Co-reporter:Pierre Guiglion, Adriano Monti, and Martijn A. Zwijnenburg
The Journal of Physical Chemistry C 2017 Volume 121(Issue 3) pp:
Publication Date(Web):December 22, 2016
DOI:10.1021/acs.jpcc.6b11133
We compare, for a range of conjugated polymers relevant to water-splitting photocatalysis, the predictions for the redox potentials associated with charge carriers and excitons by a total-energy ΔDFT approach to those measured experimentally. For solid-state potentials, of the different classes of potentials available experimentally for conjugated polymers, the class measured under conditions which are the most similar to those during water splitting, we find a good fit between the ionization potentials predicted using ΔB3LYP and those measured experimentally using photoemission spectroscopy (PES). We also observe a reasonable fit to the more limited data sets of excited-state ionization potentials, obtained from two-photon PES, and electron affinities, measured by inverse PES, respectively. Through a comparison of solid-state potentials with gas phase and solution potentials for a range of oligomers, we demonstrate how the quality of the fit to experimental solid-state data is probably the result of benign error cancellation. We discuss that the good fit for solid-state potentials in vacuum suggests that a similar accuracy can be expected for calculations on solid-state polymers interfaced with water. We also analyze the quality of approximating the ΔB3LYP potentials by orbital energies. Finally, we discuss what a comparison between experimental and predicted potentials teaches us about conjugated polymers as photocatalysts, focusing specifically on the large exciton-binding energy in these systems and the mechanism of free charge carrier generation.
Co-reporter:Pierre Guiglion;Cristina Butchosa
Macromolecular Chemistry and Physics 2016 Volume 217( Issue 3) pp:344-353
Publication Date(Web):
DOI:10.1002/macp.201500432
Co-reporter:Martijn A. Zwijnenburg, Enrico Berardo, William J. Peveler, and Kim E. Jelfs
The Journal of Physical Chemistry B 2016 Volume 120(Issue 22) pp:5063-5072
Publication Date(Web):May 5, 2016
DOI:10.1021/acs.jpcb.6b03059
We investigate using a computational approach the physical and chemical processes underlying the application of organic (macro)molecules as fluorescence quenching sensors for explosives sensing. We concentrate on the use of amine molecular cages to sense nitroaromatic analytes, such as picric acid and 2,4-dinitrophenol, through fluorescence quenching. Our observations for this model system hold for many related systems. We consider the different possible mechanisms of fluorescence quenching: Förster resonance energy transfer, Dexter energy transfer and photoinduced electron transfer, and show that in the case of our model system, the fluorescence quenching is driven by the latter and involves stable supramolecular sensor–analyte host–guest complexes. Furthermore, we demonstrate that the experimentally observed selectivity of amine molecular cages for different explosives can be explained by the stability of these host–guest complexes and discuss how this is related to the geometry of the binding site in the sensor. Finally, we discuss what our observations mean for explosive sensing by fluorescence quenching in general and how this can help in future rational design of new supramolecular detection systems.
Co-reporter:Pierre Guiglion and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 27) pp:17854-17863
Publication Date(Web):12 Jun 2015
DOI:10.1039/C5CP01916H
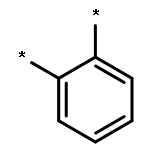
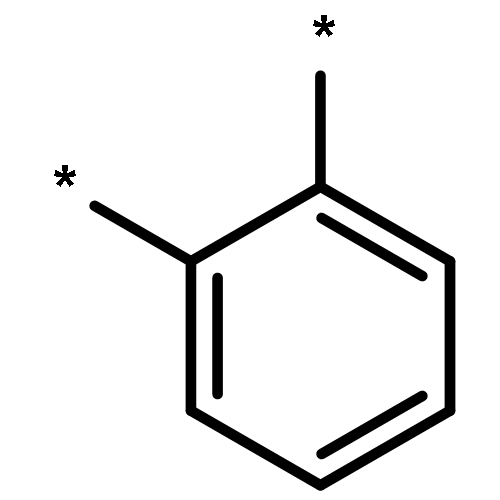
We use a combination of Time-Dependent Density Functional Theory (TD-DFT) and approximate Coupled Cluster Theory (RI-CC2) to compare trends in the optical gap and fluorescence energies of ortho-, meta- and para-oligomers of phenylene. We find that RI-CC2 and TD-DFT calculations using three different commonly employed XC-potentials (B3LYP, BHLYP and CAM-B3LYP) generally give consistent predictions. Most importantly, the fluorescence energy of m-phenylene is predicted to be independent of oligomer length, the fluorescence energy of p-phenylene to decrease with oligomer length and that of o-phenylene to increase. The origins of these differences in behaviour between the different isomers are analysed and found to stem from a subtle combination of steric and electronic factors.
Co-reporter:Milena C. C. Wobbe and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 43) pp:28892-28900
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5CP04851F
The nature and magnitude of the optical gaps of rocksalt alkaline earth (MgO, CaO, SrO, MgS, MgSe) and transition metal chalcogenide (CdO, PbS) nanoparticles are studied using time-dependent density functional theory (TD-DFT) calculations on (MX)32 nanoparticles. We demonstrate, just as we previously showed for MgO, that TD-DFT calculations on rocksalt nanoparticles require the use of hybrid exchange–correlation (XC-)functionals with a high percentage of Hartree–Fock like exchange (e.g. BHLYP) or range-separated XC-functionals to circumvent problems related to the description of charge-transfer excitations. Concentrating on the results obtained with TD-BHLYP we show that the optical gap in rocksalt nanoparticles displays a wide range of behavior; ranging from large optical gaps stemming from a localized excitation involving corner atoms in alkaline earth oxides to a delocalized excitation and small optical gaps in the transition metal chalcogenides. Finally, we rationalize this wide range of behaviour in terms of differences in the degree to which the Coulombic interaction between the excited electron and hole is screened in the different nanoparticles, and relate it to the optical dielectric constants of the bulk materials the nanoparticles are made from.
Co-reporter:Enrico Berardo
The Journal of Physical Chemistry C 2015 Volume 119(Issue 24) pp:13384-13393
Publication Date(Web):May 21, 2015
DOI:10.1021/acs.jpcc.5b01512
We explore, from a theoretical perspective, the effect of particle size on the photocatalytic water splitting activity of TiO2 rutile (nano)particles by a combination of explicit quantum chemistry calculations on a hydroxylated rutile nanoparticle in a realistic solvation environment and a comparison with the calculated properties of bulk rutile (surfaces) from the literature. Specifically, we use density functional theory (DFT) and time-dependent DFT to calculate the nanoparticle thermodynamic driving force for the water splitting half-reactions and identify in the process the crucial role of self-trapping of the free charge carriers responsible for proton reduction and water oxidation.
Co-reporter:Pierre Guiglion, Cristina Butchosa and Martijn A. Zwijnenburg
Journal of Materials Chemistry A 2014 vol. 2(Issue 30) pp:11996-12004
Publication Date(Web):11 Jun 2014
DOI:10.1039/C4TA02044H
A computational scheme to predict the thermodynamic ability of photocatalysts to drive both of the watersplitting half reactions, proton reduction and water oxidation, is discussed, and applied to a number of polymeric systems to explain their apparent inability to oxidise water. We predict that the poly(p-phenylene) (PPP) is thermodynamically unable to oxidise water and that PPP is hence unlikely to split water in the absence of an external electrical bias. For other polymers, however, for example carbon nitride, the lack of oxygen evolution activity appears kinetic in origin and hence a suitable co-catalyst could potentially transform them into true watersplitting photocatalysts.
Co-reporter:Enrico Berardo, Han-Shi Hu, Hubertus J. J. van Dam, Stephen A. Shevlin, Scott M. Woodley, Karol Kowalski, and Martijn A. Zwijnenburg
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 12) pp:5538-5548
Publication Date(Web):October 30, 2014
DOI:10.1021/ct500787x
We have investigated the description of excited state relaxation in naked and hydrated TiO2 nanoparticles using Time-Dependent Density Functional Theory (TD-DFT) with three common hybrid exchange-correlation (XC) potentials: B3LYP, CAM-B3LYP and BHLYP. Use of TD-CAM-B3LYP and TD-BHLYP yields qualitatively similar results for all structures, which are also consistent with predictions of coupled-cluster theory for small particles. TD-B3LYP, in contrast, is found to make rather different predictions; including apparent conical intersections for certain particles that are not observed with TD-CAM-B3LYP nor with TD-BHLYP. In line with our previous observations for vertical excitations, the issue with TD-B3LYP appears to be the inherent tendency of TD-B3LYP, and other XC potentials with no or a low percentage of Hartree–Fock like exchange, to spuriously stabilize the energy of charge-transfer (CT) states. Even in the case of hydrated particles, for which vertical excitations are generally well described with all XC potentials, the use of TD-B3LYP appears to result in CT problems during excited state relaxation for certain particles. We hypothesize that the spurious stabilization of CT states by TD-B3LYP even may drive the excited state optimizations to different excited state geometries from those obtained using TD-CAM-B3LYP or TD-BHLYP. Finally, focusing on the TD-CAM-B3LYP and TD-BHLYP results, excited state relaxation in small naked and hydrated TiO2 nanoparticles is predicted to be associated with a large Stokes’ shift.
Co-reporter:Enrico Berardo, Han-Shi Hu, Stephen A. Shevlin, Scott M. Woodley, Karol Kowalski, and Martijn A. Zwijnenburg
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 3) pp:1189-1199
Publication Date(Web):February 11, 2014
DOI:10.1021/ct4010273
We have investigated the suitability of Time-Dependent Density Functional Theory (TD-DFT) to describe vertical low-energy excitations in naked and hydrated titanium dioxide nanoparticles. Specifically, we compared TD-DFT results obtained using different exchange-correlation (XC) potentials with those calculated using Equation-of-Motion Coupled Cluster (EOM-CC) quantum chemistry methods. We demonstrate that TD-DFT calculations with commonly used XC potentials (e.g., B3LYP) and EOM-CC methods give qualitatively similar results for most TiO2 nanoparticles investigated. More importantly, however, we also show that, for a significant subset of structures, TD-DFT gives qualitatively different results depending upon the XC potential used and that only TD-CAM-B3LYP and TD-BHLYP calculations yield results that are consistent with those obtained using EOM-CC theory. Moreover, we demonstrate that the discrepancies for such structures originate from a particular combination of defects that give rise to charge-transfer excitations, which are poorly described by XC potentials that do not contain sufficient Hartree–Fock like exchange. Finally, we consider that such defects are readily healed in the presence of ubiquitously present water and that, as a result, the description of vertical low-energy excitations for hydrated TiO2 nanoparticles is nonproblematic.
Co-reporter:Milena C. C. Wobbe, Andrew Kerridge and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 40) pp:22052-22061
Publication Date(Web):28 Aug 2014
DOI:10.1039/C4CP03442B
The optical absorption spectra of magnesium oxide (MgO) nanoparticles, along with the atomic centres responsible, are studied using a combination of time-dependent density functional theory (TD-DFT) and coupled-cluster methods. We demonstrate that TD-DFT calculations on MgO nanoparticles require the use of range-separated exchange–correlation (XC-) functionals or hybrid XC-functionals with a high percentage of Hartree–Fock like exchange to circumvent problems related to the description of charge-transfer excitations. Furthermore, we show that the vertical excitations responsible for the experimentally studied range of the spectra of the MgO nanoparticles typically involve both 3-coordinated corner sites and 4-coordinated edge sites. We argue therefore that to label peaks in these absorption spectra exclusively as either corner or edge features does not provide insight into the full physical picture.
Co-reporter:Cristina Butchosa ; Tom O. McDonald ; Andrew I. Cooper ; Dave J. Adams
The Journal of Physical Chemistry C 2014 Volume 118(Issue 8) pp:4314-4324
Publication Date(Web):January 30, 2014
DOI:10.1021/jp411854f
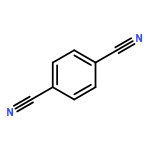
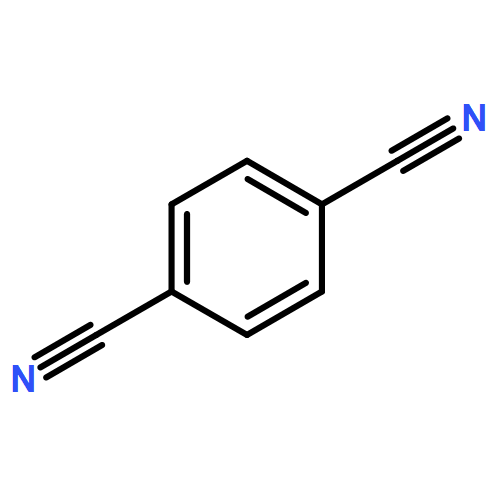
The strong interplay between the structure and optical properties of conjugated s-triazine-based framework (CTF) materials is explored in a combined experimental and computational study. The experimental absorption and fluorescence spectra of the CTF-1 material, a polymer obtained through the trimerization of 1,4-dicyanobenzene, are compared with the results of time-dependent density functional theory and approximate coupled cluster theory (CC2) calculations on cluster models of the polymer. To help explain the polymer data, we compare its optical properties with those measured and predicted for the 2,4,6-triphenyl-1,3,5-triazine model compound. Our analysis shows that CTFs, in line with experimental diffraction data, are likely to be layered materials based around flat hexagonal-like sheets and suggests that the long-wavelength part of the CTF-1 absorption spectrum displays a pronounced effect of stacking. Red-shifted peaks in the absorption spectrum appear that are absent for an isolated sheet. We also show that the experimentally observed strong fluorescence of CTF-1 and other CTF materials is further evidence of the presence of rings in the layers, as structures without rings are predicted to have extremely long excited state lifetimes and hence would display negligible fluorescence intensities. Finally, subtle differences between the experimental absorption spectra of CTF-1 samples prepared using different synthesis routes are shown to potentially arise from different relative arrangements of stacked layers.
Co-reporter:Cristina Butchosa ; Pierre Guiglion
The Journal of Physical Chemistry C 2014 Volume 118(Issue 43) pp:24833-24842
Publication Date(Web):September 30, 2014
DOI:10.1021/jp507372n
We study the thermodynamic ability of carbon nitride materials to act as water splitting photocatalysts using a computational approach that involves a combination of density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations on cluster models of both triazine- and heptazine-based structures. We first use TD-DFT to calculate the absorption spectra of the different cluster models and compare these spectra to those measured experimentally and then calculate using DFT and TD-DFT the reduction potentials of the free electron, free hole, and exciton in these models. We predict that all classes of carbon nitride structures considered should thermodynamically be able to reduce protons and oxidize water. We further provide evidence for the hypothesis that the experimental lack of overall water splitting activity for pure carbon nitride arises from the fact that water oxidation is a four-hole reaction and hence very susceptible to competition with electron–hole recombination. Finally, we propose that the recently reported overall water splitting activity of carbon nitride loaded with polypyrrole nanoparticles arises from a junction formed at the interface of both materials, which assists in keeping electrons and holes apart.
Co-reporter:Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 26) pp:11119-11127
Publication Date(Web):09 May 2013
DOI:10.1039/C3CP50800E
Excited state relaxation in zinc sulfide (ZnS) nanoparticles is studied as a model for the fate of the excited state in inorganic nanoparticles in general. A series of time-dependent density functional theory optimisations on the S1 and T1 excited states predict the existence of not merely isolated minima, as found before, but rather a connected cascade of excited state minima ending up in a conical intersection between the excited state energy surface and the ground state. The localisation of the excited state in the different minima increases down the cascade, while the barriers separating these minima, studied here for the first time for nanoparticles, are predicted to be in some cases electronic (strongly avoided crossing) in origin. The cartoon picture of excited state relaxation in inorganic nanoparticles that involves relaxation to the bottom of only one approximately harmonic well followed by photoluminescence appears for the ZnS nanoparticles studied here to be at best rather simplistic. The localisation cascade is finally found to strongly affect the excited state properties of nanoparticles and predicted to lead to the formation of defected nanoparticles after de-excitation in selected cases.
Co-reporter:Martijn A. Zwijnenburg, Ge Cheng, Tom O. McDonald, Kim. E. Jelfs, Jia-Xing Jiang, Shijie Ren, Tom Hasell, Frédéric Blanc, Andrew I. Cooper, and Dave J. Adams
Macromolecules 2013 Volume 46(Issue 19) pp:7696-7704
Publication Date(Web):September 16, 2013
DOI:10.1021/ma401311s
The photophysical properties of insoluble porous pyrene networks, which are central to their function, differ strongly from those of analogous soluble linear and branched polymers and dendrimers. This can be rationalized by the presence of strained closed rings in the networks. A combined experimental and computational approach was used to obtain atomic scale insight into the structure of amorphous conjugated microporous polymers. The optical absorption and fluorescence spectra of a series of pyrene-based materials were compared with theoretical time-dependent density functional theory predictions for model clusters. Comparison of computation and experiment sheds light on the probable structural chromophores in the various materials.
Co-reporter:Martijn A. Zwijnenburg
Nanoscale 2012 vol. 4(Issue 12) pp:3711-3717
Publication Date(Web):29 Mar 2012
DOI:10.1039/C2NR30191A
We discuss how an approach that combines global optimisation and time-dependent density functional theory (TD-DFT) calculations allows one to predict the photoluminescence (PL) signature of true nanosized semiconductor nanoparticles. As a demonstration we calculate the PL signature of both bare and water covered zinc sulfide nanoparticles and demonstrate that we can successfully reproduce their experimentally measured PL signatures. Our TD-DFT calculations suggest that the excited state, after relaxation, becomes highly localised and that the degree of localisation changes with nanoparticle size. We also show that adsorbed water molecules can strongly influence the nanoparticle's final excited state minimum and PL signature. Finally, we discuss how this approach can be used to predict the effect of doping and what the next important methodological step will be on the route to theoretically understanding processes such as photocatalysis.
Co-reporter:Martijn A. Zwijnenburg
The Journal of Physical Chemistry C 2012 Volume 116(Issue 38) pp:20191-20198
Publication Date(Web):August 29, 2012
DOI:10.1021/jp305146p
A combination of Time-Dependent Density Functional Theory (TD-DFT) and approximate coupled cluster theory (CC2) is used to elucidate the microscopic origin of the experimental observation that the absorption and fluorescence spectra of 1,3-polypyrene display a much smaller shift with chain length than other conjugated polymers. The optical absorption and fluorescence spectra of a large range of oligomers are calculated using TD-DFT and CC2 and successfully compared with available experimental data. The calculations show that the lowest singlet excitation is excitonic in nature and that this exciton becomes strongly localized upon excited state relaxation. This strong localization explains the negligible shift in fluorescence energy between the dimer/trimer and polymer, observed in experiment.
Co-reporter:Martijn A. Zwijnenburg
Nanoscale 2011 vol. 3(Issue 9) pp:3780-3787
Publication Date(Web):09 Aug 2011
DOI:10.1039/C1NR10486A
We calculate the optical absorption spectra of low-energy uncapped zinc sulfide nanostructures found by global optimisation (basin-hopping/simulated annealing) using time–dependent density functional theory (TD-DFT) and compare the results with experimental spectra. We predict that for all nanostructures studied the lowest excited state found by TD-DFT corresponds to an exciton with an exciton binding energy that is much larger than that of excitons in bulk zinc sulfide. We further show that for the more symmetrical nanostructures some of the excitons are dark and that the absorption on-sets, the energy of the lowest exciton, for the different nanostructures show no clear evidence of quantum confinement. We propose that this apparent lack of quantum confinement finds its origin in the fact that the lowest exciton is not evenly spread over the whole nanostructure but shows large contributions for specific groups of atoms. Finally, we show that the predicted optical absorption spectra fit with those reported experimentally.
Co-reporter:Martijn A. Zwijnenburg, Francesc Illas and Stefan T. Bromley
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 20) pp:9311-9317
Publication Date(Web):07 Apr 2011
DOI:10.1039/C1CP20298G
In this paper we explore the effect of water on the excited state properties of ZnS nanostructures by means of time-dependent density functional theory (TD-DFT) calculations. Using these TD-DFT calculations we show that the effect of water on the optical absorption spectra is primarily a small blue-shift and that a secondary effect is that spectroscopic features that correspond to dark excitations for the anhydrous nanostructures gain intensity and new absorption peaks are predicted to appear. The effect of adsorbed water on the localisation of excited states is to produce small shifts in the values of the excited stabilisation energies but, more importantly, it results in the formation of extra minima when compared with the case for anhydrous ZnS. Finally, the effect of water on photoluminescence (PL) energies is predicted to be small but the appearance of extra minima induced by the presence of adsorbed water is expected to lead to a splitting/broadening of the PL signal.
Co-reporter:Martijn A. Zwijnenburg and Robert G. Bell
Chemistry of Materials 2008 Volume 20(Issue 9) pp:3008
Publication Date(Web):April 17, 2008
DOI:10.1021/cm702175q
Through atomistic optimizations of recently enumerated hypothetical frameworks, we demonstrate that in contrast with the experimental observation by Brunner and Meier (Brunner, G. O.; Meier, W. M. Nature 1989, 337, 146), there is theoretically no evidence for any topological, geometric, or energetic constraint on framework density and pore size in siliceous zeolites. More specifically, we show that there are numerous very low-density and/or very large-pore siliceous materials with comparable energetics to currently synthesized materials. The experimentally observed limitations thus seem instead to stem from a far from complete experimental exploration of the siliceous zeolite structural landscape and might be related to the lack of suitably large and rigid template molecules to fill the enormous void volume. Self-assembly of organic molecules to form nanosized templates might therefore hold the key to low density and/or extra-large-pore frameworks.
Co-reporter:Martijn A. Zwijnenburg, Robert G. Bell, Furio Corà
Journal of Solid State Chemistry 2008 Volume 181(Issue 9) pp:2480-2487
Publication Date(Web):September 2008
DOI:10.1016/j.jssc.2008.06.006
The energetics, structure and physical properties of tetragonal and orthorhombic SiS2 were calculated by periodic density functional theory (DFT) calculations, using both localized orbital and projected augmented wave basis-sets. All methods applied agree upon the relative energies of the different polymorphs but show differences in the predicted geometries, which are minimized upon improving the basis-set quality. The hybrid PBE0 functional was found to give the best match between experimental and calculated structures. When comparing SiS2 with its much better studied oxide analog silica, we observe that upon substituting sulphur for oxygen, the energy landscape changes dramatically. Other effects of changing S for O are found to be smaller Si–X–Si angles, a broader distribution of X–Si–X angles, a more flexible framework and a significantly reduced band gap. The latter is in line with the experimental observation of photoluminescence in related GaGeS2 compounds and suggests that SiS2 might find application in UV light emitting diodes. Finally, a comparison of the maximally localized Wannier functions demonstrates that the Si–S bonds in SiS2 have a considerably more covalent character than the Si–O bonds in silica.Periodic DFT calculations were employed to study the (physical) properties of tetragonal and orthorhombic SiS2. The results obtained were compared with those for SiS2 better studied oxide analog silica and demonstrate large changes in the materials’ energy landscape, nature of bonding, flexibility and band gap, upon substitution of sulphur for oxygen.
Co-reporter:Martijn A. Zwijnenburg, Francesc Illas and Stefan T. Bromley
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 20) pp:NaN9317-9317
Publication Date(Web):2011/04/07
DOI:10.1039/C1CP20298G
In this paper we explore the effect of water on the excited state properties of ZnS nanostructures by means of time-dependent density functional theory (TD-DFT) calculations. Using these TD-DFT calculations we show that the effect of water on the optical absorption spectra is primarily a small blue-shift and that a secondary effect is that spectroscopic features that correspond to dark excitations for the anhydrous nanostructures gain intensity and new absorption peaks are predicted to appear. The effect of adsorbed water on the localisation of excited states is to produce small shifts in the values of the excited stabilisation energies but, more importantly, it results in the formation of extra minima when compared with the case for anhydrous ZnS. Finally, the effect of water on photoluminescence (PL) energies is predicted to be small but the appearance of extra minima induced by the presence of adsorbed water is expected to lead to a splitting/broadening of the PL signal.
Co-reporter:Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 26) pp:NaN11127-11127
Publication Date(Web):2013/05/09
DOI:10.1039/C3CP50800E
Excited state relaxation in zinc sulfide (ZnS) nanoparticles is studied as a model for the fate of the excited state in inorganic nanoparticles in general. A series of time-dependent density functional theory optimisations on the S1 and T1 excited states predict the existence of not merely isolated minima, as found before, but rather a connected cascade of excited state minima ending up in a conical intersection between the excited state energy surface and the ground state. The localisation of the excited state in the different minima increases down the cascade, while the barriers separating these minima, studied here for the first time for nanoparticles, are predicted to be in some cases electronic (strongly avoided crossing) in origin. The cartoon picture of excited state relaxation in inorganic nanoparticles that involves relaxation to the bottom of only one approximately harmonic well followed by photoluminescence appears for the ZnS nanoparticles studied here to be at best rather simplistic. The localisation cascade is finally found to strongly affect the excited state properties of nanoparticles and predicted to lead to the formation of defected nanoparticles after de-excitation in selected cases.
Co-reporter:Milena C. C. Wobbe, Andrew Kerridge and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 40) pp:NaN22061-22061
Publication Date(Web):2014/08/28
DOI:10.1039/C4CP03442B
The optical absorption spectra of magnesium oxide (MgO) nanoparticles, along with the atomic centres responsible, are studied using a combination of time-dependent density functional theory (TD-DFT) and coupled-cluster methods. We demonstrate that TD-DFT calculations on MgO nanoparticles require the use of range-separated exchange–correlation (XC-) functionals or hybrid XC-functionals with a high percentage of Hartree–Fock like exchange to circumvent problems related to the description of charge-transfer excitations. Furthermore, we show that the vertical excitations responsible for the experimentally studied range of the spectra of the MgO nanoparticles typically involve both 3-coordinated corner sites and 4-coordinated edge sites. We argue therefore that to label peaks in these absorption spectra exclusively as either corner or edge features does not provide insight into the full physical picture.
Co-reporter:Pierre Guiglion and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 27) pp:NaN17863-17863
Publication Date(Web):2015/06/12
DOI:10.1039/C5CP01916H
We use a combination of Time-Dependent Density Functional Theory (TD-DFT) and approximate Coupled Cluster Theory (RI-CC2) to compare trends in the optical gap and fluorescence energies of ortho-, meta- and para-oligomers of phenylene. We find that RI-CC2 and TD-DFT calculations using three different commonly employed XC-potentials (B3LYP, BHLYP and CAM-B3LYP) generally give consistent predictions. Most importantly, the fluorescence energy of m-phenylene is predicted to be independent of oligomer length, the fluorescence energy of p-phenylene to decrease with oligomer length and that of o-phenylene to increase. The origins of these differences in behaviour between the different isomers are analysed and found to stem from a subtle combination of steric and electronic factors.
Co-reporter:Pierre Guiglion, Cristina Butchosa and Martijn A. Zwijnenburg
Journal of Materials Chemistry A 2014 - vol. 2(Issue 30) pp:NaN12004-12004
Publication Date(Web):2014/06/11
DOI:10.1039/C4TA02044H
A computational scheme to predict the thermodynamic ability of photocatalysts to drive both of the watersplitting half reactions, proton reduction and water oxidation, is discussed, and applied to a number of polymeric systems to explain their apparent inability to oxidise water. We predict that the poly(p-phenylene) (PPP) is thermodynamically unable to oxidise water and that PPP is hence unlikely to split water in the absence of an external electrical bias. For other polymers, however, for example carbon nitride, the lack of oxygen evolution activity appears kinetic in origin and hence a suitable co-catalyst could potentially transform them into true watersplitting photocatalysts.
Co-reporter:Milena C. C. Wobbe and Martijn A. Zwijnenburg
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 43) pp:NaN28900-28900
Publication Date(Web):2015/09/25
DOI:10.1039/C5CP04851F
The nature and magnitude of the optical gaps of rocksalt alkaline earth (MgO, CaO, SrO, MgS, MgSe) and transition metal chalcogenide (CdO, PbS) nanoparticles are studied using time-dependent density functional theory (TD-DFT) calculations on (MX)32 nanoparticles. We demonstrate, just as we previously showed for MgO, that TD-DFT calculations on rocksalt nanoparticles require the use of hybrid exchange–correlation (XC-)functionals with a high percentage of Hartree–Fock like exchange (e.g. BHLYP) or range-separated XC-functionals to circumvent problems related to the description of charge-transfer excitations. Concentrating on the results obtained with TD-BHLYP we show that the optical gap in rocksalt nanoparticles displays a wide range of behavior; ranging from large optical gaps stemming from a localized excitation involving corner atoms in alkaline earth oxides to a delocalized excitation and small optical gaps in the transition metal chalcogenides. Finally, we rationalize this wide range of behaviour in terms of differences in the degree to which the Coulombic interaction between the excited electron and hole is screened in the different nanoparticles, and relate it to the optical dielectric constants of the bulk materials the nanoparticles are made from.