Co-reporter:Jakob Skjold-Jørgensen, Jesper Vind, Olga V. Moroz, Elena Blagova, Vikram K. Bhatia, Allan Svendsen, Keith S. Wilson, Morten J. Bjerrum
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2017 Volume 1865(Issue 1) pp:20-27
Publication Date(Web):January 2017
DOI:10.1016/j.bbapap.2016.09.016
•Rational design and expression of a lipase variant containing a switch controlling opening and closing of the lid.•The closed form of the lipase displayed very low enzymatic activity and interfacial binding compared to wild-type TlL.•Release of the lock on the lid by reducing agent restored enzymatic activity.•The crystal structure shows formation of a disulfide bond in the locked form which causes strained closure of the lid-domain.•The crystal structure reveal sites of interest for rational design of lipases that activate under controlled conditions.Here, we present a lipase mutant containing a biochemical switch allowing a controlled opening and closing of the lid independent of the environment. The closed form of the TlL mutant shows low binding to hydrophobic surfaces compared to the binding observed after activating the controlled switch inducing lid-opening. We directly show that lipid binding of this mutant is connected to an open lid conformation demonstrating the impact of the exposed amino acid residues and their participation in binding at the water-lipid interface. The switch was created by introducing two cysteine residues into the protein backbone at sites 86 and 255. The crystal structure of the mutant shows the successful formation of a disulfide bond between C86 and C255 which causes strained closure of the lid-domain. Control of enzymatic activity and binding was demonstrated on substrate emulsions and natural lipid layers. The locked form displayed low enzymatic activity (~ 10%) compared to wild-type. Upon release of the lock, enzymatic activity was fully restored. Only 10% binding to natural lipid substrates was observed for the locked lipase compared to wild-type, but binding was restored upon adding reducing agent. QCM-D measurements revealed a seven-fold increase in binding rate for the unlocked lipase. The TlL_locked mutant shows structural changes across the protein important for understanding the mechanism of lid-opening and closing. Our experimental results reveal sites of interest for future mutagenesis studies aimed at altering the activation mechanism of TlL and create perspectives for generating tunable lipases that activate under controlled conditions.Simplified model of the intrinsic switch function in TlL_locked. The disulfide bond (S‐S) spanning the active site pocket (between residues C86 and C255) locks the lid in a closed conformation (red pie) causing low enzymatic activity and interfacial binding levels (short arrow). Upon adding a reducing agent (TCEP), the disulfide bond breaks (light grey pie) and unlocks the lid increasing the rate of interfacial binding and enzymatic activity (large arrows) on the triglyceride substrate. Di- and triglycerides (black line; glycerol backbone, blue line; acyl chains). Produced fatty acid is shown with a negative charge (red line).
Co-reporter:Jon Agirre;Antonio Ariza;Wendy A. Offen;Johan P. Turkenburg;Shirley M. Roberts;Stuart McNicholas;Paul V. Harris;Brett McBrayer;Jan Dohnalek;Kevin D. Cowtan;Gideon J. Davies
Acta Crystallographica Section D 2016 Volume 72( Issue 2) pp:
Publication Date(Web):
DOI:10.1107/S2059798315024237
The industrial conversion of cellulosic plant biomass into useful products such as biofuels is a major societal goal. These technologies harness diverse plant degrading enzymes, classical exo- and endo-acting cellulases and, increasingly, cellulose-active lytic polysaccharide monooxygenases, to deconstruct the recalcitrant β-d-linked polysaccharide. A major drawback with this process is that the exo-acting cellobiohydrolases suffer from severe inhibition from their cellobiose product. β-d-Glucosidases are therefore important for liberating glucose from cellobiose and thereby relieving limiting product inhibition. Here, the three-dimensional structures of two industrially important family GH3 β-d-glucosidases from Aspergillus fumigatus and A. oryzae, solved by molecular replacement and refined at 1.95 Å resolution, are reported. Both enzymes, which share 78% sequence identity, display a three-domain structure with the catalytic domain at the interface, as originally shown for barley β-d-glucan exohydrolase, the first three-dimensional structure solved from glycoside hydrolase family GH3. Both enzymes show extensive N-glycosylation, with only a few external sites being truncated to a single GlcNAc molecule. Those glycans N-linked to the core of the structure are identified purely as high-mannose trees, and establish multiple hydrogen bonds between their sugar components and adjacent protein side chains. The extensive glycans pose special problems for crystallographic refinement, and new techniques and protocols were developed especially for this work. These protocols ensured that all of the d-pyranosides in the glycosylation trees were modelled in the preferred minimum-energy 4C1 chair conformation and should be of general application to refinements of other crystal structures containing O- or N-glycosylation. The Aspergillus GH3 structures, in light of other recent three-dimensional structures, provide insight into fungal β-d-glucosidases and provide a platform on which to inform and inspire new generations of variant enzymes for industrial application.
Co-reporter:Keith S. Wilson;Olga V. Moroz;Anne-K. Duhme-Klair;Johan P. Turkenburg;Daniel J. Raines;Elena V. Blagova
PNAS 2016 Volume 113 (Issue 21 ) pp:5850-5855
Publication Date(Web):2016-05-24
DOI:10.1073/pnas.1520829113
To acquire essential Fe(III), bacteria produce and secrete siderophores with high affinity and selectivity for Fe(III) to
mediate its uptake into the cell. Here, we show that the periplasmic binding protein CeuE of Campylobacter jejuni, which was previously thought to bind the Fe(III) complex of the hexadentate siderophore enterobactin (Kd ∼ 0.4 ± 0.1 µM), preferentially binds the Fe(III) complex of the tetradentate enterobactin hydrolysis product bis(2,3-dihydroxybenzoyl-l-Ser) (H5-bisDHBS) (Kd = 10.1 ± 3.8 nM). The protein selects Λ-configured [Fe(bisDHBS)]2− from a pool of diastereomeric Fe(III)-bisDHBS species that includes complexes with metal-to-ligand ratios of 1:1 and 2:3.
Cocrystal structures show that, in addition to electrostatic interactions and hydrogen bonding, [Fe(bisDHBS)]2− binds through coordination of His227 and Tyr288 to the iron center. Similar binding is observed for the Fe(III) complex of
the bidentate hydrolysis product 2,3-dihydroxybenzoyl-l-Ser, [Fe(monoDHBS)2]3−. The mutation of His227 and Tyr288 to noncoordinating residues (H227L/Y288F) resulted in a substantial loss of affinity for
[Fe(bisDHBS)]2− (Kd ∼ 0.5 ± 0.2 µM). These results suggest a previously unidentified role for CeuE within the Fe(III) uptake system of C. jejuni, provide a molecular-level understanding of the underlying binding pocket adaptations, and rationalize reports on the use
of enterobactin hydrolysis products by C. jejuni, Vibrio cholerae, and other bacteria with homologous periplasmic binding proteins.
Co-reporter:Olga V. Moroz;Michelle Maranta;Tarana Shaghasi;Paul V. Harris, ;Gideon J. Davies
Acta Crystallographica Section F 2015 Volume 71( Issue 1) pp:114-120
Publication Date(Web):
DOI:10.1107/S2053230X14027307
The enzymatic degradation of plant cell-wall cellulose is central to many industrial processes, including second-generation biofuel production. Key players in this deconstruction are the fungal cellobiohydrolases (CBHs), notably those from family GH7 of the carbohydrate-active enzymes (CAZY) database, which are generally known as CBHI enzymes. Here, three-dimensional structures are reported of the Aspergillus fumigatus CBHI Cel7A solved in uncomplexed and disaccharide-bound forms at resolutions of 1.8 and 1.5 Å, respectively. The product complex with a disaccharide in the +1 and +2 subsites adds to the growing three-dimensional insight into this family of industrially relevant biocatalysts.
Co-reporter:Jennifer Timm;Dolores González-Pacanowska
Acta Crystallographica Section F 2014 Volume 70( Issue 1) pp:34-39
Publication Date(Web):
DOI:10.1107/S2053230X13033621
Trypanosoma brucei is a single-cellular parasite of the genus Kinetoplastida and is the causative agent of African sleeping sickness in humans. Adenosine kinase is a key enzyme in the purine-salvage pathway, phosphorylating adenosine to AMP, and also activates cytotoxic analogues such as cordycepin and Ara-A by their phosphorylation. The structures of T. brucei brucei adenosine kinase (TbAK) in its unliganded open conformation and complexed with adenosine and ADP in the closed conformation are both reported to 2.6 Å resolution. The structures give insight into the binding mode of the substrates and the conformational change induced upon substrate binding. This information can be used to guide the improvement of cytotoxic substrate analogues as potential antitrypanosomal drugs.
Co-reporter:Huaqing Cui ; Juana Carrero-Lérida ; Ana P. G. Silva ; Jean L. Whittingham ; James A. Brannigan ; Luis M. Ruiz-Pérez ; Kevin D. Read ; Keith S. Wilson ; Dolores González-Pacanowska ;Ian H. Gilbert
Journal of Medicinal Chemistry 2012 Volume 55(Issue 24) pp:10948-10957
Publication Date(Web):December 14, 2012
DOI:10.1021/jm301328h
Plasmodium falciparum thymidylate kinase (PfTMPK) is a key enzyme in pyrimidine nucleotide biosynthesis. 3-Trifluoromethyl-4-chloro-phenyl-urea-α-thymidine has been reported as an inhibitor of Mycobacterium tuberculosis TMPK (MtTMPK). Starting from this point, we designed, synthesized and evaluated a number of thymidine analogues as antimalarials. Both 5′-urea-α- and β-thymidine derivatives were moderate inhibitors of PfTMPK and furthermore showed moderate inhibition of parasite growth. The structure of several enzyme–inhibitor complexes provides a basis for improved inhibitor design. However, we found that certain 5′-urea-α-thymidine analogues had antimalarial activity where inhibition of PfTMPK is not the major mode of action. Optimization of this series resulted in a compound with potent antimalarial activity (EC50 = 28 nM; CC50 = 29 μM).

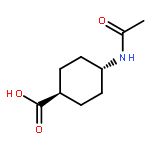
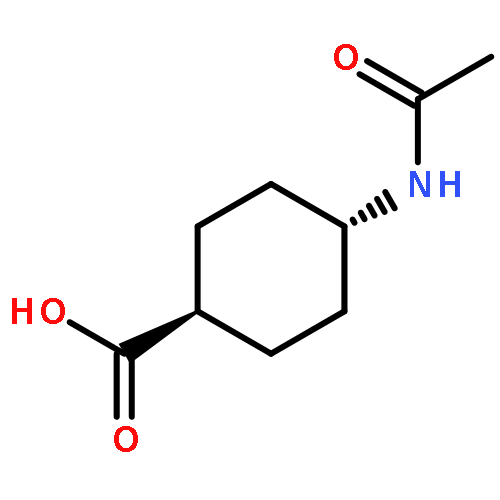

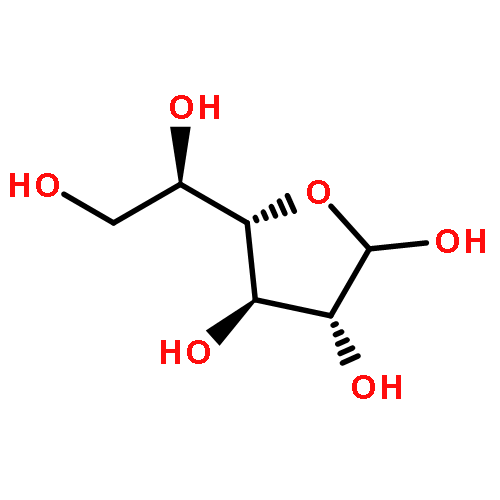


![2,5-Pyrrolidinedione, 1-[[2,3-bis(phenylmethoxy)benzoyl]oxy]-](http://img.cochemist.com/ccimg/97000/96920-81-9.png)
![2,5-Pyrrolidinedione, 1-[[2,3-bis(phenylmethoxy)benzoyl]oxy]-](http://img.cochemist.com/ccimg/97000/96920-81-9_b.png)
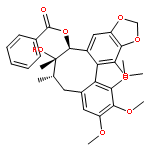
















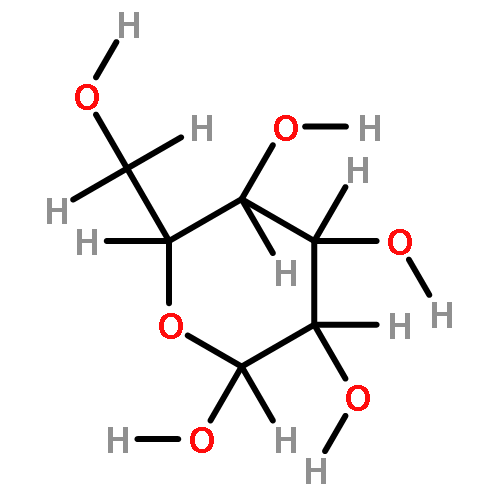
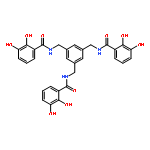
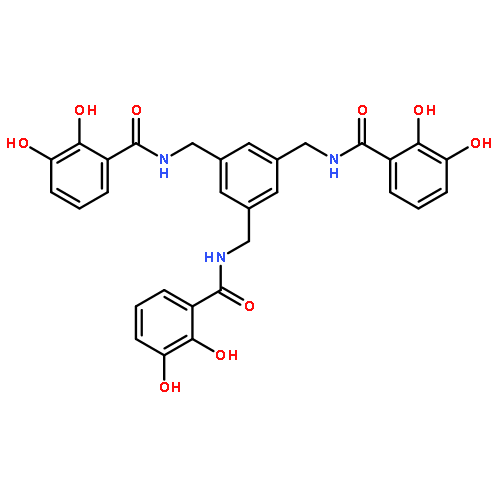


![(6S,7R)-2,3,10,11,12-pentamethoxy-6,7-dimethyl-5,6,7,8-tetrahydrodibenzo[a,c][8]annulen-1-ol](http://img.cochemist.com/ccimg/69400/69363-14-0.png)
![(6S,7R)-2,3,10,11,12-pentamethoxy-6,7-dimethyl-5,6,7,8-tetrahydrodibenzo[a,c][8]annulen-1-ol](http://img.cochemist.com/ccimg/69400/69363-14-0_b.png)








![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 3,3-dimethyl-7-oxo-6-[(phenylacetyl)amino]- (2S,5R,6R)-, 4-oxide](http://img.cochemist.com/ccimg/4100/4052-54-4.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 3,3-dimethyl-7-oxo-6-[(phenylacetyl)amino]- (2S,5R,6R)-, 4-oxide](http://img.cochemist.com/ccimg/4100/4052-54-4_b.png)








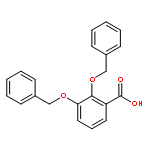
















![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)

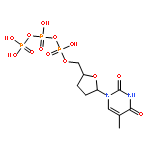

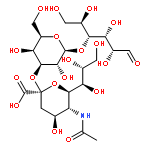
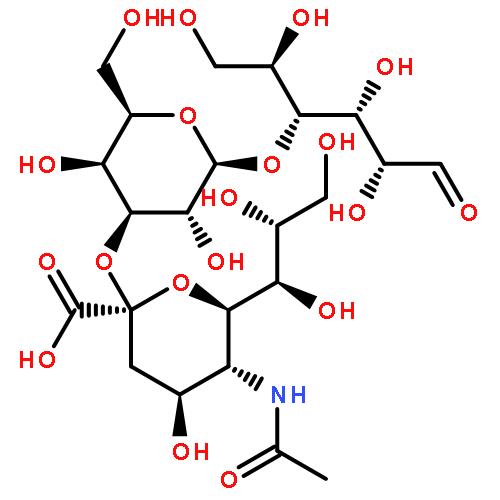


![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5_b.png)


![[(2s,3s,5r)-3-azido-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/29800/29706-85-2.png)
![[(2s,3s,5r)-3-azido-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/29800/29706-85-2_b.png)