functional group compatibility were examined. The reaction features relay catalysis by Rh(III) and Zn(II).
Co-reporter:Lingheng Kong, Xukai Zhou, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 24) pp:6320-6323
Publication Date(Web):December 5, 2016
DOI:10.1021/acs.orglett.6b03203
A cobalt(III)-catalyzed α-fluoroalkenylation of different arenes with readily available gem-difluorostyrenes has been realized under mild and redox-neutral conditions. This reaction occurs via a C–H activation pathway and offers a step-economical access to various 1,2-diaryl-substituted monofluoroalkenes in excellent Z selectivity in moderate to excellent yields.
Co-reporter:Zisong Qi, Songjie Yu, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 4) pp:700-703
Publication Date(Web):January 29, 2016
DOI:10.1021/acs.orglett.5b03669
The synthesis of N-unprotected indoles has been realized via Rh(III)-catalyzed C–H activation/annulation of imidamides with α-diazo β-ketoesters. The reaction occurs with the release of an amide coproduct, which originates from both the imidamide and the diazo as a result of C═N cleavage of the imidamide and C–C(acyl) cleavage of the diazo. A rhodacyclic intermediate has been isolated and a plausible mechanism has been proposed.
Co-reporter:Fen Wang, He Wang, Qiang Wang, Songjie Yu, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 6) pp:1306-1309
Publication Date(Web):March 8, 2016
DOI:10.1021/acs.orglett.6b00227
C–H activation of arenes has been established as an important strategy for heterocycle synthesis via annulations between arenes and unsaturated coupling partners. However, nitriles failed to act as such a coupling partner. Dioxazolones have been employed as a synthon of nitriles, and subsequent coupling with arenes such as N-sulfinylimines and benzimidates bearing a functionalizable directing group provided facile access to two classes of quinazolines under Co(III)-catalysis.
Co-reporter:Qiang Wang and Xingwei Li
Organic Letters 2016 Volume 18(Issue 9) pp:2102-2105
Publication Date(Web):April 15, 2016
DOI:10.1021/acs.orglett.6b00727
Nitrosobenzenes have been used as a convenient aminating reagent for the efficient synthesis of 1H-indazoles via rhodium and copper catalyzed C–H activation and C–N/N–N coupling. The reaction occurred under redox-neutral conditions with high efficiency and functional group tolerance. Moreover, a rhodacyclic imidate complex has been identified as a key intermediate.
Co-reporter:He Wang, Lei Li, Songjie Yu, Yunyun Li, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 12) pp:2914-2917
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.orglett.6b01284
Imidate esters and diazo compounds have been established as bifunctional substrates for the construction of biologically active fused heterocycles via rhodium-catalyzed C–H activation and C–C/C–N coupling. This reaction occurs under mild conditions with high efficiency, step economy, and low catalyst loading.
Co-reporter:Songjie Yu, Yunyun Li, Xukai Zhou, He Wang, Lingheng Kong, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 12) pp:2812-2815
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.orglett.6b01032
Rhodium(III)-catalyzed C–H activation of heteroarenes and functionalization with bifunctional substrates such as anthranils allows facile construction of quinoline-fused heterocycles under redox-neutral conditions. The couplings feature broad substrate scope and provide step-economical access to two classes of quinoline-fused condensed heterocycles.
Co-reporter:Lingheng Kong, Songjie Yu, Guodong Tang, He Wang, Xukai Zhou, and Xingwei Li
Organic Letters 2016 Volume 18(Issue 15) pp:3802-3805
Publication Date(Web):July 8, 2016
DOI:10.1021/acs.orglett.6b01806
Co(III)-catalyzed mild C–C couplings of arenes with strained rings such as 7-oxabenzonorbornadienes and 2-vinyloxirane have been realized. The transformation is proposed to undergo ortho C–H activation, olefin insertion, and subsequent β-oxygen elimination. A broad range of synthetically useful functional groups are compatible, thus providing a new entry to access diversely 2-functionalized indoles.
Co-reporter:Yunyun Li;Fen Wang;Songjie Yu
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 6) pp:880-886
Publication Date(Web):
DOI:10.1002/adsc.201500709
Co-reporter:Xifa Yang, He Wang, Xukai Zhou and Xingwei Li
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 23) pp:5233-5237
Publication Date(Web):17 May 2016
DOI:10.1039/C6OB00825A
Mild and efficient synthesis of benzophenones via Ir(III)- and Rh(III)-catalyzed, directing group-assisted formyl C–H arylation of benzaldehydes has been achieved using diaryliodonium salts, in which Rh(III) and Ir(III) catalysts exhibited a complementary substrate scope.
Co-reporter:Lei Li, He Wang, Xifa Yang, Lingheng Kong, Fen Wang, and Xingwei Li
The Journal of Organic Chemistry 2016 Volume 81(Issue 23) pp:12038-12045
Publication Date(Web):November 7, 2016
DOI:10.1021/acs.joc.6b02356
Rh(III)-catalyzed synthesis of mesoionic heterocycles has been achieved via C–H activation of sydnones and oxidative coupling with internal alkynes. This reaction occurred under mild conditions with high efficiency, broad substrate scope, and low catalyst loading. Moreover, synthetic applications of a coupled product have been demonstrated in the late-stage derivatization into a variety of highly functionalized scaffolds.
Co-reporter:Fen Wang;Xinzhang Yu;Zisong Qi ;Dr. Xingwei Li
Chemistry - A European Journal 2016 Volume 22( Issue 2) pp:511-516
Publication Date(Web):
DOI:10.1002/chem.201504179
Abstract
Rhodium-catalyzed sulfonylation, thioetherification, thiocyanation, and other heterofunctionalizations of arenes bearing a heterocyclic directing group have been realized. The reaction proceeds by initial RhIII-catalyzed CH hyperiodination of arene at room temperature followed by uncatalyzed nucleophilic functionalization. A diaryliodonium salt is isolated as an intermediate, which represents umpolung of the arene substrate, in contrast to previous studies that suggested umpolung of the coupling partner.
Co-reporter:Xukai Zhou, Songjie Yu, Zisong Qi, Lingheng Kong, and Xingwei Li
The Journal of Organic Chemistry 2016 Volume 81(Issue 11) pp:4869-4875
Publication Date(Web):May 11, 2016
DOI:10.1021/acs.joc.6b00650
The rhodium(III)-catalyzed regioselective alkylation of (hetero)arenes using cyclopropanols as a reactive and efficient coupling partner under oxidative conditions has been developed. This coupling occurred at room temperature via C–H activation of arenes and C–C cleavage of cyclopropanols. Various types of (hetero)arenes (indolines, carbazole, tetrahydrocarbazole, pyrrole, thiophene, etc.) were all successfully reacted under the present conditions. This protocol provides the facile and efficient construction of C7-alkylated indoline scaffolds.
Co-reporter:Yunyun Li, Fang Xie, and Xingwei Li
The Journal of Organic Chemistry 2016 Volume 81(Issue 2) pp:715-722
Publication Date(Web):December 28, 2015
DOI:10.1021/acs.joc.5b02410
Formal regiodivergent C–H alkynylation of 2-pyridones bearing different N-substituents has been realized under Au(I) and Rh(III) catalysis using a hypervalent iodine alkyne reagent. When catalyzed by Au(I), the alkynylation occurred at the most electron-rich 5-position via an electrophilic alkynylation pathway. The selectivity was switched to the 6-position under assistance of an N-chelation group when a Rh(III) catalyst was employed. A rhodacylic complex has been isolated as a key intermediate.
Co-reporter:Guoyong Song and Xingwei Li
Accounts of Chemical Research 2015 Volume 48(Issue 4) pp:1007
Publication Date(Web):April 6, 2015
DOI:10.1021/acs.accounts.5b00077
The possibility of developing new methods for the efficient construction of organic molecules via disconnections other than traditional functional group transformations has driven the interest in direct functionalization of C–H bonds. The ubiquity of C–H bonds makes such transformations attractive, but they also pose several challenges. The first is the reactivity and selectivity of C–H bonds. To achieve this, directing groups (DGs) are often installed that can enhance the effective concentration of the catalyst, leading to thermodynamically stable metallacyclic intermediates. However, the presence of a pendant directing group in the product is often undesirable and unnecessary. This may account for the limitation of applications of C–H functionalization reactions in more common and general uses. Thus, the development of removable or functionalizable directing groups is desirable. Another key problem is that the reactivity of the resulting M–C bond can be low, which may limit the scope of the coupling partners and hence limit the reaction patterns of C–H activation reactions.While the first Cp*Rh(III)-catalyzed C–H activation of arenes was reported only 7 years ago, significant progress has been made in this area in the past few years. We began our studies in this area in 2010, and we and others have demonstrated that diversified catalytic functionalization of arenes can be realized using Cp*Rh(III) complexes with high reactivity, stability, and functional group compatibility. This Account describes our efforts to solve some of these challenges using Rh(III) catalysis.We fulfilled our design and activation of the arene substrates by taking advantage of the nucleophilicity, electrophilicity, oxidizing potential, and properties of a participating ligand of the directing groups when the arenes are coupled with relatively reactive unsaturated partners such as alkenes and alkynes. These in situ funtionalizable roles of the DG allowed extensive chemical manipulation of the initial coupled product, especially in the construction of a diverse array of heterocycles. In the coupling of arenes with polar coupling partners, the polar Rh(III)–C(aryl) bond showed higher reactivity as both an organometallic reagent and a nucleophilic aryl source. The polar coupling partners were accordingly activated by virtue of umpolung, ring strain, and rearomatization. All of these transformations have been made possible by integration of the higher reactivity, stability, and compatibility of Rh(III)–C bonds into catalytic systems. We have demonstrated that to date some of these transformations can be achieved only under rhodium catalysis. In addition, by means of stoichiometric reactions, we have gained mechanistic insights into the interactions between the Rh–C bond and the other coupling partners, which have opened new avenues in future direct C–H functionalization reactions.
Co-reporter:Songjie Yu; Song Liu; Yu Lan; Boshun Wan
Journal of the American Chemical Society 2015 Volume 137(Issue 4) pp:1623-1631
Publication Date(Web):January 8, 2015
DOI:10.1021/ja511796h
Rh(III)-catalyzed C–H activation assisted by an oxidizing directing group has evolved to a mild and redox-economic strategy for the construction of heterocycles. Despite the success, these coupling systems are currently limited to cleavage of an oxidizing N–O or N–N bond. Cleavage of an oxidizing C–N bond, which allows for complementary carbocycle synthesis, is unprecedented. In this article, α-ammonium acetophenones with an oxidizing C–N bond have been designed as substrates for Rh(III)-catalyzed C–H activation under redox-neutral conditions. The coupling with α-diazo esters afforded benzocyclopentanones, and the coupling with unactivated alkenes such as styrenes and aliphatic olefins gave ortho-olefinated acetophenoes. In both systems the reactions proceeded with a broad scope, high efficiency, and functional group tolerance. Moreover, efficient one-pot coupling of diazo esters has been realized starting from α-bromoacetophenones and triethylamine. The reaction mechanism for the coupling with diazo esters has been studied by a combination of experimental and theoretical methods. In particular, three distinct mechanistic pathways have been scrutinized by DFT studies, which revealed that the C–H activation occurs via a C-bound enolate-assisted concerted metalation–deprotonation mechanism and is rate-limiting. In subsequent C–C formation steps, the lowest energy pathway involves two rhodium carbene species as key intermediates.
Co-reporter:Jie Zhou, Jingjing Shi, Zisong Qi, Xingwei Li, H. Eric Xu, and Wei Yi
ACS Catalysis 2015 Volume 5(Issue 11) pp:6999
Publication Date(Web):October 22, 2015
DOI:10.1021/acscatal.5b01571
Ir(III)-catalyzed direct C–H alkynylation of arenes has been developed using commercially available TIPS-acetylene as an efficient alkynylating reagent, where O-NHAc was employed as an autocleavable oxidizing-directing group (ODGauto), thus giving rise to ortho-alkynylated phenols under mild reaction conditions in a highly efficient and redox-neutral manner. The reaction proceeded with high regioselectivity and broad substrate/functional group (FG) tolerance. The synthetic application of the products has been briefly exemplified. Preliminary mechanistic studies have been conducted, and a five-membered iridacycle has also been identified as a key intermediate.Keywords: C−H alkynylation; Ir(III); N-phenoxyacetamides; reaction mechanism; TIPS-acetylene
Co-reporter:He Wang, Fang Xie, Zisong Qi, and Xingwei Li
Organic Letters 2015 Volume 17(Issue 4) pp:920-923
Publication Date(Web):February 4, 2015
DOI:10.1021/acs.orglett.5b00027
Mild and efficient synthesis of ynones via Ir(III)- and Rh(III)-catalyzed, chelation-assisted formyl C–H alkynylation of benzaldehydes has been achieved using hypervalent iodine–alkyne reagents. Rhodium and iridium catalysis exhibited complementary substrate scope.
Co-reporter:He Wang, Songjie Yu, Zisong Qi, and Xingwei Li
Organic Letters 2015 Volume 17(Issue 11) pp:2812-2815
Publication Date(Web):May 14, 2015
DOI:10.1021/acs.orglett.5b01232
Rhodium(III)-catalyzed direct alkylation of arenes using commercially available alkyltrifluoroborates is disclosed. Oximes, heteroarenes, azomethines, N-nitrosoamines, and amides are viable directing groups to entail this transformation. The alkyl group in the boron reagent can be extended to primary alkyls, benzyl, and cycloalkyls, and the reaction proceeded with controllable mono- and dialkylation selectivity when both ortho C–H sites are accessible.
Co-reporter:Songjie Yu, Boshun Wan, and Xingwei Li
Organic Letters 2015 Volume 17(Issue 1) pp:58-61
Publication Date(Web):December 17, 2014
DOI:10.1021/ol503231p
Rh(III)-catalyzed, chelation-assisted C–H activation and selenylation of arenes has been achieved. Arenes bearing oxime, azo, pyridyl, and N-oxide chelating groups are viable substrates, and electrophilic selenyl chlorides and diselenides are used as selenylating reagents. The catalytic system is highly efficient under mild conditions over a broad range of substrates with excellent functional group tolerance.
Co-reporter:Qiang Wang;Zisong Qi;Fang Xie
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 2-3) pp:355-360
Publication Date(Web):
DOI:10.1002/adsc.201400717
Co-reporter:Zisong Qi, Guo-Dong Tang, Cheng-Ling Pan and Xingwei Li
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 45) pp:10977-10980
Publication Date(Web):13 Oct 2015
DOI:10.1039/C5OB01886B
Rh(III)-catalyzed C–H activation of 3-aryl-dihydroisoxazoles in the coupling with diarylacetylenes has been developed under redox-neutral conditions. This reaction occurred under mild conditions with no by-product, and the N–O bond functions as an oxidizing directing group, leading to efficient synthesis of isoquinolines functionalized with a proximal secondary alcohol.
Co-reporter:Qiang Wang, Fang Xie, and Xingwei Li
The Journal of Organic Chemistry 2015 Volume 80(Issue 16) pp:8361-8366
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.joc.5b00940
Cp*Rh(III) complexes have been applied as efficient catalysts for the C–H activation and trifluoromethylthiolation of indoles functionalized with a heterocycle. With N-trifluoromethylthiosaccharin being an electrophilic SCF3 reagent, this C–S coupling occurred selectively at the 2-position with good functional group tolerance.
Co-reporter:Zisong Qi, Songjie Yu, and Xingwei Li
The Journal of Organic Chemistry 2015 Volume 80(Issue 7) pp:3471-3479
Publication Date(Web):March 13, 2015
DOI:10.1021/acs.joc.5b00059
Rh(III)-catalyzed C–H activation of 2-phenylimidazo[1,2-a]pyridines in divergent oxidative coupling with alkynes has been achieved. Selective mono versus 2-fold C–H activation has been attained under condition control. When AgOAc was used as an oxidant, the coupling afforded 5,6-disubstituted naphtho[1′,2′:4,5]imidazo[1,2-a]pyridines as a result of initial nitrogen chelation-assisted C–H activation at the benzene ring followed by rollover C–H activation. In contrast, the reaction afforded a fused isoquinolinium salt as a result of C–C and C–N coupling when AgBF4 was employed as a co-oxidant. A rhodacyclic intermediate has been isolated.
Co-reporter:Yingzi Li;Song Liu;Zisong Qi;Xiaotian Qi;Dr. Xingwei Li;Dr. Yu Lan
Chemistry - A European Journal 2015 Volume 21( Issue 28) pp:10131-10137
Publication Date(Web):
DOI:10.1002/chem.201500290
Abstract
Metal-catalyzed CH activation not only offers important strategies to construct new bonds, it also allows the merge of important research areas. When quinoline N-oxide is used as an arene source in CH activation studies, the NO bond can act as a directing group as well as an O-atom donor. The newly reported density functional theory method, M11L, has been used to elucidate the mechanistic details of the coupling between quinoline NO bond and alkynes, which results in CH activation and O-atom transfer. The computational results indicated that the most favorable pathway involves an electrophilic deprotonation, an insertion of an acetylene group into a RhC bond, a reductive elimination to form an oxazinoquinolinium-coordinated RhI intermediate, an oxidative addition to break the NO bond, and a protonation reaction to regenerate the active catalyst. The regioselectivity of the reaction has also been studied by using prop-1-yn-1-ylbenzene as a model unsymmetrical substrate. Theoretical calculations suggested that 1-phenyl-2-quinolinylpropanone would be the major product because of better conjugation between the phenyl group and enolate moiety in the corresponding transition state of the regioselectivity-determining step. These calculated data are consistent with the experimental observations.
Co-reporter:Dr. Fang Xie;Zhipeng Zhang;Dr. Xinzhang Yu;Guodong Tang;Dr. Xingwei Li
Angewandte Chemie International Edition 2015 Volume 54( Issue 25) pp:7405-7409
Publication Date(Web):
DOI:10.1002/anie.201502278
Abstract
Diaryliodonium salts play an increasingly important role as an aryl source. Reported is the first synthesis of diaryliodoniums by rhodium(III)-catalyzed CH hyperiodination of electron-poor arenes under chelation assistance. This CI coupling reaction occurred at room temperature with high regio-selectivity and functional-group compatibility. Subsequent diversified nucleophilic functionalization of a diaryliodonium allowed facile construction of CC, CN, CO, CS, CP and CBr bonds, and in all cases the initial functionalization occurred at the arene containing the chelating-group.
Co-reporter:Dr. He Wang;Guodong Tang ;Dr. Xingwei Li
Angewandte Chemie International Edition 2015 Volume 54( Issue 44) pp:13049-13052
Publication Date(Web):
DOI:10.1002/anie.201506323
Abstract
Nitrogenation by direct functionalization of CH bonds represents an important strategy for constructing CN bonds. Rhodium(III)-catalyzed direct amidation of unactivated C(sp3)H bonds is rare, especially under mild reaction conditions. Herein, a broad scope of C(sp3)H bonds are amidated under rhodium catalysis in high efficiency using 3-substituted 1,4,2-dioxazol-5-ones as the amide source. The protocol broadens the scope of rhodium(III)-catalyzed C(sp3)H activation chemistry, and is applicable to the late-stage functionalization of natural products.
Co-reporter:Dr. He Wang;Guodong Tang ;Dr. Xingwei Li
Angewandte Chemie 2015 Volume 127( Issue 44) pp:13241-13244
Publication Date(Web):
DOI:10.1002/ange.201506323
Abstract
Nitrogenation by direct functionalization of CH bonds represents an important strategy for constructing CN bonds. Rhodium(III)-catalyzed direct amidation of unactivated C(sp3)H bonds is rare, especially under mild reaction conditions. Herein, a broad scope of C(sp3)H bonds are amidated under rhodium catalysis in high efficiency using 3-substituted 1,4,2-dioxazol-5-ones as the amide source. The protocol broadens the scope of rhodium(III)-catalyzed C(sp3)H activation chemistry, and is applicable to the late-stage functionalization of natural products.
Co-reporter:Dr. Fang Xie;Zhipeng Zhang;Dr. Xinzhang Yu;Guodong Tang;Dr. Xingwei Li
Angewandte Chemie 2015 Volume 127( Issue 25) pp:7513-7517
Publication Date(Web):
DOI:10.1002/ange.201502278
Abstract
Diaryliodonium salts play an increasingly important role as an aryl source. Reported is the first synthesis of diaryliodoniums by rhodium(III)-catalyzed CH hyperiodination of electron-poor arenes under chelation assistance. This CI coupling reaction occurred at room temperature with high regio-selectivity and functional-group compatibility. Subsequent diversified nucleophilic functionalization of a diaryliodonium allowed facile construction of CC, CN, CO, CS, CP and CBr bonds, and in all cases the initial functionalization occurred at the arene containing the chelating-group.
Co-reporter:Xukai Zhou;Songjie Yu;Zisong Qi
Science China Chemistry 2015 Volume 58( Issue 8) pp:1297-1301
Publication Date(Web):2015 August
DOI:10.1007/s11426-015-5408-8
Rhodium(III)-catalyzed coupling between ketoximes and alkynes via C-H activation and annulation typically followed the [4+2] selectivity to afford isoquinolines. By designing alkynes bearing a highly electron-withdrawing group and under substrate control, we have successfully switched the selectivity of the coupling between oximes and alkynes to the alternative [3+2] annulation, leading to the efficient synthesis of indenamines. This process features good regioselectivity for both substrates, high efficiency, broad substrate scope, and excellent functional group tolerance.
Co-reporter:Fang Xie ; Zisong Qi ; Songjie Yu
Journal of the American Chemical Society 2014 Volume 136(Issue 12) pp:4780-4787
Publication Date(Web):March 4, 2014
DOI:10.1021/ja501910e
An efficient Rh(III)- and Ir(III)-catalyzed, chelation-assisted C–H alkynylation of a broad scope of (hetero)arenes has been developed using hypervalent iodine-alkyne reagents. Heterocycles, N-methoxy imines, azomethine imines, secondary carboxamides, azo compounds, N-nitrosoamines, and nitrones are viable directing groups to entail ortho C–H alkynylation. The reaction proceeded under mild conditions and with controllable mono- and dialkynylation selectivity when both mono- and dialkynylation was observed. Rh(III) and Ir(III) catalysts exhibited complementary substrate scope in this reaction. The synthetic applications of the coupled products have been demonstrated in subsequent derivatization reactions. Some mechanistic studies have been conducted, and two Rh(III) complexes have been established as key reaction intermediates. The current C–H alkynylation system complements those previously reported under gold or palladium catalysis using hypervalent iodine reagents.
Co-reporter:Songjie Yu and Xingwei Li
Organic Letters 2014 Volume 16(Issue 4) pp:1220-1223
Publication Date(Web):February 11, 2014
DOI:10.1021/ol500140e
A Rh(III)-catalyzed aryl C–H bond insertion into cyclopropenones via a C–H activation pathway has been reported. A series of arenes bearing directing groups such as 2-pyridyl, 2-pyrimidyl, N-pyrazyl, and oxime can be applicable, providing chalcones in excellent yields under mild conditions. Several possible Rh(III) intermediates in this reaction were investigated.
Co-reporter:Songjie Yu and Xingwei Li
Organic Letters 2014 Volume 16(Issue 4) pp:1200-1203
Publication Date(Web):February 7, 2014
DOI:10.1021/ol5000764
A rhodium(III)-catalyzed C–C coupling between 2-vinyloxiranes and arenes directed by different chelating groups has been realized via a C–H activation pathway. This reaction proceeded under conditions with a low catalyst loading, and allylic alcohols were isolated as the coupling products. A series of benzoazepanes has been synthesized by following this coupling.
Co-reporter:Xueyun Zhang, Fen Wang, Zisong Qi, Songjie Yu, and Xingwei Li
Organic Letters 2014 Volume 16(Issue 6) pp:1586-1589
Publication Date(Web):March 3, 2014
DOI:10.1021/ol500186j
Rhodium(III)-catalyzed arylation of arenes bearing a chelating group has been realized via a redox-economy process using 4-hydroxycyclohexa-2,5-dienones as the arylating reagents, leading to the synthesis of 3-arylated phenols. This redox-neutral process proceeds via a C–H activation pathway with rearomatization being the driving force.
Co-reporter:Zisong Qi, Mei Wang and Xingwei Li
Chemical Communications 2014 vol. 50(Issue 68) pp:9776-9778
Publication Date(Web):08 Jul 2014
DOI:10.1039/C4CC03627A
A new rhodium-catalyzed synthesis of sultones via the oxidative coupling of sulfonic acids with internal alkynes is described. The reaction proceeds via aryl C–H activation assisted by a sulfonic acid group.
Co-reporter:Fen Wang, Xiaoneng Cui, Zhangrong Lou, Jianzhang Zhao, Ming Bao and Xingwei Li
Chemical Communications 2014 vol. 50(Issue 98) pp:15627-15630
Publication Date(Web):27 Oct 2014
DOI:10.1039/C4CC07603F
Acid-switching of the triplet excited state in rhodamine-C60 dyads was achieved. The rhodamine moiety acts as an acid-activated visible light-harvesting antenna and C60 as the singlet energy acceptor and the spin converter, and production of the triplet state was enhanced in the presence of acid.
Co-reporter:Xueyun Zhang, Zisong Qi, Jian Gao and Xingwei Li
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 46) pp:9329-9332
Publication Date(Web):30 Sep 2014
DOI:10.1039/C4OB01596G
Rh(III)-catalyzed efficient C–H alkynylation of azomethine imines with alkynylated hypervalent iodine is developed under mild conditions. A broad scope of azomethine imines and alkyne substrates is established. The azomethine acts as a masked aldehyde and circumvents its poor directing effect.
Co-reporter:Tingting Yang, Tao Zhang, Shangdong Yang, Shanshan Chen and Xingwei Li
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 25) pp:4290-4294
Publication Date(Web):29 Apr 2014
DOI:10.1039/C4OB00704B
Rh(III)-catalyzed coupling of N-sulfonyl 2-aminobenzaldehydes with oxygenated allylic olefins via C–H bond activation is described. Diarylketones were obtained through coupling of N-sulfonyl 2-aminobenzaldehydes with 7-oxabenzonorbornadienes. On the other hand, the coupling with allyl carbonate yielded a six-membered sulfonyl lactam via a sequence of allylation–isomerization–Michael cyclization.
Co-reporter:Fen Wang, Yuchen Liu, Zisong Qi, Wei Dai, Xingwei Li
Tetrahedron Letters 2014 Volume 55(Issue 47) pp:6399-6402
Publication Date(Web):19 November 2014
DOI:10.1016/j.tetlet.2014.09.093
A simple catalytic, redox-neutral access to 3-methylcyclohexenones has been developed via rhodium catalysis in the presence of an amine additive and Ag2CO3. This process utilized simple aldehydes and acetone as substrates and tolerates a variety of functional groups. Disubstituted phenols were isolated in moderate yields when Cu(OAc)2 was employed as an oxidant.
Co-reporter:Xueyun Zhang;Zisong Qi; Xingwei Li
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:10794-10798
Publication Date(Web):
DOI:10.1002/anie.201406747
Abstract
[Cp*RhIII]-catalyzed CH activation of arenes assisted by an oxidizing NO or NN directing group has allowed the construction of a number of hetercycles. In contrast, a polar NO bond is well-known to undergo O-atom transfer (OAT) to alkynes. Despite the liability of NO bonds in both CH activation and OAT, these two important areas evolved separately. In this report, [Cp*RhIII] catalysts integrate both areas in an efficient redox-neutral coupling of quinoline N-oxides with alkynes to afford α-(8-quinolyl)acetophenones. In this process the NO bond acts as both a directing group for CH activation and as an O-atom donor.
Co-reporter:Xueyun Zhang;Zisong Qi; Xingwei Li
Angewandte Chemie 2014 Volume 126( Issue 40) pp:10970-10974
Publication Date(Web):
DOI:10.1002/ange.201406747
Abstract
[Cp*RhIII]-catalyzed CH activation of arenes assisted by an oxidizing NO or NN directing group has allowed the construction of a number of hetercycles. In contrast, a polar NO bond is well-known to undergo O-atom transfer (OAT) to alkynes. Despite the liability of NO bonds in both CH activation and OAT, these two important areas evolved separately. In this report, [Cp*RhIII] catalysts integrate both areas in an efficient redox-neutral coupling of quinoline N-oxides with alkynes to afford α-(8-quinolyl)acetophenones. In this process the NO bond acts as both a directing group for CH activation and as an O-atom donor.
Co-reporter:Tao Zhang;Zisong Qi;Xueyun Zhang; Lamei Wu;Dr. Xingwei Li
Chemistry - A European Journal 2014 Volume 20( Issue 12) pp:3283-3287
Publication Date(Web):
DOI:10.1002/chem.201400022
Abstract
Metal-catalyzed hydroacylation of olefins represents an important atom-economic synthetic process in CH activation. For the first time highly efficient RhIIICp*-catalyzed hydroacylation was realized in the coupling of N-sulfonyl 2-aminobenzaldehydes with both conjugated and aliphatic olefins, leading to the synthesis of various aryl ketones. Occasionally, oxidative coupling occurred when a silver(I) oxidant was used.
Co-reporter:Tao Zhang;Zisong Qi;Xueyun Zhang; Lamei Wu;Dr. Xingwei Li
Chemistry - A European Journal 2014 Volume 20( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/chem.201490049
Co-reporter:Jie Ma, Xiaoneng Cui, Fen Wang, Xueyan Wu, Jianzhang Zhao, and Xingwei Li
The Journal of Organic Chemistry 2014 Volume 79(Issue 22) pp:10855-10866
Publication Date(Web):October 23, 2014
DOI:10.1021/jo5018662
Dithienylethene (DTE)-2,6-diiodoBodipy triads were prepared with the aim to photoswitch the triplet excited state of the 2,6-diiodoBodipy moiety. Bodipy was selected due to its low T1 state energy level to avoid sensitized photocyclization of DTE, which is very often encountered in DTE photoswitches, so that the photochemistry of DTE and the organic chromophore can be addressed independently. This is the first time that DTE was covalently connected with an organic triplet photosensitizer. For the triad with DTE-o structure, selective photoexcitation into the diiodoBodipy part did not initiate photocyclization of DTE-o. Upon photoirradiation at 254 nm, thus the DTE-o → DTE-c transformation, the intersystem crossing (ISC) of 2,6-diiodoBodipy moiety was competed by the photoactivated resonance energy transfer (RET), with Bodipy as the intramolecular energy donor and DTE-c as energy acceptor. The fluorescence of Bodipy was quenched and the triplet state lifetime of Bodipy was reduced from 105.1 to 40.9 μs. The photoreversion is O2-independent, but can be greatly accelerated upon selective photoexcitation into the diiodoBodipy absorption band (at 535 nm). We concluded that ISC is not outcompeted by RET. The photoswitching of the triplet state was transduced to the singlet oxygen photosensitizing, as well as triplet–triplet annihilation upconversion.
Co-reporter:Yuchen Liu, Wencui Zhen, Wei Dai, Fen Wang, and Xingwei Li
Organic Letters 2013 Volume 15(Issue 4) pp:874-877
Publication Date(Web):February 7, 2013
DOI:10.1021/ol4000108
AgOTf can catalyze an addition-cyclization tandem between alkyne-azomethine and a nucleophile such as ketone, nitroalkane, water, and terminal alkyne to give a polycyclic amide via six-exo-trig selectivity.
Co-reporter:Zisong Qi, Mei Wang, and Xingwei Li
Organic Letters 2013 Volume 15(Issue 21) pp:5440-5443
Publication Date(Web):October 11, 2013
DOI:10.1021/ol4025309
Under redox-neutral conditions, rhodium(III)-catalyzed C–H annulation of N-tert-butyl-α-arylnitrones with internal alkynes has been realized for the synthesis of indenones under mild conditions. This reaction proceeded in moderate to high yields and with good functional group tolerance.
Co-reporter:Tao Zhang, Lamei Wu, and Xingwei Li
Organic Letters 2013 Volume 15(Issue 24) pp:6294-6297
Publication Date(Web):November 26, 2013
DOI:10.1021/ol403178a
Rh(III)-catalyzed olefination of N-sulfonyl imines using acrylates and styrenes has been achieved for the synthesis of ortho-olefinated benaldehydes. This reaction proceeds via a chelation assisted C–H olefination/in situ hydrolysis process.
Co-reporter:Fen Wang, Zisong Qi, Jiaqiong Sun, Xuelin Zhang, and Xingwei Li
Organic Letters 2013 Volume 15(Issue 24) pp:6290-6293
Publication Date(Web):November 26, 2013
DOI:10.1021/ol403166p
Rh(III)-catalyzed C–H activation and annulation of 1-benzoylpyrrolidine with propargyl alcohols has been achieved for an efficient synthesis of (4-benzylidene)isochroman-1-one. Highly enantioenriched products were obtained starting from optically pure propargyl alcohols.
Co-reporter:Songjie Yu, Boshun Wan, and Xingwei Li
Organic Letters 2013 Volume 15(Issue 14) pp:3706-3709
Publication Date(Web):July 3, 2013
DOI:10.1021/ol401569u
Rhodium(III)-catalyzed C–H activation–amidation of arenes bearing chelating groups has been achieved using N-arenesulfonated imides as efficient amidating reagents without using any base additive. Pyridine, oxime, and pyrimidine proved to be viable directing groups.
Co-reporter:Yuye Chen;Fen Wang;Wencui Zhen
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 2-3) pp:353-359
Publication Date(Web):
DOI:10.1002/adsc.201200924
Abstract
The rhodium(III)-catalyzed coupling of azomethine ylides with alkynes via CH activation has been developed for the synthesis of indenamines in moderate to high yields. The coupled products can be further oxidized to indenones and derivatives.
Co-reporter:Shui Hu, Dongqi Wang, Jiexiang Liu and Xingwei Li
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 17) pp:2761-2765
Publication Date(Web):12 Mar 2013
DOI:10.1039/C3OB40272J
Rhodium(III)-catalyzed oxidative couplings between N-sulfonyl allylamines and activated olefins have been achieved. Only olefination occurred for acrylates, and the butadiene product can be further cyclized under palladium-catalyzed aerobic conditions. The coupling with N,N-dimethylacrylamide followed a cyclization pathway.
Co-reporter:Dr. Fang Xie;Zisong Qi;Dr. Xingwei Li
Angewandte Chemie International Edition 2013 Volume 52( Issue 45) pp:11862-11866
Publication Date(Web):
DOI:10.1002/anie.201305902
Co-reporter:Dr. Fang Xie;Zisong Qi;Dr. Xingwei Li
Angewandte Chemie 2013 Volume 125( Issue 45) pp:12078-12082
Publication Date(Web):
DOI:10.1002/ange.201305902
Co-reporter:Xinzhang Yu, Songjie Yu, Jian Xiao, Boshun Wan, and Xingwei Li
The Journal of Organic Chemistry 2013 Volume 78(Issue 11) pp:5444-5452
Publication Date(Web):May 10, 2013
DOI:10.1021/jo400572h
Cp*Rh(III)-catalyzed intermolecular C–C couplings between activated α-diazocarbonyl compounds and arenes bearing a range of azacyclic directing groups have been achieved. This catalytic alkylation reaction operates under mild conditions with good functional group tolerance.
Co-reporter:Xinzhang Yu, Xiaoyi Xin, Boshun Wan, and Xingwei Li
The Journal of Organic Chemistry 2013 Volume 78(Issue 10) pp:4895-4904
Publication Date(Web):April 26, 2013
DOI:10.1021/jo4004635
The reaction of N-sulfonyl propargylamides in the presence of a base catalyst selectively affords 5-sulfonylmethyl oxazoles via 1,4-sulfonyl migration. Allenes have been established as the key intermediates. Experimental evidence has been provided to support a two-step mechanism in the cyclization.
Co-reporter:Dr. Xingwei Li;Songjie Yu;Fen Wang;Dr. Boshun Wan;Xinzhang Yu
Angewandte Chemie 2013 Volume 125( Issue 9) pp:2637-2640
Publication Date(Web):
DOI:10.1002/ange.201209887
Co-reporter:Zisong Qi ;Dr. Xingwei Li
Angewandte Chemie 2013 Volume 125( Issue 34) pp:9165-9170
Publication Date(Web):
DOI:10.1002/ange.201303507
Co-reporter:Zisong Qi ;Dr. Xingwei Li
Angewandte Chemie International Edition 2013 Volume 52( Issue 34) pp:8995-9000
Publication Date(Web):
DOI:10.1002/anie.201303507
Co-reporter:Dr. Xingwei Li;Songjie Yu;Fen Wang;Dr. Boshun Wan;Xinzhang Yu
Angewandte Chemie International Edition 2013 Volume 52( Issue 9) pp:2577-2580
Publication Date(Web):
DOI:10.1002/anie.201209887
Co-reporter:Guoyong Song, Fen Wang and Xingwei Li
Chemical Society Reviews 2012 vol. 41(Issue 9) pp:3651-3678
Publication Date(Web):29 Feb 2012
DOI:10.1039/C2CS15281A
Rhodium(III)-catalyzed direct functionalization of C–H bonds under oxidative conditions leading to C–C, C–N, and C–O bond formation is reviewed. Various arene substrates bearing nitrogen and oxygen directing groups are covered in their coupling with unsaturated partners such as alkenes and alkynes. The facile construction of C–E (E = C, N, S, or O) bonds makes Rh(III) catalysis an attractive step-economic approach to value-added molecules from readily available starting materials. Comparisons and contrasts between rhodium(III) and palladium(II)-catalyzed oxidative coupling are made. The remarkable diversity of structures accessible is demonstrated with various recent examples, with a proposed mechanism for each transformation being briefly summarized (critical review, 138 references).
Co-reporter:Yuye Chen, Fen Wang, Aiqun Jia and Xingwei Li
Chemical Science 2012 vol. 3(Issue 11) pp:3231-3236
Publication Date(Web):07 Aug 2012
DOI:10.1039/C2SC20869E
Substrate-controlled selective oxidative olefination of N-protected 2-pyridones has been achieved under palladium catalysis. The 5-position selectivity was followed for N-protected simple pyridones. Introduction of substituents into the 4- or the 6-position switched the site selectivity to the 3-position. Diolefination can also be achieved with high efficiency. Oxidative arylation with polyfluorobenzenes followed a similar selectivity except that the system is more sterically and electronically demanding.
Co-reporter:Rui Niu, Jian Xiao, Tao Liang, and Xingwei Li
Organic Letters 2012 Volume 14(Issue 3) pp:676-679
Publication Date(Web):January 24, 2012
DOI:10.1021/ol2030982
Brønsted acid catalyzed functionalization of sp3 C–H bonds in 2-methyl azaarenes has been achieved in the reaction with isatins. This method provides facile synthesis of biologically important azaarene-substituted 3-hydroxy-2-oxindoles in one step in moderate to good yields.
Co-reporter:Miao Zhao, Fen Wang, and Xingwei Li
Organic Letters 2012 Volume 14(Issue 6) pp:1412-1415
Publication Date(Web):February 27, 2012
DOI:10.1021/ol300147t
Tetrasubstituted pyrroles have been synthesized via the cross-dehydrogenative coupling between enamino esters and acetone. Silver carbonate proved to be an effective oxidant, and no transition metal catalyst is necessary.
Co-reporter:Peng Zhao, Rui Niu, Fen Wang, Keli Han, and Xingwei Li
Organic Letters 2012 Volume 14(Issue 16) pp:4166-4169
Publication Date(Web):July 31, 2012
DOI:10.1021/ol3018352
NH and N-protected isoquinolones undergo Rh(III)-catalyzed oxidative olefination at the 8-position. Complementary redox-neutral olefination of such isoquinolones using internal alkynes was achieved under ruthenium catalysis.
Co-reporter:Peng Zhao, Fen Wang, Keli Han, and Xingwei Li
Organic Letters 2012 Volume 14(Issue 21) pp:5506-5509
Publication Date(Web):October 23, 2012
DOI:10.1021/ol302594w
Ruthenium(II)-catalyzed redox-neutral annulative coupling of N-sulfonyl imines with alkynes has been achieved for the synthesis of indenamines, where a sulfonamide cocatalyst is necessary.
Co-reporter:Xinzhang Yu, Xingwei Li and Boshun Wan
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 37) pp:7479-7482
Publication Date(Web):02 Aug 2012
DOI:10.1039/C2OB26270C
Palladium-catalyzed desulfitative and denitrogenative arylation of azoles with arylsulfonyl hydrazides has been achieved. A broad scope of azoles and arylsulfonyl hydrazides has been used to produce arylated azoles in high yields.
Co-reporter:Dongqi Wang;Fen Wang;Dr. Guoyong Song; Xingwei Li
Angewandte Chemie 2012 Volume 124( Issue 49) pp:12514-12518
Publication Date(Web):
DOI:10.1002/ange.201206918
Co-reporter:Wencui Zhen;Fen Wang;Miao Zhao;Dr. Zhengyin Du;Dr. Xingwei Li
Angewandte Chemie 2012 Volume 124( Issue 47) pp:11989-11993
Publication Date(Web):
DOI:10.1002/ange.201207204
Co-reporter:Peng Zhao, Dan Chen, Guoyong Song, Keli Han, and Xingwei Li
The Journal of Organic Chemistry 2012 Volume 77(Issue 3) pp:1579-1584
Publication Date(Web):December 27, 2011
DOI:10.1021/jo202228k
Palladium(II) can catalyze the oxidative coupling of tert-butyl 2-alkynylbenzoates with olefins such as acrylates and styrenes, leading to isocoumarines. The reaction was carried out under simple aerobic conditions, and in most cases, high selectivity has been attained.
Co-reporter:Tao Liang, Jian Xiao, Zhiyi Xiong, and Xingwei Li
The Journal of Organic Chemistry 2012 Volume 77(Issue 7) pp:3583-3588
Publication Date(Web):March 5, 2012
DOI:10.1021/jo2025114
The organocatalytic enantioselective 1,4-addition of aldehydes to acridiniums catalyzed by diarylprolinol silyl ether was achieved to furnish chiral acridanes in both high yields (82–96%) and excellent enantioselectivities (up to 99% ee), which also provides the highly enantioselective intermolecular α-alkylation of aldehydes with acridiniums salt.
Co-reporter:Dongqi Wang;Fen Wang;Dr. Guoyong Song; Xingwei Li
Angewandte Chemie International Edition 2012 Volume 51( Issue 49) pp:12348-12352
Publication Date(Web):
DOI:10.1002/anie.201206918
Co-reporter:Wencui Zhen;Fen Wang;Miao Zhao;Dr. Zhengyin Du;Dr. Xingwei Li
Angewandte Chemie International Edition 2012 Volume 51( Issue 47) pp:11819-11823
Publication Date(Web):
DOI:10.1002/anie.201207204
Co-reporter:Xiaohong Wei, Miao Zhao, Zhengyin Du, and Xingwei Li
Organic Letters 2011 Volume 13(Issue 17) pp:4636-4639
Publication Date(Web):August 3, 2011
DOI:10.1021/ol2018505
[RhCp*Cl2]2 can catalyze the oxidative coupling of N-aryl and N-alkyl benzamidines with alkynes to give N-substituted 1-aminoisoquinolines in high selectivity.
Co-reporter:Xuting Li, Xue Gong, Miao Zhao, Guoyong Song, Jian Deng, and Xingwei Li
Organic Letters 2011 Volume 13(Issue 21) pp:5808-5811
Publication Date(Web):October 11, 2011
DOI:10.1021/ol2023856
Rhodium(III)-catalyzed oxidative olefination of N-(1-naphthyl)sulfonamides has been achieved at the peri position. Three categories of olefins have been successfully applied. Activated olefins reacted to afford five-membered azacycles as a result of oxidative olefination–hydroamination. Unactivated olefins reacted to give the olefination product. 2-fold oxidative C–C and C–N coupling was achieved for allylbenzenes.
Co-reporter:Xingping Zhang;Dan Chen;Miao Zhao;Jing Zhao;Aiqun Jia
Advanced Synthesis & Catalysis 2011 Volume 353( Issue 5) pp:719-723
Publication Date(Web):
DOI:10.1002/adsc.201000887
Abstract
Isoquinolines have been synthesized from the redox-neutral dehydrative CN and CC cross-coupling between oximines and alkynes using a catalytic amount of (pentamethylcyclopentadiene)rhodium dichloride dimer {[RhCp*Cl2]2} and cesium acetate (CsOAc), a process that involves ortho CH activation of oximines and subsequent functionalization with alkynes. This redox-neutral catalytic isoquinoline synthesis operates under mild conditions, and is insensitive to moisture or air. A broad scope of coupling partners has been established, and a likely mechanism has been suggested.
Co-reporter:Xue Gong, Hong Zhang, Xingwei Li
Tetrahedron Letters 2011 Volume 52(Issue 43) pp:5596-5600
Publication Date(Web):26 October 2011
DOI:10.1016/j.tetlet.2011.08.058
Several iridium complexes bearing chelating abnormal N-heterocyclic carbenes (NHCs) are shown to be active catalysts for transfer hydrogenation of ketones or enones, dehydrative C–C coupling between primary and secondary alcohols, and dehydrogenation of benzyl alcohol to benzyl benzoate. In the transfer hydrogenation of acetophenone, abnormal NHC complexes give higher activity than a normal analogue. Dehydrative C–C coupling reactions between primary and secondary alcohols result in β-alkylation of the secondary alcohols, using primary alcohols as the apparent alkylating reagents, and such reactions proceed with high yield and selectivity. These catalytic processes are known to involve metal-mediated temporary borrowing of hydrogen from alcohols and subsequent delivery of the hydrogen to CC and /or CO bonds.
Co-reporter:Dr. Jian Xiao ; Xingwei Li
Angewandte Chemie 2011 Volume 123( Issue 32) pp:7364-7375
Publication Date(Web):
DOI:10.1002/ange.201100148
Abstract
Reaktive Gold-α-Oxocarbenoide können als Zwischenstufen in der goldkatalysierten Funktionalisierung von Alkinen auftreten. Derartige Zwischenstufen lassen sich anhand einer inter- und intramolekularen Oxidation von Alkinen mit nukleophilen, Sauerstoff übertragenden Gruppen wie Amin-N-oxiden, Pyridin-N-oxiden, N-Oxiden einer Schiffschen Base (Nitronen), Nitroverbindungen, Sulfoxiden und Epoxiden synthetisieren. Diese Sauerstoffübertragungsprozesse erfolgen als goldvermittelte Additions-Eliminierungs-Reaktionen. In katalytischen Prozessen werden α-Oxocarbenoide von Iminen und Arenen sowie von wandernden Hydriden und Alkylgruppen angegriffen. Diese Reaktionssequenzen führen zu Produkten mit neuen Molekülstrukturen. Der einfache Aufbau von C-E-Einfachbindungen (E=C, N, S oder O) ist ein direkter Weg zu wertvollen Verbindungen aus leicht zugänglichen Ausgangsmaterialien.
Co-reporter:Dr. Guoyong Song;Dan Chen;Yan Su;Dr. Keli Han;Dr. Cheng-Ling Pan;Dr. Aiqun Jia;Dr. Xingwei Li
Angewandte Chemie 2011 Volume 123( Issue 34) pp:7937-7942
Publication Date(Web):
DOI:10.1002/ange.201102561
Co-reporter:Dr. Jian Xiao ; Xingwei Li
Angewandte Chemie International Edition 2011 Volume 50( Issue 32) pp:7226-7236
Publication Date(Web):
DOI:10.1002/anie.201100148
Abstract
An overview of reactive gold α-oxo carbenoid intermediates in the gold-catalyzed functionalization of alkynes is presented. Such intermediates can be generated from inter- and intramolecular oxidation of alkynes by nucleophilic oxygen-atom donor groups, such as amine N-oxides, pyridine N-oxides, nitrones, nitro compounds, sulfoxides, and epoxides. These O-atom transfer processes occur by gold-mediated addition–elimination reactions. In catalytic systems, α-oxo carbenoids can undergo nucleophilic attack by imine, arene, and migrating hydride as well as alkyl groups, leading to cascade reactions and the construction of new skeletons. The facile construction of CE (E=C, N, S, or O) bonds makes it an attractive step-economic approach to value-added molecules from readily available starting materials. The scope, mechanisms, and reactivity of such α-oxo carbenoid species are discussed. The remarkable diversity of structures accessible is demonstrated with various recent examples.
Co-reporter:Fen Wang, Guoyong Song, Zhengyin Du, and Xingwei Li
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2926-2932
Publication Date(Web):March 17, 2011
DOI:10.1021/jo2002209
Rh(III)-catalyzed oxidative coupling reactions between isoquinolones with 3-aryl groups and activated olefins have been achieved using anhydrous Cu(OAc)2 as an oxidant to give tetracyclic products. The nitrogen atom acts as a directing group to facilitate ortho C−H activation. This reaction can be one-pot starting from methyl benzohydroxamates, without the necessity of the isolation of isoquinolone products. A broad scope of substrates has been demonstrated, and both terminal and internal activated olefins can be applied. In the coupling of N-methylmaleimide, a Wacker-like mechanism was proposed, where backside attack of the NH group in isoquinolones is suggested as a key step. Selective C−H activation has also been achieved at the 8-position of 1-naphthol, leading to an olefination product.
Co-reporter:Guoyong Song, Xue Gong, and Xingwei Li
The Journal of Organic Chemistry 2011 Volume 76(Issue 18) pp:7583-7589
Publication Date(Web):August 5, 2011
DOI:10.1021/jo201266u
Selective synthesis of quinolines has been achieved via oxidative annulation of functionalized pyridines with two alkyne molecules under Rh(III)-catalyzed cascade C–H activation of pyridines using Cu(OAc)2 as an oxidant. The selectivity of this reaction is oxidant-dependent, particularly on the anion of the oxidant.
Co-reporter:Xingwei Li and Miao Zhao
The Journal of Organic Chemistry 2011 Volume 76(Issue 20) pp:8530-8536
Publication Date(Web):September 13, 2011
DOI:10.1021/jo201530r
[RhCp*Cl2]2-catalyzed oxidative coupling of 5-aryl-1H-pyrazoles with alkynes and acrylates has been achieved using Cu(OAc)2 as an oxidant. Coupling with alkynes afforded six-membered azacycles as a result of C–C and C–N coupling. Coupling with acrylates followed a process of diolefination and a subsequent aza-Michael cyclization.
Co-reporter:Dan Chen, Guoyong Song, Aiqun Jia, and Xingwei Li
The Journal of Organic Chemistry 2011 Volume 76(Issue 20) pp:8488-8494
Publication Date(Web):September 7, 2011
DOI:10.1021/jo201347r
Intramolecular oxygen transfer of nitrone– and sulfoxide–alkynes was achieved using a catalytic amount of Au(I) and a stoichiometric amount of iodine. The Au(I)-catalyzed cyclization of a nitrone–terminal alkyne afforded a cyclic iminoester, while cyclization of analogous nitrone–internal alkynes yielded aldehyde–enones. The I2-mediated cyclization of nitrone–alkynes afforded iodinated γ-lactams and the I2-mediated internal redox of the closely related sulfoxide–alkynes gave diketones functionalized with a thoiether.
Co-reporter:Dr. Guoyong Song;Dan Chen;Yan Su;Dr. Keli Han;Dr. Cheng-Ling Pan;Dr. Aiqun Jia;Dr. Xingwei Li
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7791-7796
Publication Date(Web):
DOI:10.1002/anie.201102561
Co-reporter:Jinlei Chen, Qingyu Pang, Yanbo Sun, and Xingwei Li
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3523-3526
Publication Date(Web):March 22, 2011
DOI:10.1021/jo1025546
Readily available Pd(II) chloride catalysts can catalyze selective and efficient oxidative coupling between N-aryl-2-aminopyridines and internal alkynes to yield N-(2-pyridyl)indoles. This process involves the ortho C−H activation of N-aryl-2-aminopyridines, and CuCl2 was used as an oxidant. Compared to our previously reported Rh(III)-catalyzed synthesis of this class of product, this method is advantageous with a wider scope of alkynes and cost-effective Pd(II) catalysts. Molecular oxygen can be used as a terminal oxidant.
Co-reporter:Yan Su, Miao Zhao, Keli Han, Guoyong Song, and Xingwei Li
Organic Letters 2010 Volume 12(Issue 23) pp:5462-5465
Publication Date(Web):October 29, 2010
DOI:10.1021/ol102306c
Catalytic oxidative coupling between acrylamides and alkynes was achieved using 0.5 mol % loading of [RhCp*Cl2]2 with Cu(OAc)2 as an oxidant. 2-Pyridones, iminoesters, and substituted indoles could be obtained as a result of the electronic and steric effects of the substituents in the acrylamides.
Co-reporter:Fen Wang, Guoyong Song, and Xingwei Li
Organic Letters 2010 Volume 12(Issue 23) pp:5430-5433
Publication Date(Web):November 1, 2010
DOI:10.1021/ol102241f
Rh(III)-catalyzed oxidative coupling reactions between benzamides or heteroaryl carboxamides and olefins have been developed. The vinylation product can further undergo a Michael reaction leading to γ-lactam in the case of electron-withdrawing olefins.
Co-reporter:Jinlei Chen, Guoyong Song, Cheng-Ling Pan, and Xingwei Li
Organic Letters 2010 Volume 12(Issue 23) pp:5426-5429
Publication Date(Web):November 1, 2010
DOI:10.1021/ol1022596
[RhCp*Cl2]2 (1−2 mol %) can catalyze the oxidative coupling of N-aryl-2-aminopyridines with alkynes and arylates to give N-(2-pyridyl)indoles and N-(2-pyridyl)quinolones, respectively, using Cu(OAc)2 as an oxidant. Coupling with styrenes gave mono- and/or disubstituted olefination products.
Co-reporter:Yao Zhang;Soon Hyeok Hong
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 10) pp:1779-1783
Publication Date(Web):
DOI:10.1002/adsc.201000083
Abstract
A novel nitrone-based pincer ligand was developed by a single-step synthesis from N-(tert-butyl)hydroxylamine acetate and 2,6-pyridinedicarboxaldehyde. The developed ligand allowed us to synthesize a cationic ruthenium pincer complex. A distorted octahedral coordination environment around the ruthenium center was observed. The complex showed excellent catalytic activity in transfer hydrogenation reactions with turnover numbers up to 590,000.
Co-reporter:Lingheng Kong; Songjie Yu; Xukai Zhou
Organic Letters () pp:
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.orglett.5b03629
C–H activation assisted by a bifunctional directing group has allowed the construction of heterocycles. This is ideally catalyzed by earth-abundant and eco-friendly transition metals. We report Co(III)-catalyzed redox-neutral coupling between arenes and alkynes using an NH amide as an electrophilic directing group. The redox-neutral C–H activation/coupling afforded quinolines with water as the sole byproduct.
Co-reporter:Peng Zhao ; Fen Wang ; Keli Han
Organic Letters () pp:
Publication Date(Web):June 12, 2012
DOI:10.1021/ol301371p
N-Acetoxyl ketoimine-alkynes undergo Rh(III)-catalyzed oxidative olefination to afford (2-acetoxymethyl)isoquinolines bearing an ortho-olefinated aryl group via a sequence that involves (1) Rh(III)-catalyzed alkyne cyclization with intramolecular 1,3-acetoxyl migration and (2) isoquinoline-directed ortho C–H olefination.
Co-reporter:Fen Wang, Songjie Yu and Xingwei Li
Chemical Society Reviews 2016 - vol. 45(Issue 23) pp:NaN6477-6477
Publication Date(Web):2016/09/29
DOI:10.1039/C6CS00371K
Organic transformations that involve direct functionalization of C–H bonds represent an attractive synthetic strategy that maximizes atom- and step-economy. With the generally high stability of C–H bonds, these processes have mostly required harsh reaction conditions, in combination with the necessity of activation of the C–H substrates and/or the coupling partners. As a class of activated coupling partners, strained or reactive rings exhibited high activity in the coupling with aryl and alkyl C–H bonds. Such a high reactivity of the rings allowed the facile construction of various new structural platforms via coupling with scission of the ring structures. The combination of C–H activation and scission of the rings allowed for applications of a broader scope of C–H bonds, including those less reactive alkyl ones. This synthetic diversity of these rings has been realized owing to the intrinsically different mechanisms of the interactions of transition metal catalysts and the strained/reactive rings.
Co-reporter:Guoyong Song, Fen Wang and Xingwei Li
Chemical Society Reviews 2012 - vol. 41(Issue 9) pp:NaN3678-3678
Publication Date(Web):2012/02/29
DOI:10.1039/C2CS15281A
Rhodium(III)-catalyzed direct functionalization of C–H bonds under oxidative conditions leading to C–C, C–N, and C–O bond formation is reviewed. Various arene substrates bearing nitrogen and oxygen directing groups are covered in their coupling with unsaturated partners such as alkenes and alkynes. The facile construction of C–E (E = C, N, S, or O) bonds makes Rh(III) catalysis an attractive step-economic approach to value-added molecules from readily available starting materials. Comparisons and contrasts between rhodium(III) and palladium(II)-catalyzed oxidative coupling are made. The remarkable diversity of structures accessible is demonstrated with various recent examples, with a proposed mechanism for each transformation being briefly summarized (critical review, 138 references).
Co-reporter:Fen Wang, Xiaoneng Cui, Zhangrong Lou, Jianzhang Zhao, Ming Bao and Xingwei Li
Chemical Communications 2014 - vol. 50(Issue 98) pp:NaN15630-15630
Publication Date(Web):2014/10/27
DOI:10.1039/C4CC07603F
Acid-switching of the triplet excited state in rhodamine-C60 dyads was achieved. The rhodamine moiety acts as an acid-activated visible light-harvesting antenna and C60 as the singlet energy acceptor and the spin converter, and production of the triplet state was enhanced in the presence of acid.
Co-reporter:Zisong Qi, Mei Wang and Xingwei Li
Chemical Communications 2014 - vol. 50(Issue 68) pp:NaN9778-9778
Publication Date(Web):2014/07/08
DOI:10.1039/C4CC03627A
A new rhodium-catalyzed synthesis of sultones via the oxidative coupling of sulfonic acids with internal alkynes is described. The reaction proceeds via aryl C–H activation assisted by a sulfonic acid group.
Co-reporter:Xueyun Zhang, Zisong Qi, Jian Gao and Xingwei Li
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 46) pp:NaN9332-9332
Publication Date(Web):2014/09/30
DOI:10.1039/C4OB01596G
Rh(III)-catalyzed efficient C–H alkynylation of azomethine imines with alkynylated hypervalent iodine is developed under mild conditions. A broad scope of azomethine imines and alkyne substrates is established. The azomethine acts as a masked aldehyde and circumvents its poor directing effect.
Co-reporter:Zisong Qi, Guo-Dong Tang, Cheng-Ling Pan and Xingwei Li
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 45) pp:NaN10980-10980
Publication Date(Web):2015/10/13
DOI:10.1039/C5OB01886B
Rh(III)-catalyzed C–H activation of 3-aryl-dihydroisoxazoles in the coupling with diarylacetylenes has been developed under redox-neutral conditions. This reaction occurred under mild conditions with no by-product, and the N–O bond functions as an oxidizing directing group, leading to efficient synthesis of isoquinolines functionalized with a proximal secondary alcohol.
Co-reporter:Xinzhang Yu, Xingwei Li and Boshun Wan
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 37) pp:NaN7482-7482
Publication Date(Web):2012/08/02
DOI:10.1039/C2OB26270C
Palladium-catalyzed desulfitative and denitrogenative arylation of azoles with arylsulfonyl hydrazides has been achieved. A broad scope of azoles and arylsulfonyl hydrazides has been used to produce arylated azoles in high yields.
Co-reporter:Yuye Chen, Fen Wang, Aiqun Jia and Xingwei Li
Chemical Science (2010-Present) 2012 - vol. 3(Issue 11) pp:NaN3236-3236
Publication Date(Web):2012/08/07
DOI:10.1039/C2SC20869E
Substrate-controlled selective oxidative olefination of N-protected 2-pyridones has been achieved under palladium catalysis. The 5-position selectivity was followed for N-protected simple pyridones. Introduction of substituents into the 4- or the 6-position switched the site selectivity to the 3-position. Diolefination can also be achieved with high efficiency. Oxidative arylation with polyfluorobenzenes followed a similar selectivity except that the system is more sterically and electronically demanding.
Co-reporter:Xifa Yang, He Wang, Xukai Zhou and Xingwei Li
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 23) pp:NaN5237-5237
Publication Date(Web):2016/05/17
DOI:10.1039/C6OB00825A
Mild and efficient synthesis of benzophenones via Ir(III)- and Rh(III)-catalyzed, directing group-assisted formyl C–H arylation of benzaldehydes has been achieved using diaryliodonium salts, in which Rh(III) and Ir(III) catalysts exhibited a complementary substrate scope.
Co-reporter:Shui Hu, Dongqi Wang, Jiexiang Liu and Xingwei Li
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 17) pp:NaN2765-2765
Publication Date(Web):2013/03/12
DOI:10.1039/C3OB40272J
Rhodium(III)-catalyzed oxidative couplings between N-sulfonyl allylamines and activated olefins have been achieved. Only olefination occurred for acrylates, and the butadiene product can be further cyclized under palladium-catalyzed aerobic conditions. The coupling with N,N-dimethylacrylamide followed a cyclization pathway.
Co-reporter:Tingting Yang, Tao Zhang, Shangdong Yang, Shanshan Chen and Xingwei Li
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 25) pp:NaN4294-4294
Publication Date(Web):2014/04/29
DOI:10.1039/C4OB00704B
Rh(III)-catalyzed coupling of N-sulfonyl 2-aminobenzaldehydes with oxygenated allylic olefins via C–H bond activation is described. Diarylketones were obtained through coupling of N-sulfonyl 2-aminobenzaldehydes with 7-oxabenzonorbornadienes. On the other hand, the coupling with allyl carbonate yielded a six-membered sulfonyl lactam via a sequence of allylation–isomerization–Michael cyclization.
Co-reporter:Shanshan Chen, Yan Su, Keli Han and Xingwei Li
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 7) pp:NaN791-791
Publication Date(Web):2015/04/29
DOI:10.1039/C5QO00049A
A series of five-coordinate Rh(III) vinyl complexes [Rh(N^C)(PAr3)2CHCHR]PF6 have been isolated as an intermediate in the coupling of a Rh(III) hydride with terminal alkynes. These Rh(III) vinyl complexes underwent aryl–vinyl reductive coupling to afford the Rh(I) chelating complex [Rh(N^C–CHCHR)–(PAr3)2]PF6 in high yields. Kinetic studies on the C–C reductive elimination revealed that the reaction kinetics is first order for a Rh(III)(4-trifluoromethyl)styryl complex with activation parameters of ΔH≠ = 20.9 kcal mol−1 and ΔS≠ = −6.1 eu. The electronic effects of the styryl group and the phosphine ligands on the rate of C–C reductive elimination were studied, and the rate constant decreases for a more electron-poor styryl group but increases for a less donating phosphine. The inhibitive effect of the added phosphine indicates that the dissociation of phosphine to afford a four-coordinate intermediate is involved, which was further supported by DFT calculations. Although intermediacy of a 4-coordinate species has been suggested, the active intermediate that directly undergoes C–C coupling was pinpointed to a five-coordinate cis phosphine complex on the basis of DFT studies. Significant accelerating effects were observed for oxygen donor solvents (THF-d8 and acetone-d6), possibly via efficient stabilization of the four-coordinate intermediate. However coordination of CO forms an inert six-coordinate Rh(III) complex. Thus an overall detailed mechanism of alkyne insertion and subsequent aryl–vinyl reductive elimination from the Rh(III) center has been proposed based on the experimental and theoretical data.
Co-reporter:Lingheng Kong, Xifa Yang, Xukai Zhou, Songjie Yu and Xingwei Li
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 7) pp:NaN816-816
Publication Date(Web):2016/04/28
DOI:10.1039/C6QO00134C
Cobalt(III)-catalyzed C–H activation of simple benzoate esters has been achieved, and redox-neutral annulative coupling with internal alkynes allowed efficient synthesis of indenones. The employment of the weak and simple ester directing group makes it an attractive protocol in carbocycle synthesis.
Co-reporter:Qiang Wang and Xingwei Li
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 9) pp:NaN1162-1162
Publication Date(Web):2016/07/26
DOI:10.1039/C6QO00287K
Benzylamine has been applied as an arene source in C–H activation and coupling with different types of diazo compounds, leading to the synthesis of fused isoquinolines. This occurs via a mild synergistic rhodium- and copper-catalyzed process. Moreover, ecofriendly O2 has been used as a terminal oxidant with high efficiency.



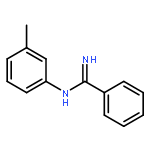


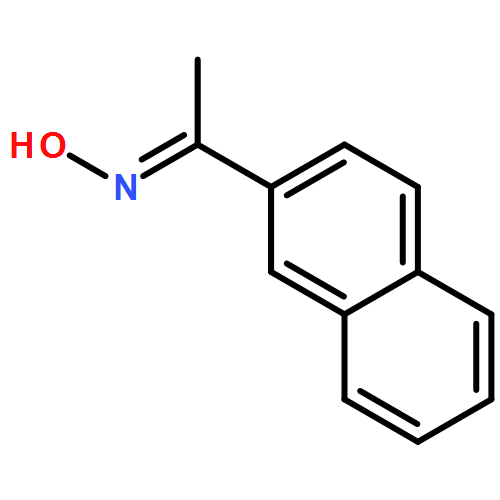
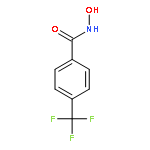
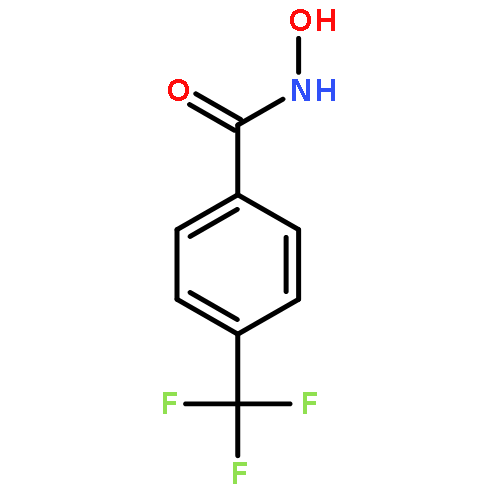




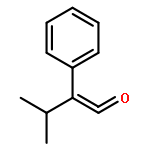
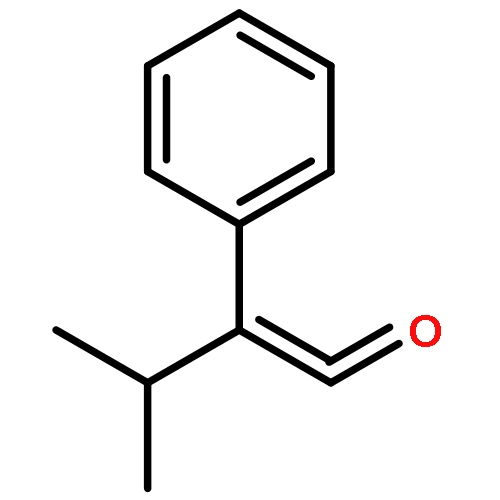



![Benzenamine, N-[(4-chlorophenyl)methylene]-, N-oxide, (Z)-](http://img.cochemist.com/ccimg/32300/32213-75-5.png)
![Benzenamine, N-[(4-chlorophenyl)methylene]-, N-oxide, (Z)-](http://img.cochemist.com/ccimg/32300/32213-75-5_b.png)
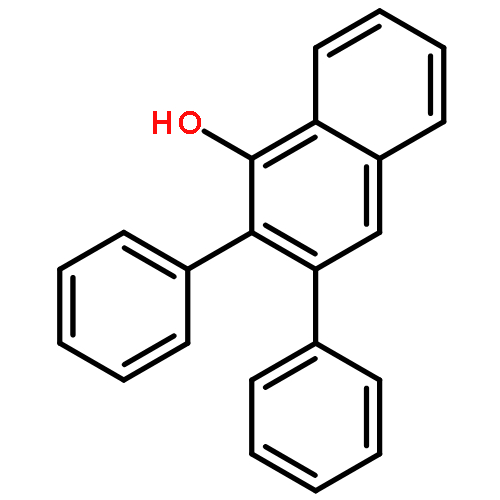



















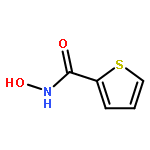



![Ethanone, 1-[4-(trifluoromethyl)phenyl]-, oxime](/data/chemimg/523900/15996-83-5.png)
![Ethanone, 1-[4-(trifluoromethyl)phenyl]-, oxime](/data/chemimg/523900/15996-83-5_b.png)



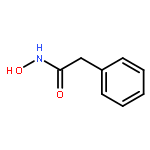
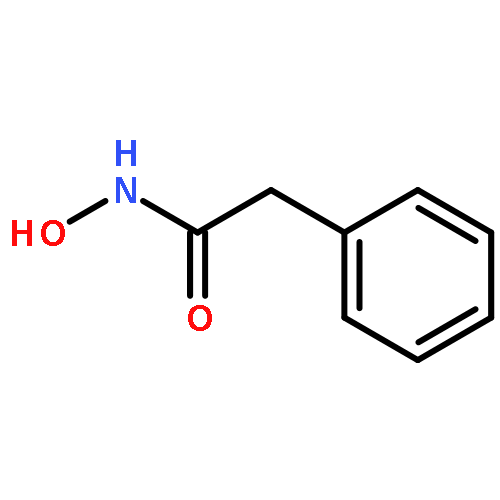
![11H-INDENO[1,2-C]ISOQUINOLIN-11-ONE](http://img.cochemist.com/ccimg/5300/5264-22-2.png)
![11H-INDENO[1,2-C]ISOQUINOLIN-11-ONE](http://img.cochemist.com/ccimg/5300/5264-22-2_b.png)





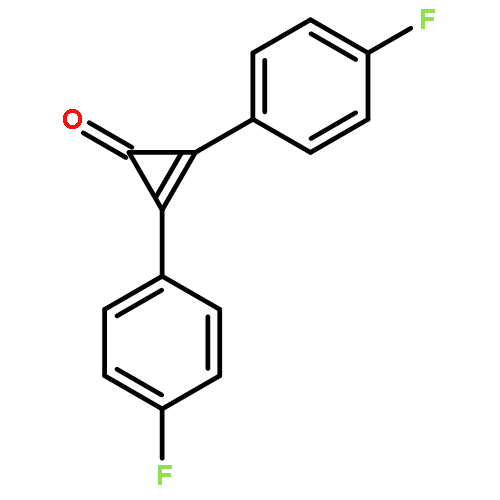
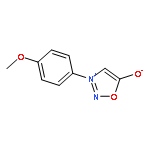
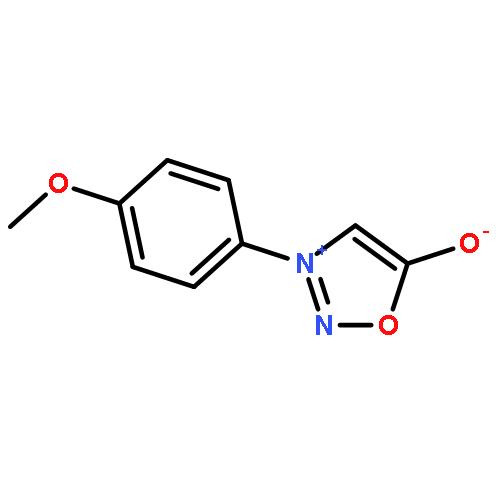


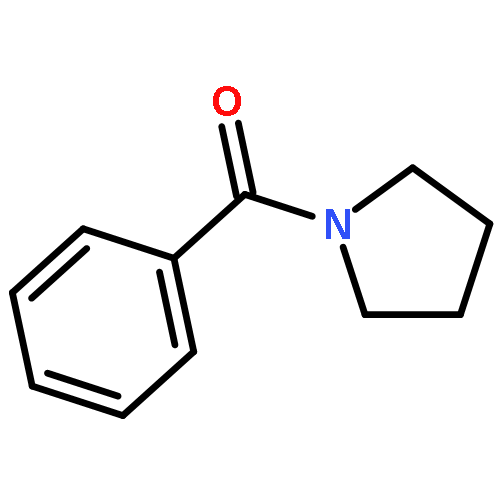

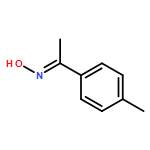

![(nz)-n-[1-(4-ethylphenyl)ethylidene]hydroxylamine](http://img.cochemist.com/ccimg/2100/2089-32-9.png)
![(nz)-n-[1-(4-ethylphenyl)ethylidene]hydroxylamine](http://img.cochemist.com/ccimg/2100/2089-32-9_b.png)












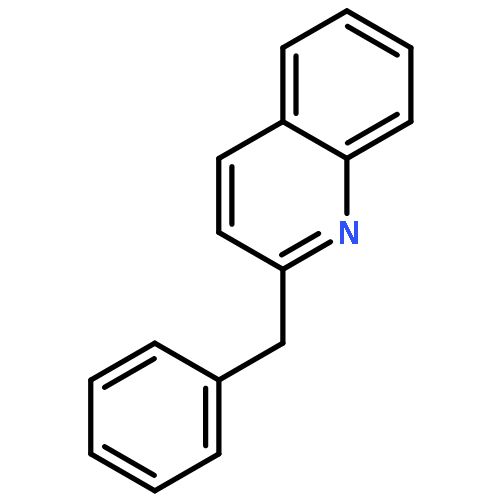
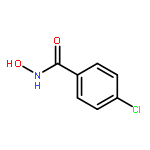



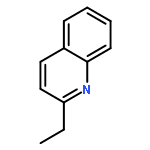
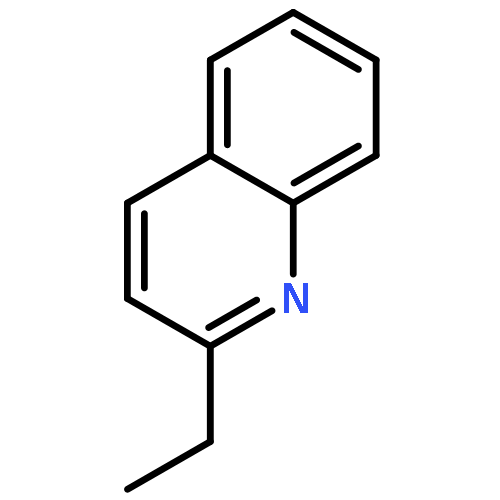
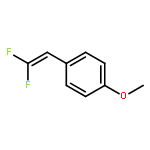
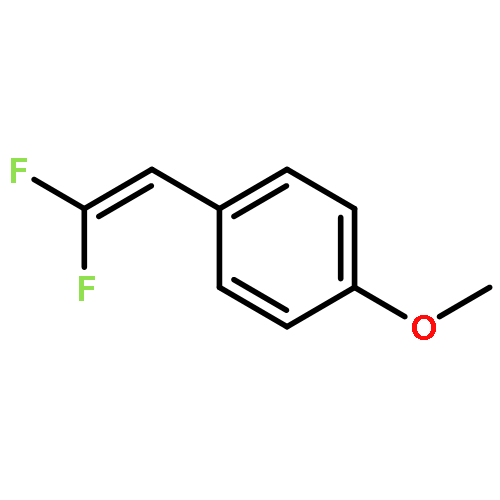

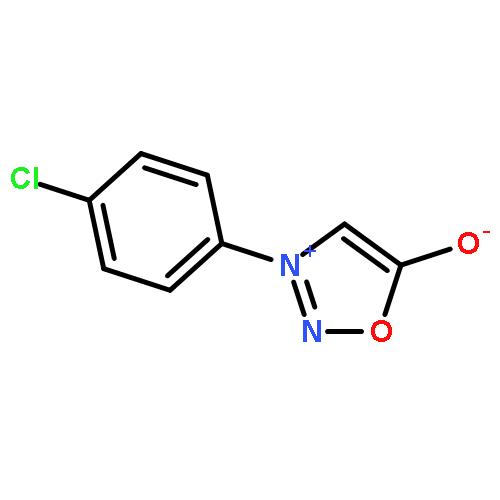



![11H-INDENO[1,2-C]ISOQUINOLINE](http://img.cochemist.com/ccimg/300/238-92-6.png)
![11H-INDENO[1,2-C]ISOQUINOLINE](http://img.cochemist.com/ccimg/300/238-92-6_b.png)





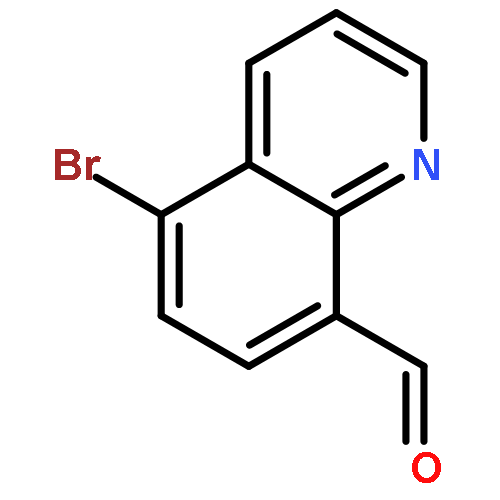



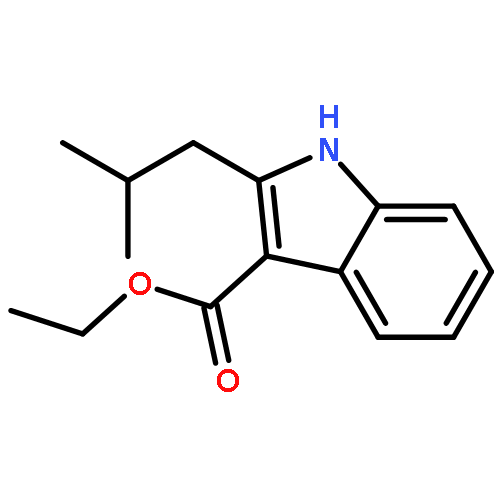


![PYRIDINE, 2-[2-[(TRIMETHYLSILYL)ETHYNYL]PHENYL]-](http://img.cochemist.com/ccimg/485400/485385-78-2.png)
![PYRIDINE, 2-[2-[(TRIMETHYLSILYL)ETHYNYL]PHENYL]-](http://img.cochemist.com/ccimg/485400/485385-78-2_b.png)
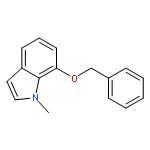







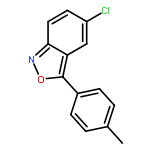







![ETHANONE, 1-[4-(2-PYRIMIDINYL)PHENYL]- (9CI)](http://img.cochemist.com/ccimg/259600/259541-90-7.png)
![ETHANONE, 1-[4-(2-PYRIMIDINYL)PHENYL]- (9CI)](http://img.cochemist.com/ccimg/259600/259541-90-7_b.png)




![Imidazo[1,2-a]pyridine, 2-(3-methoxyphenyl)-](http://img.cochemist.com/ccimg/205700/205655-15-8.png)
![Imidazo[1,2-a]pyridine, 2-(3-methoxyphenyl)-](http://img.cochemist.com/ccimg/205700/205655-15-8_b.png)






![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6.png)
![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6_b.png)


![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2.png)
![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2_b.png)
![7-Azabicyclo[4.1.0]hept-2-ene, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/172800/172791-02-5.png)
![7-Azabicyclo[4.1.0]hept-2-ene, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/172800/172791-02-5_b.png)


![Benzene, 1-chloro-4-[[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/150700/150619-48-0.png)
![Benzene, 1-chloro-4-[[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/150700/150619-48-0_b.png)
![Pyridine, 2-[2-(phenylsulfonyl)phenyl]-](http://img.cochemist.com/ccimg/147900/147879-73-0.png)
![Pyridine, 2-[2-(phenylsulfonyl)phenyl]-](http://img.cochemist.com/ccimg/147900/147879-73-0_b.png)

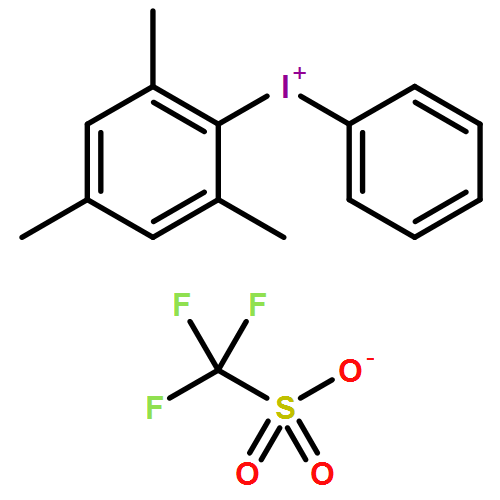
![Benzenesulfonamide, 4-methyl-N-[2-(4-methylbenzoyl)phenyl]-](http://img.cochemist.com/ccimg/141400/141368-26-5.png)
![Benzenesulfonamide, 4-methyl-N-[2-(4-methylbenzoyl)phenyl]-](http://img.cochemist.com/ccimg/141400/141368-26-5_b.png)
![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2.png)
![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2_b.png)



![Benzene, 1-methyl-4-[[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/121300/121289-99-4.png)
![Benzene, 1-methyl-4-[[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/121300/121289-99-4_b.png)


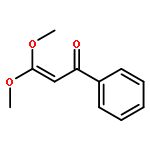


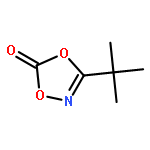
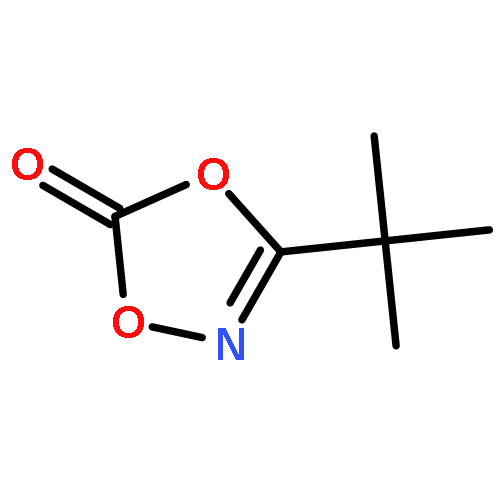












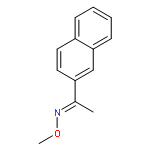
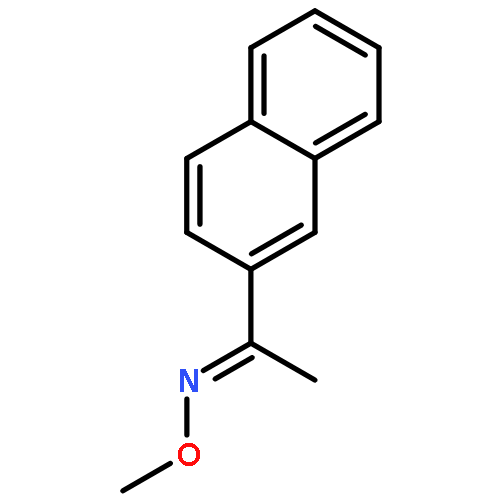







![5,8-Epoxynaphtho[2,3-d]-1,3-dioxole, 5,8-dihydro-](http://img.cochemist.com/ccimg/94700/94670-77-6.png)
![5,8-Epoxynaphtho[2,3-d]-1,3-dioxole, 5,8-dihydro-](http://img.cochemist.com/ccimg/94700/94670-77-6_b.png)
![4-[2-(acetyloxy)ethyl]-4-hydroxy-2,5-cyclohexadien-1-one](http://img.cochemist.com/ccimg/94500/94414-03-6.png)
![4-[2-(acetyloxy)ethyl]-4-hydroxy-2,5-cyclohexadien-1-one](http://img.cochemist.com/ccimg/94500/94414-03-6_b.png)





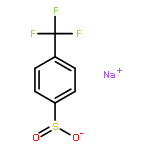
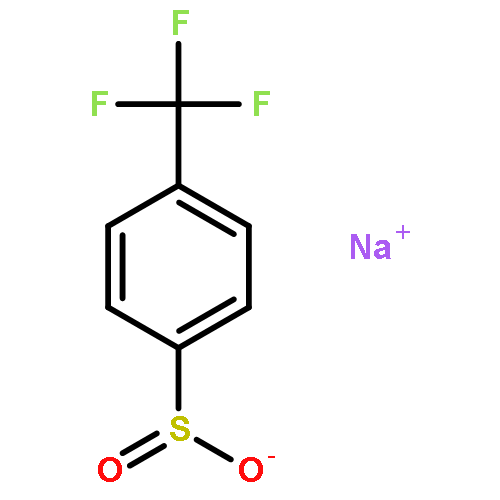


![BENZENESULFONAMIDE, 4-METHYL-N-[4-METHYL-2-(4-METHYLBENZOYL)PHENYL]-](http://img.cochemist.com/ccimg/88000/87995-70-8.png)
![BENZENESULFONAMIDE, 4-METHYL-N-[4-METHYL-2-(4-METHYLBENZOYL)PHENYL]-](http://img.cochemist.com/ccimg/88000/87995-70-8_b.png)


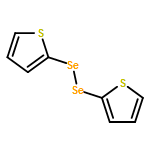
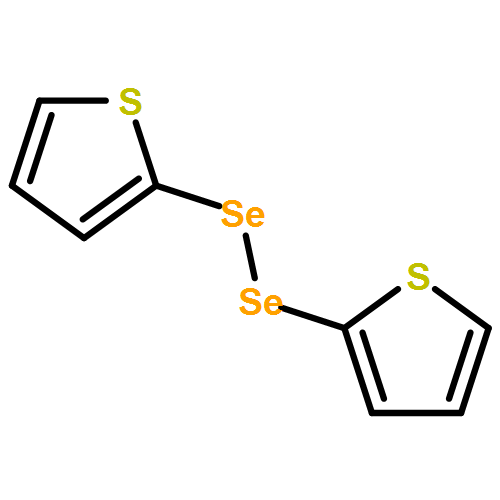
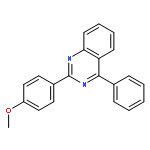
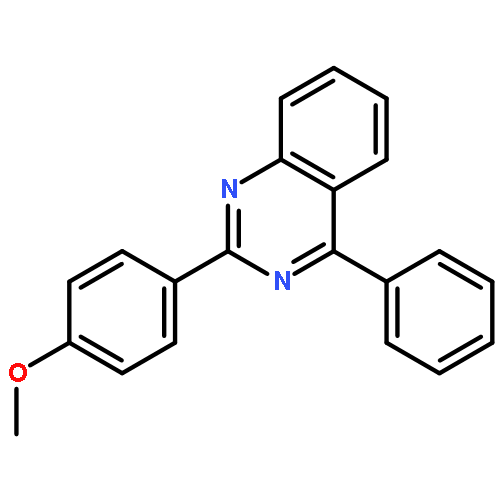









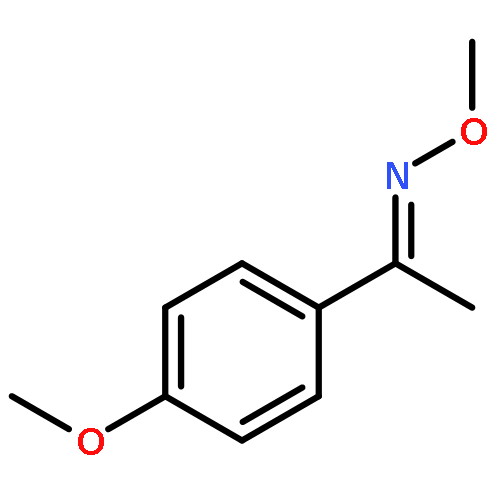






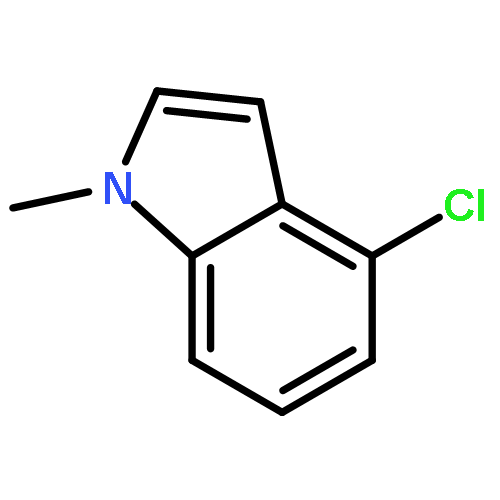


![[1(4H),2'-Bipyridin]-4-one](http://img.cochemist.com/ccimg/76600/76520-27-9.png)
![[1(4H),2'-Bipyridin]-4-one](http://img.cochemist.com/ccimg/76600/76520-27-9_b.png)

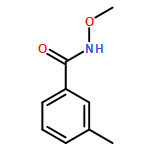
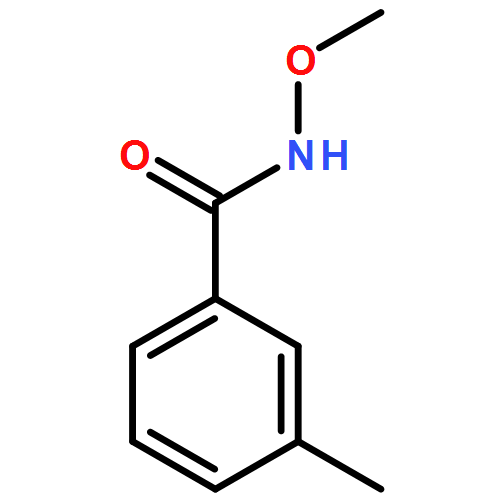


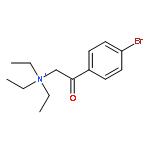


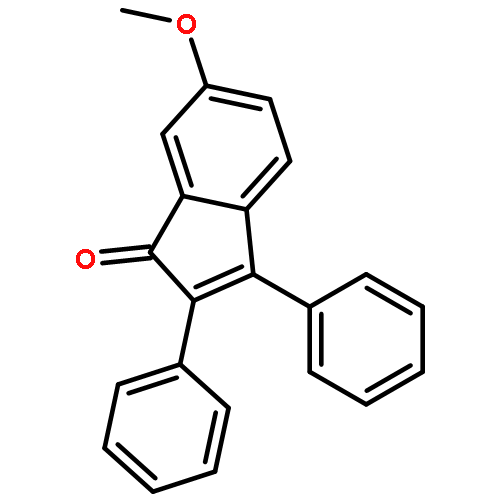

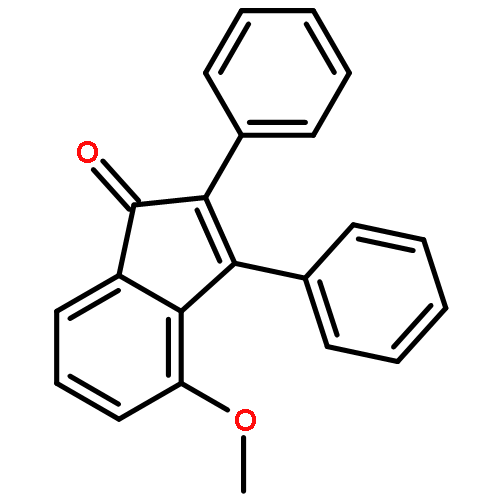
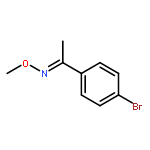

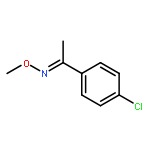








![Benzene, 2,4-dimethoxy-1-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/66500/66476-29-7.png)
![Benzene, 2,4-dimethoxy-1-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/66500/66476-29-7_b.png)



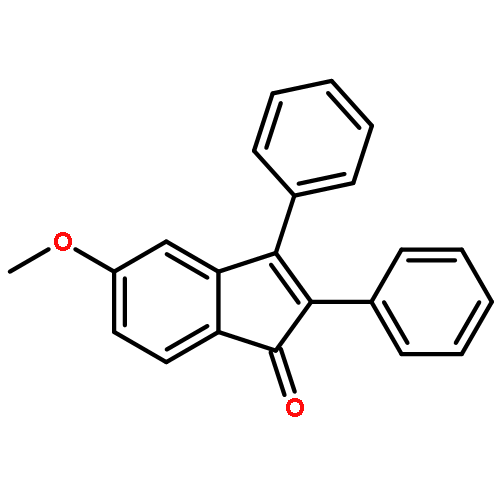





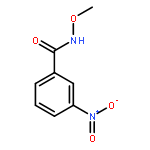
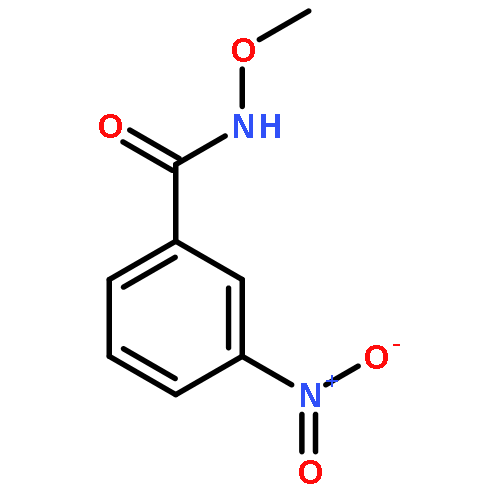


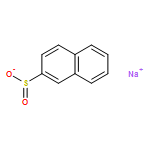
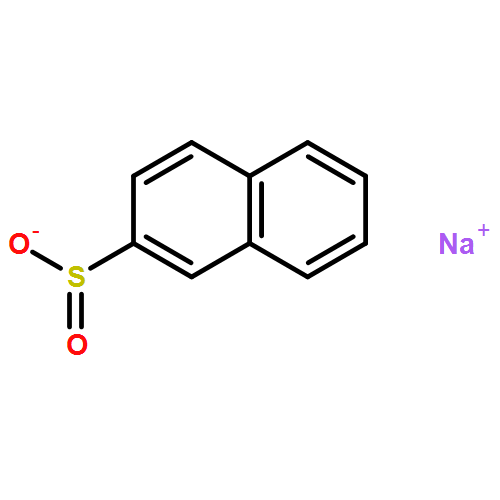
![5H-Indolo[2,3-c]isoquinoline, 5-methyl-7-phenyl-](http://img.cochemist.com/ccimg/62800/62729-13-9.png)
![5H-Indolo[2,3-c]isoquinoline, 5-methyl-7-phenyl-](http://img.cochemist.com/ccimg/62800/62729-13-9_b.png)
![1H-Indole, 3-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/62700/62665-49-0.png)
![1H-Indole, 3-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/62700/62665-49-0_b.png)
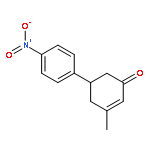






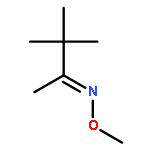



![Diselenide, bis[4-(trifluoromethyl)phenyl]](/data/chemimg/589700/57204-86-1.png)
![Diselenide, bis[4-(trifluoromethyl)phenyl]](/data/chemimg/589700/57204-86-1_b.png)





![ethyl [(2)H5]-benzoate](http://img.cochemist.com/ccimg/54400/54354-03-9.png)
![ethyl [(2)H5]-benzoate](http://img.cochemist.com/ccimg/54400/54354-03-9_b.png)
![Benzene,[2-[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/52900/52843-77-3.png)
![Benzene,[2-[(trifluoromethyl)sulfonyl]ethynyl]-](http://img.cochemist.com/ccimg/52900/52843-77-3_b.png)
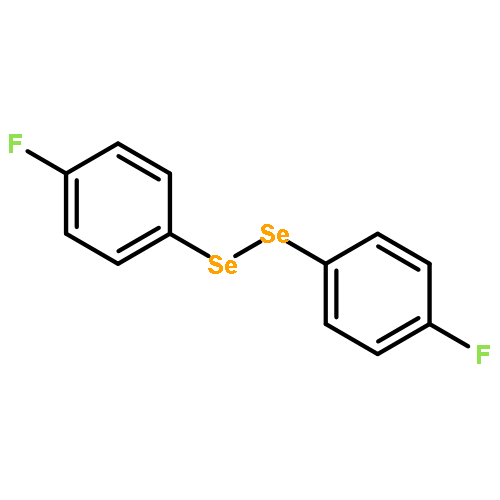






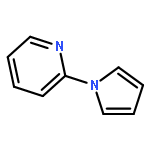
![5-Methyl-[1,1'-biphenyl]-3-ol](http://img.cochemist.com/ccimg/50800/50715-82-7.png)
![5-Methyl-[1,1'-biphenyl]-3-ol](http://img.cochemist.com/ccimg/50800/50715-82-7_b.png)

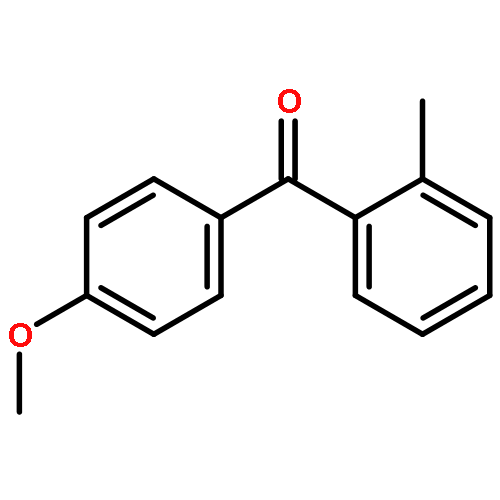
![Benzo[1,3]dioxole-5-carboxylic acid phenylamide](http://img.cochemist.com/ccimg/40200/40141-72-8.png)
![Benzo[1,3]dioxole-5-carboxylic acid phenylamide](http://img.cochemist.com/ccimg/40200/40141-72-8_b.png)


![2-NAPHTHALEN-2-YLIMIDAZO[1,2-A]PYRIDINE](http://img.cochemist.com/ccimg/39000/38922-71-3.png)
![2-NAPHTHALEN-2-YLIMIDAZO[1,2-A]PYRIDINE](http://img.cochemist.com/ccimg/39000/38922-71-3_b.png)



![Acetamide, N-[2-(4-methylbenzoyl)phenyl]-](http://img.cochemist.com/ccimg/36200/36188-57-5.png)
![Acetamide, N-[2-(4-methylbenzoyl)phenyl]-](http://img.cochemist.com/ccimg/36200/36188-57-5_b.png)






![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1.png)
![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1_b.png)
![Methanone, (4-methylphenyl)[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/34400/34328-30-8.png)
![Methanone, (4-methylphenyl)[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/34400/34328-30-8_b.png)















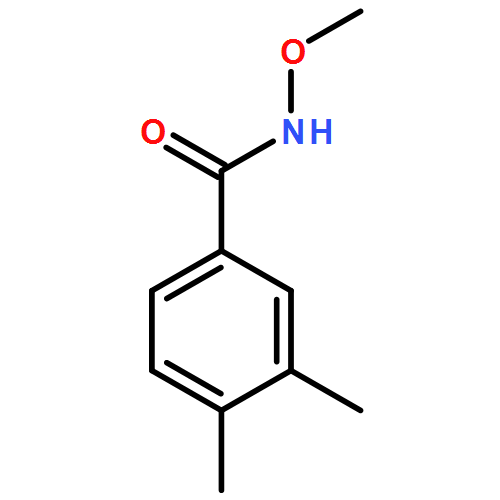







![N-[3-(TRIFLUOROMETHYL)PHENYL]PYRIDIN-2-AMINE](http://img.cochemist.com/ccimg/24100/24020-61-9.png)
![N-[3-(TRIFLUOROMETHYL)PHENYL]PYRIDIN-2-AMINE](http://img.cochemist.com/ccimg/24100/24020-61-9_b.png)









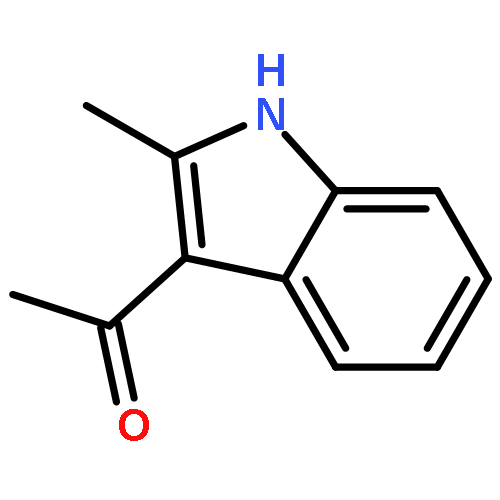
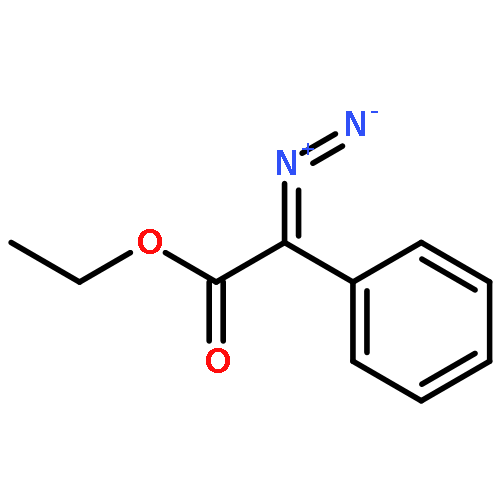
![Methanone, bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/21300/21221-91-0.png)
![Methanone, bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/21300/21221-91-0_b.png)







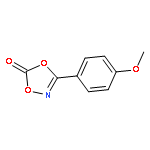
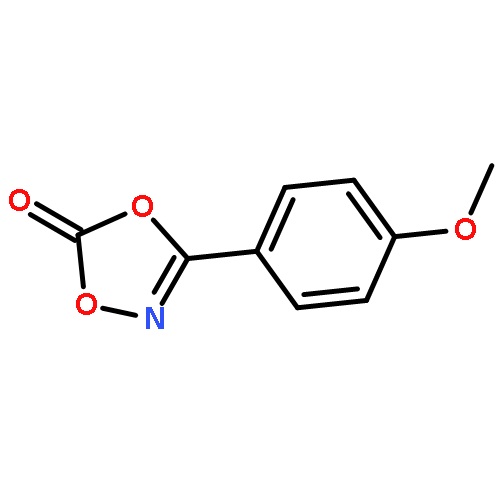
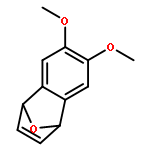
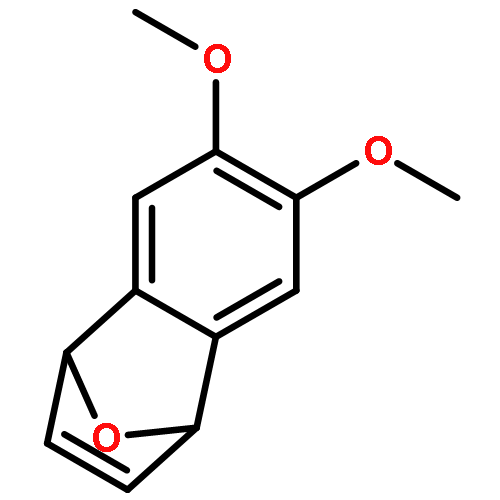


![1H-Pyrrole, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/18000/17900-53-7.png)
![1H-Pyrrole, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/18000/17900-53-7_b.png)
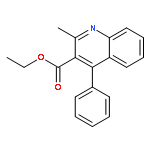
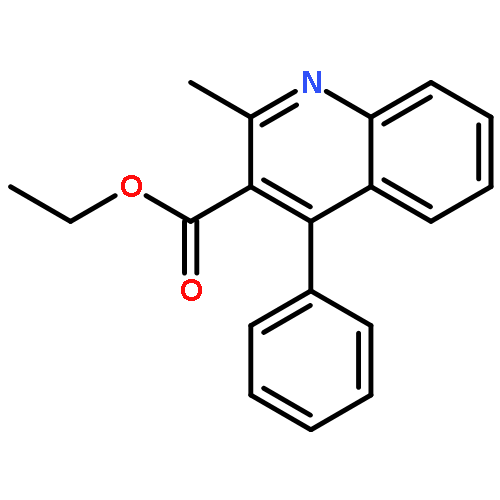
![Benzo[f]quinoline, 4-oxide](http://img.cochemist.com/ccimg/17200/17104-69-7.png)
![Benzo[f]quinoline, 4-oxide](http://img.cochemist.com/ccimg/17200/17104-69-7_b.png)
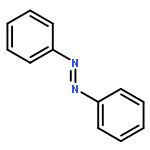
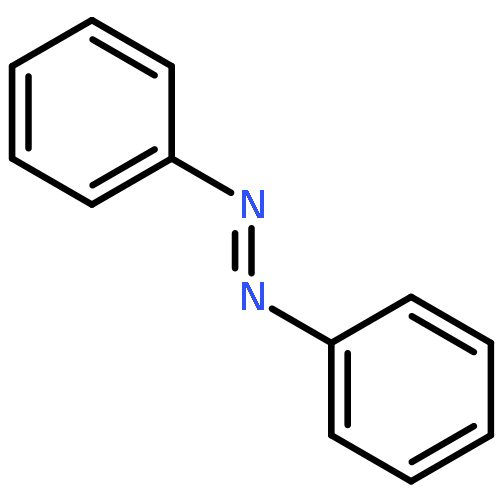







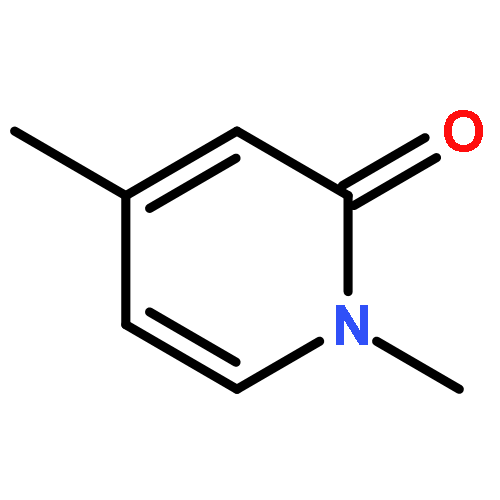
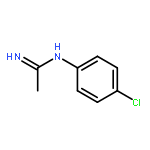

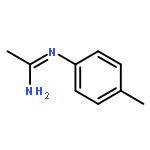

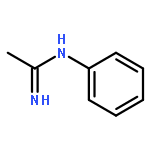
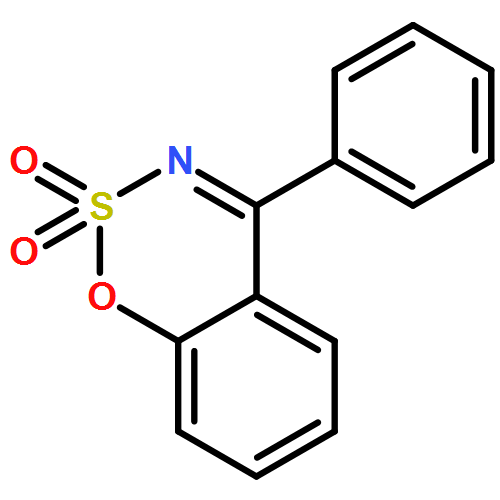


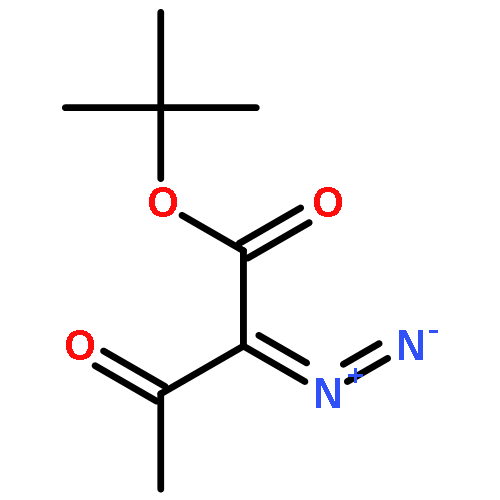





![1H-Benz[e]inden-1-one, 2,3-diphenyl-](http://img.cochemist.com/ccimg/10500/10408-65-8.png)
![1H-Benz[e]inden-1-one, 2,3-diphenyl-](http://img.cochemist.com/ccimg/10500/10408-65-8_b.png)



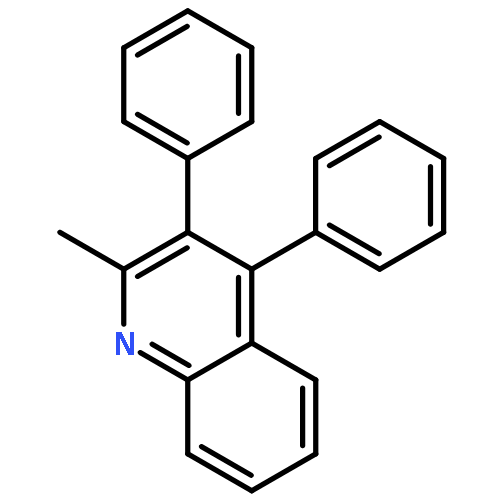




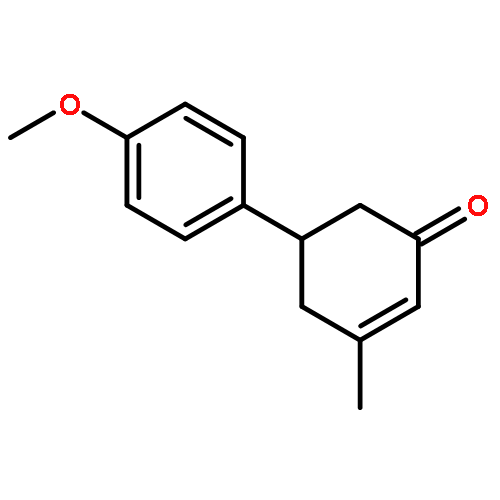


![9-oxabicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/6700/6690-12-6.png)
![9-oxabicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/6700/6690-12-6_b.png)
![8-Oxabicyclo[5.1.0]oct-2-ene](http://img.cochemist.com/ccimg/6700/6669-45-0.png)
![8-Oxabicyclo[5.1.0]oct-2-ene](http://img.cochemist.com/ccimg/6700/6669-45-0_b.png)







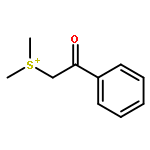
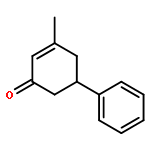
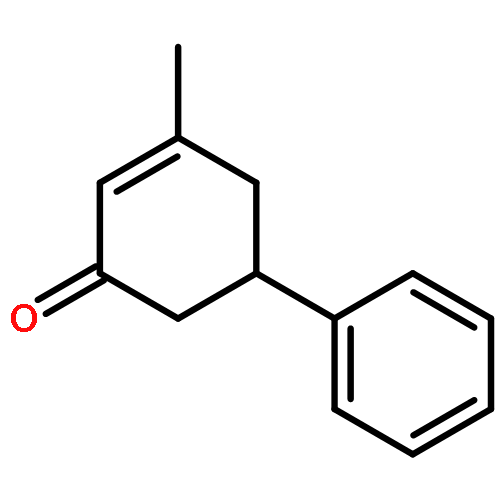
![[(phenylethynyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5400/5324-64-1.png)
![[(phenylethynyl)sulfonyl]benzene](http://img.cochemist.com/ccimg/5400/5324-64-1_b.png)





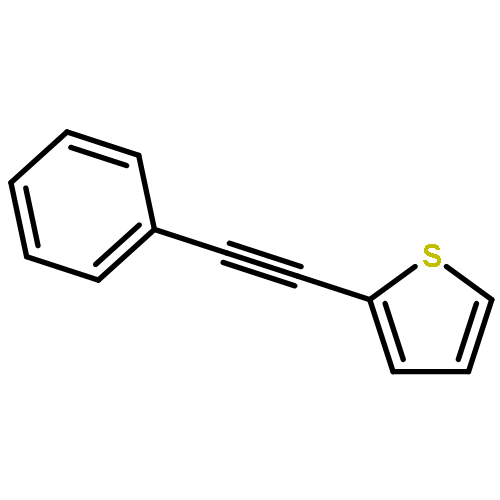
![5-Chlorobenzo[c]isoxazole](http://img.cochemist.com/ccimg/4600/4596-92-3.png)
![5-Chlorobenzo[c]isoxazole](http://img.cochemist.com/ccimg/4600/4596-92-3_b.png)






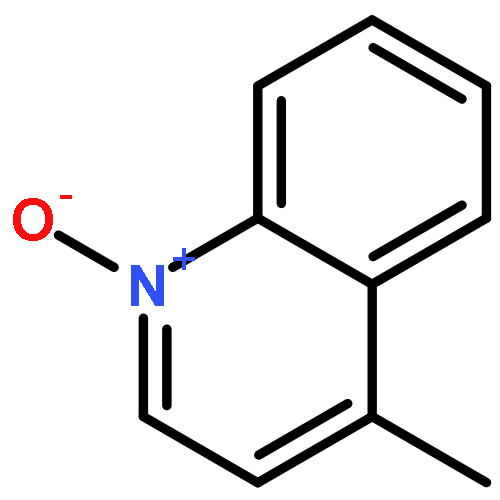







![Benzenamine,N,N-dimethyl-4-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/2700/2677-71-6.png)
![Benzenamine,N,N-dimethyl-4-[(trifluoromethyl)thio]-](http://img.cochemist.com/ccimg/2700/2677-71-6_b.png)



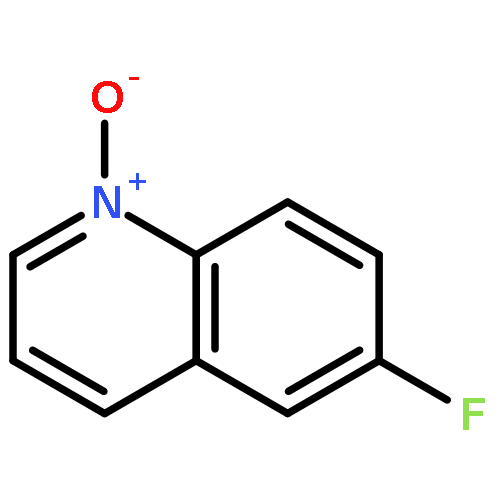
![6-Chloro-2-(4-fluorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/2100/2069-47-8.png)
![6-Chloro-2-(4-fluorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/2100/2069-47-8_b.png)


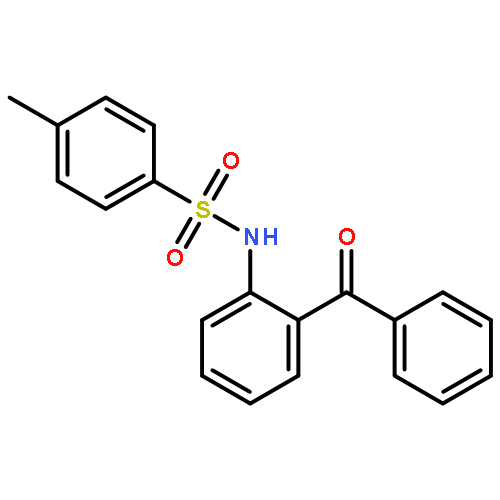






![(R)-Methyl 4-((3R,5R,8R,9S,10S,13R,14S,17R)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate](http://img.cochemist.com/ccimg/1300/1249-75-8.png)
![(R)-Methyl 4-((3R,5R,8R,9S,10S,13R,14S,17R)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate](http://img.cochemist.com/ccimg/1300/1249-75-8_b.png)










![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2.png)
![8-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-89-2_b.png)




![[1,3]Dioxolo[4,5-f]-2,1-benzisoxazole(8CI,9CI)](http://img.cochemist.com/ccimg/300/267-54-9.png)
![[1,3]Dioxolo[4,5-f]-2,1-benzisoxazole(8CI,9CI)](http://img.cochemist.com/ccimg/300/267-54-9_b.png)
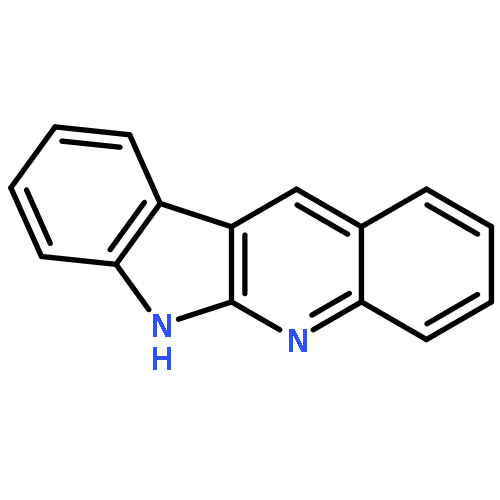
















![Ethanone, 1-[3-(trifluoromethyl)phenyl]-, O-methyloxime](http://img.cochemist.com/ccimg/127800/127733-36-2.png)
![Ethanone, 1-[3-(trifluoromethyl)phenyl]-, O-methyloxime](http://img.cochemist.com/ccimg/127800/127733-36-2_b.png)


![5-Methoxybenzo[c]isoxazole](http://img.cochemist.com/ccimg/122600/122528-39-6.png)
![5-Methoxybenzo[c]isoxazole](http://img.cochemist.com/ccimg/122600/122528-39-6_b.png)


![1,4,2-DIOXAZOL-5-ONE, 3-[4-(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/117100/117032-68-5.png)
![1,4,2-DIOXAZOL-5-ONE, 3-[4-(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/117100/117032-68-5_b.png)
![Imidazo[1,2-a]pyridine, 2-(4-chlorophenyl)-6-methyl-](http://img.cochemist.com/ccimg/103900/103844-28-6.png)
![Imidazo[1,2-a]pyridine, 2-(4-chlorophenyl)-6-methyl-](http://img.cochemist.com/ccimg/103900/103844-28-6_b.png)
![BENZENESULFONAMIDE, N-[2-(HYDROXYMETHYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/90400/90312-02-0.png)
![BENZENESULFONAMIDE, N-[2-(HYDROXYMETHYL)PHENYL]-4-METHYL-](http://img.cochemist.com/ccimg/90400/90312-02-0_b.png)
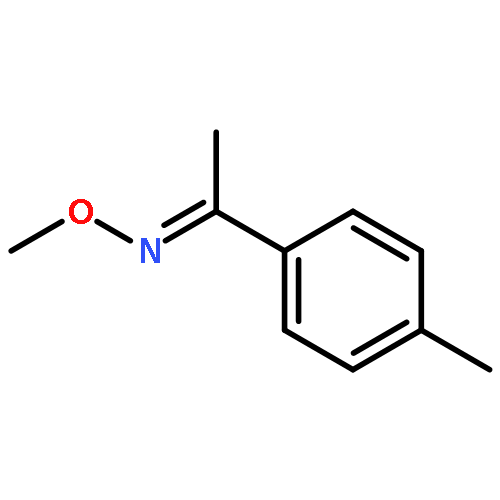
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6.png)
![IMIDAZO[1,2-A]PYRIDINE, 7-METHYL-2-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/66000/65964-61-6_b.png)


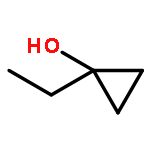



![(2E)-2-CYANO-3-{1-[3-(DIMETHYLAMINO)PROPYL]-1H-INDOL-3-YL}-N-OCTYLACRYLAMIDE](http://img.cochemist.com/ccimg/54700/54610-47-8.png)
![(2E)-2-CYANO-3-{1-[3-(DIMETHYLAMINO)PROPYL]-1H-INDOL-3-YL}-N-OCTYLACRYLAMIDE](http://img.cochemist.com/ccimg/54700/54610-47-8_b.png)


![[1,1'-Bicyclopropyl]-1-ol](http://img.cochemist.com/ccimg/54300/54251-80-8.png)
![[1,1'-Bicyclopropyl]-1-ol](http://img.cochemist.com/ccimg/54300/54251-80-8_b.png)

![2-(4-Biphenyl)imidazolo[1,2-a]pyridine](http://img.cochemist.com/ccimg/39000/38922-75-7.png)
![2-(4-Biphenyl)imidazolo[1,2-a]pyridine](http://img.cochemist.com/ccimg/39000/38922-75-7_b.png)
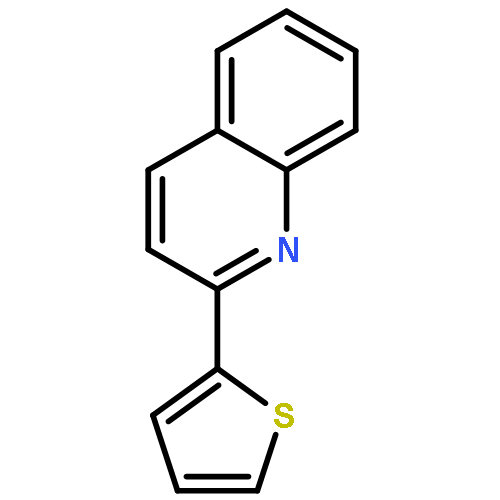







![[1(2H),2'-Bipyridin]-2-one, 5'-methyl-](http://img.cochemist.com/ccimg/10300/10201-68-0.png)
![[1(2H),2'-Bipyridin]-2-one, 5'-methyl-](http://img.cochemist.com/ccimg/10300/10201-68-0_b.png)
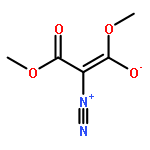
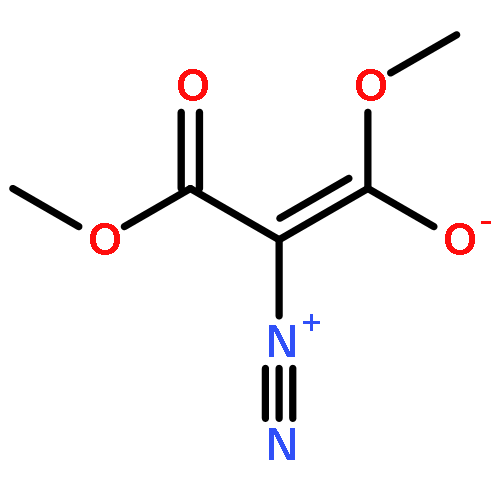






![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9.png)
![2-Phenylimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/4200/4105-21-9_b.png)
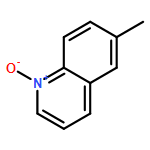

![Imidazo[1,2-a]pyridine, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/4100/4045-03-8.png)
![Imidazo[1,2-a]pyridine, 2-(2-thienyl)-](http://img.cochemist.com/ccimg/4100/4045-03-8_b.png)


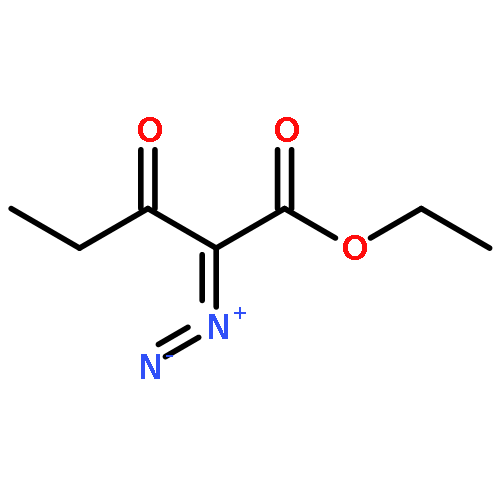
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6.png)
![7-Methyl-2-phenyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/900/885-91-6_b.png)




![2-(4-Fluorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/400/347-12-6.png)
![2-(4-Fluorophenyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/400/347-12-6_b.png)

![Benzoic acid, 4-[1-(methoxyimino)ethyl]-, methyl ester](/data/chemimg/185100/1262134-28-0.png)
![Benzoic acid, 4-[1-(methoxyimino)ethyl]-, methyl ester](/data/chemimg/185100/1262134-28-0_b.png)


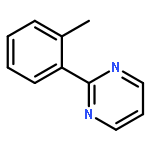
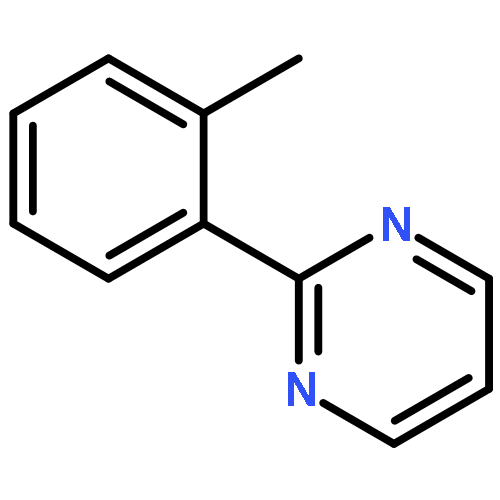




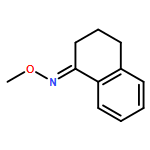







![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7.png)
![2-(4-Chloro-phenyl)-7-methyl-1H-imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-62-7_b.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5.png)
![2-(p-Tolyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/66000/65964-60-5_b.png)


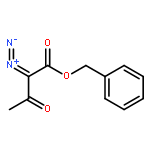
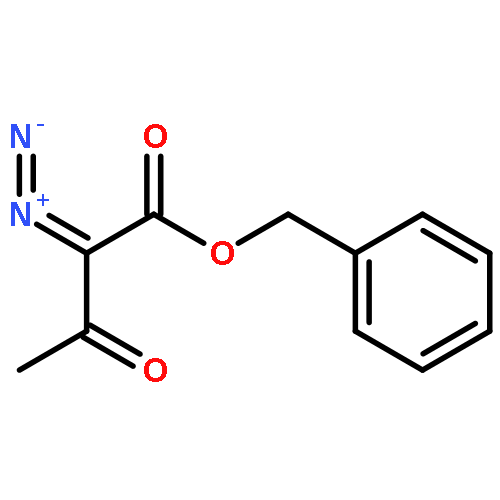


![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9_b.png)
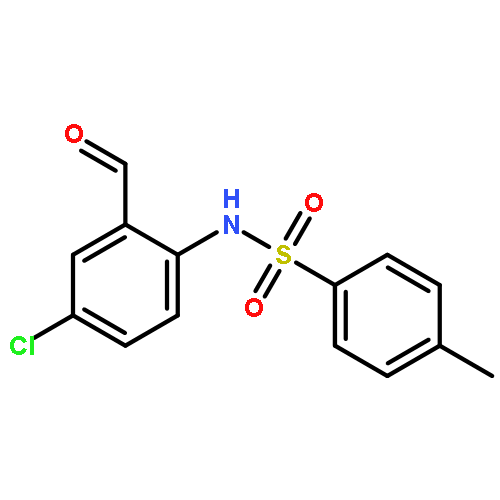






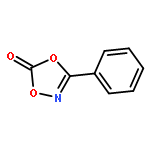
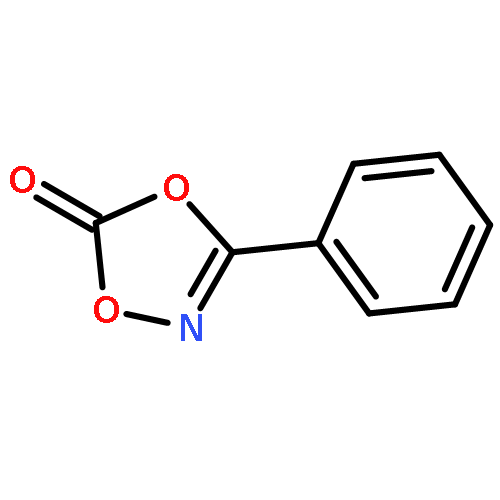


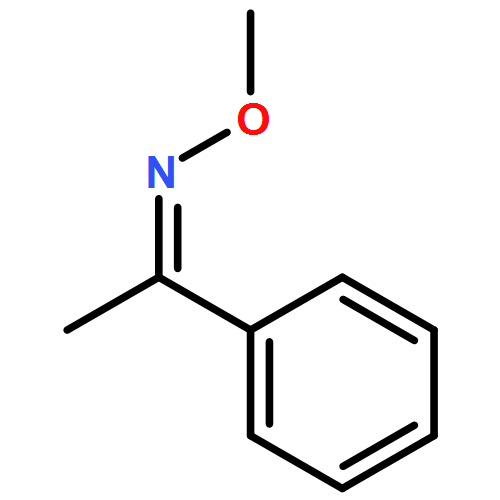












![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2.png)
![6-methyl-2-phenyl-Imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/1100/1019-89-2_b.png)


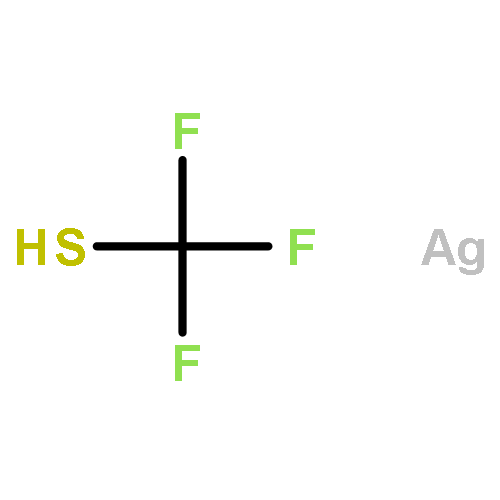


![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5.png)
![1-[2-tri(propan-2-yl)silylethynyl]-1λ3,2-benziodoxol-3-one](/data/chemimg/1772700/181934-30-5_b.png)


![Pyridine, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/93400/93324-66-4.png)
![Pyridine, 2-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/93400/93324-66-4_b.png)


![2-[5-[(4-METHYLPHENYL)METHYL]FURAN-2-YL]-1,3,4-OXADIAZOLE](http://img.cochemist.com/ccimg/57900/57870-49-2.png)
![2-[5-[(4-METHYLPHENYL)METHYL]FURAN-2-YL]-1,3,4-OXADIAZOLE](http://img.cochemist.com/ccimg/57900/57870-49-2_b.png)









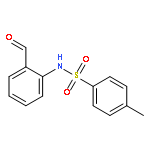
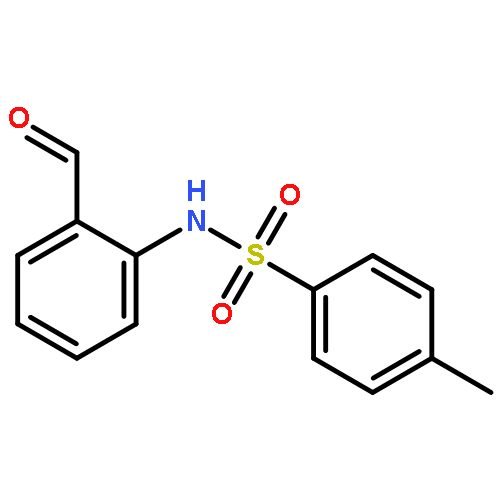
![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8.png)
![4-tert-butyl-1-[(2,2-dichlorocyclopropyl)methyl]-2-nitrobenzene](http://img.cochemist.com/ccimg/5300/5216-30-8_b.png)












![N-[3-[4-(3-acetamidopropylamino)butylamino]propyl]acetamide,dihydrochloride](http://img.cochemist.com/ccimg/900/825-60-5.png)
![N-[3-[4-(3-acetamidopropylamino)butylamino]propyl]acetamide,dihydrochloride](http://img.cochemist.com/ccimg/900/825-60-5_b.png)
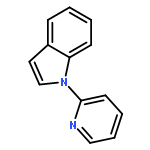
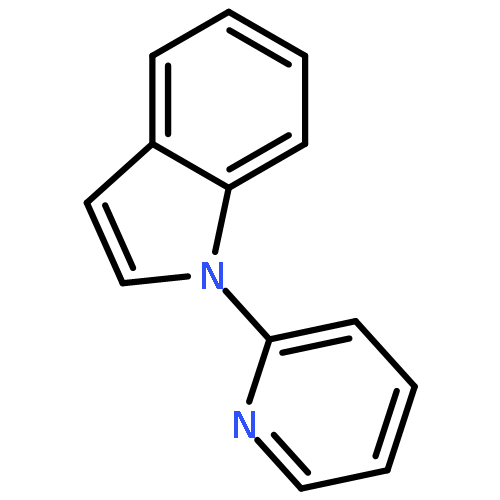








![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)