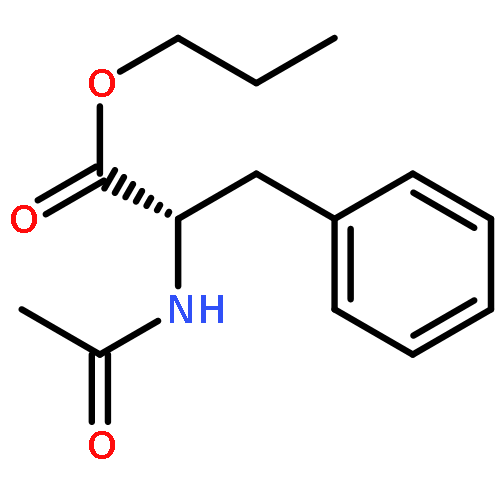
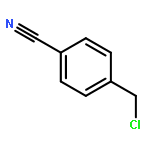
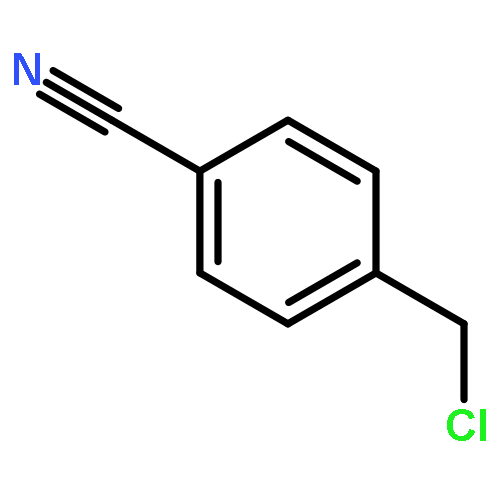
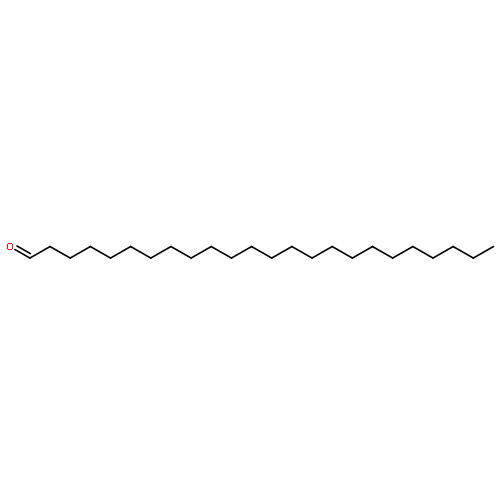
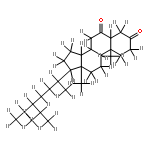
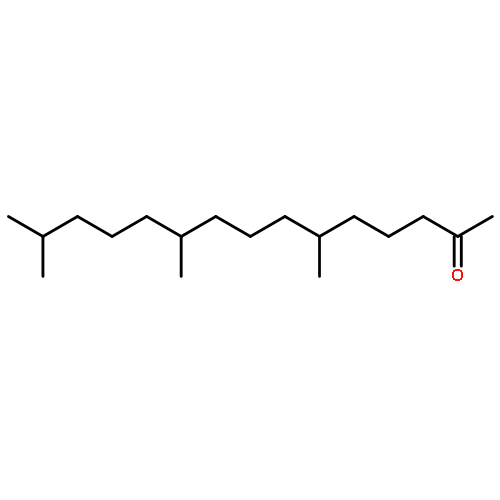
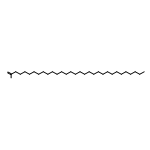
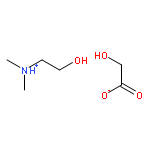
Co-reporter:Joshua E. S. J. Reid;Filipe Agapito;Carlos E. S. Bernardes;Filomena Martins;Adam J. Walker;Manuel E. Minas da Piedade
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 30) pp:19928-19936
Publication Date(Web):2017/08/02
DOI:10.1039/C7CP02230A
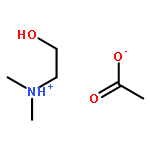
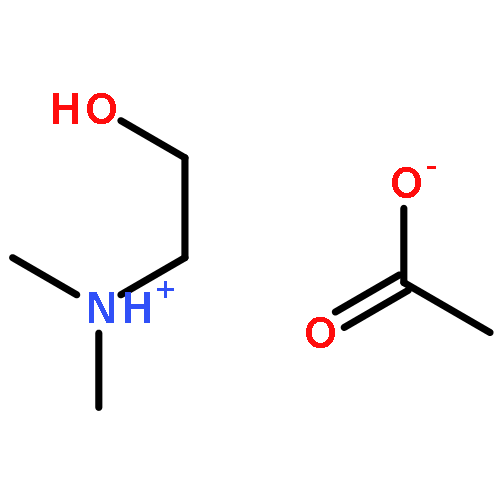
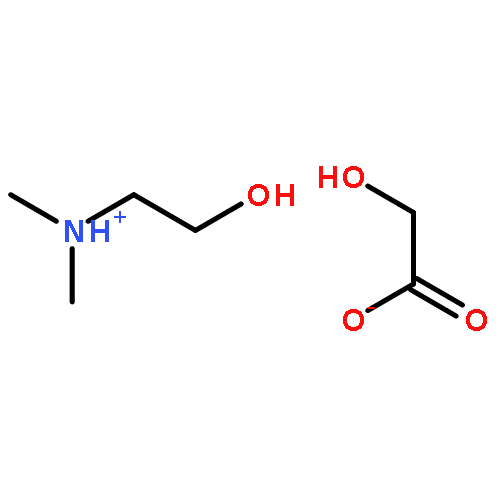
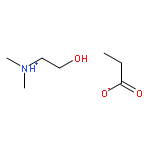
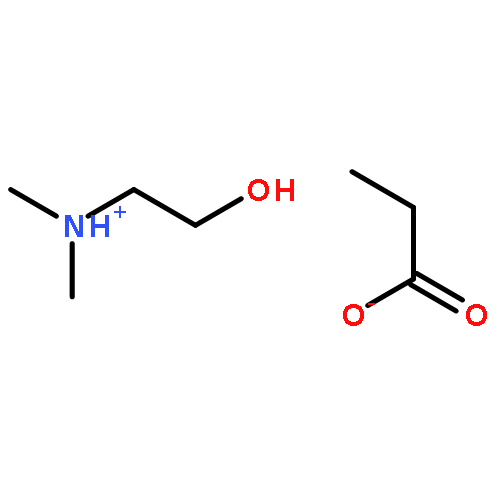
How does cation functionality influence the strength of intermolecular interactions in protic ionic liquids (PILs)? Quantifying the energetics of PILs can be an invaluable tool to answer this fundamental question. With this in view, we have determined the standard molar enthalpy of vaporization, , and the standard molar enthalpy of formation, , of three tertiary ammonium acetate PILs with varying cation functionality, and of their corresponding precursor amines, through a combination of Calvet-drop microcalorimetry, solution calorimetry, and ab initio calculations. The obtained results suggest that these PILs vaporize as their neutral acid and base precursors. We also found a strong correlation between of the PILs and of their corresponding amines. This suggests that, within this series of PILs, the influence of cation modification on their cohesive energies follows a group additivity rule. Finally, no correlation between the of PILs and the extent of proton transfer, as estimated from the difference in aqueous pKa between the precursor acid and the conjugate acid of the precursor base, was observed.
Co-reporter:Seishi Shimizu;Nobuyuki Matubayasi
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 35) pp:23597-23605
Publication Date(Web):2017/09/13
DOI:10.1039/C7CP02132A
The task of elucidating the mechanism of solubility enhancement using hydrotropes has been hampered by the wide variety of phase behaviour that hydrotropes can exhibit, encompassing near-ideal aqueous solution, self-association, micelle formation, and micro-emulsions. Instead of taking a field guide or encyclopedic approach to classify hydrotropes into different molecular classes, we take a rational approach aiming at constructing a unified theory of hydrotropy based upon the first principles of statistical thermodynamics. Achieving this aim can be facilitated by the two key concepts: (1) the Gibbs phase rule as the basis of classifying the hydrotropes in terms of the degrees of freedom and the number of variables to modulate the solvation free energy; (2) the Kirkwood–Buff integrals to quantify the interactions between the species and their relative contributions to the process of solubilization. We demonstrate that the application of the two key concepts can in principle be used to distinguish the different molecular scenarios at work under apparently similar solubility curves observed from experiments. In addition, a generalization of our previous approach to solutes beyond dilution reveals the unified mechanism of hydrotropy, driven by a strong solute–hydrotrope interaction which overcomes the apparent per-hydrotrope inefficiency due to hydrotrope self-clustering.
Co-reporter:Thomas W. J. Nicol;Noriyuki Isobe;James H. Clark
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 34) pp:23106-23112
Publication Date(Web):2017/08/30
DOI:10.1039/C7CP04647B
In the study of the cellulose dissolution mechanism opinion is still divided. Here, the solution interaction components of the most prominent hypotheses for the driving force of cellulose dissolution were evaluated quantitatively. Combining a rigorous statistical thermodynamic theory and cellobiose solubility data in the presence of chloride salts, whose cations progress in the Hofmeister series (KCl, NaCl, LiCl and ZnCl2), we have shown that cellobiose solubilization is driven by the preferential accumulation of salts around the solutes which is stronger than cellobiose hydration. Yet contrary to the classical chaotropy hypothesis, increasing salt concentration leads to cellobiose dehydration in the presence of the strongest solubilizer ZnCl2. However, thanks to cellobiose dehydration, cellobiose–salt interaction still remains preferential despite weakening salt accumulation. Based on such insights, the previous hypotheses based on hydrophobicity and polymer charging have also been evaluated quantitatively. Thus, our present study successfully paved a way towards identifying the basic driving forces for cellulose solubilization in a quantitative manner for the first time. When combined with unit additivity methods this quantitative information could lead to a full understanding of cellulose solubility.
Co-reporter:Seishi Shimizu;Nobuyuki Matubayasi
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 39) pp:26734-26742
Publication Date(Web):2017/10/11
DOI:10.1039/C7CP04990K
Solubilization or mixing in the presence of hydrotropes is often accompanied by the increase of scattering intensity. When the scattering corresponding to mesoscale structuring grows, the mixed state is called the “pre-ouzo” aggregate, which appears often and is distinct from critical density fluctuation, yet its precise mechanism of appearance is still obscure. Combining the results from the theories of scattering and thermodynamic phase stability with differential geometry, we have constructed a theory which can account for hydrotrope mixing thermodynamics and the pre-ouzo effect in a unified manner. In addition, another well-known signature of hydrotropy, the minimum hydrotrope concentration (MHC) at which solubility of sparingly soluble hydrophobic solutes suddenly increases, has also been linked to the scattering increase. The thermodynamic signatures of the pre-ouzo effect and MHC reveal a mechanistic difference between them, which manifests in the thermodynamic order of derivatives.
Co-reporter:Steven Abbott;Jonathan J. Booth
Green Chemistry (1999-Present) 2017 vol. 19(Issue 1) pp:68-75
Publication Date(Web):2017/01/03
DOI:10.1039/C6GC03002E
We all know that to enhance solubility using greener chemistry we should harness sound principles of molecular-based thermodynamics. The problem is that even for simple systems it can be hard to know how to use fundamental tools for formulation benefit, and for the more complex systems that we must often use, calculations required for molecular thermodynamics can often be quite involved. In this paper we show that a fundamental, assumption-free statistical thermodynamics approach, the Kirkwood–Buff theory, can be used in practical, complex aqueous systems to provide the insights we need to optimise formulations. The theory itself is not that difficult, but its implementation, which requires many steps of thermodynamic calculations, has up to now not been straightforward. Taking full advantage of an interactive approach, here we review what the Kirkwood–Buff theory can provide for formulators; we use the power of modern web browsers to provide open-source, user-friendly, responsive-design apps to do the hard work of data analysis, leaving formulators to focus on the interpretation of the results for their specific optimisation task. Indeed the apps are intended to be used by researchers and formulators for specific systems of interest to them.
Co-reporter:Seishi Shimizu;Steven Abbott;Nobuyuki Matubayasi
Food & Function (2010-Present) 2017 vol. 8(Issue 9) pp:2999-3009
Publication Date(Web):2017/09/20
DOI:10.1039/C7FO00313G
The ways in which flavour molecules interact with proteins in foods have an impact on flavour and aroma and on the (in)stability of the proteins. There is a long history of analysing these interactions using a “specific binding model” that gives values for the number of molecules, n, bound to the proteins with a binding constant Kb. However, recent progress in molecular thermodynamics forced us to reconsider this approach. In addition, there are a number of methods for determining these values and it is not at all clear whether the various assumptions behind the various methods allow legitimate comparisons between techniques. By adopting an assumption-free molecular thermodynamics approach, Kirkwood–Buff theory, we find that we gain a welcome universality, simplicity and deep understanding of what is happening at the molecular level. Here we look at three different methods for examining flavour–protein interactions (vapour pressure, dialysis equilibrium and melting temperature changes), show how historical data can be re-cast into the universal language of Kirkwood–Buff and provide a free, open-source app that can both re-analyze historical data and be a platform for analyzing fresh data. In each case, the fundamental theory is described along with a pragmatic implementation accepting the realities of experimentation. One key insight is that the n and Kb parameters of the classical binding models can be turned directly, via simple arithmetic, into the Kirkwood–Buff integrals that accurately capture non-specific flavour–protein interactions.
Co-reporter:Joshua E. S. J. Reid, Richard J. Gammons, John M. SlatteryAdam J. Walker, Seishi Shimizu
The Journal of Physical Chemistry B 2017 Volume 121(Issue 3) pp:
Publication Date(Web):January 13, 2017
DOI:10.1021/acs.jpcb.6b10562
The sensitivity of ionic liquids (ILs) to water affects their physical and chemical properties, even at relatively low concentrations, yet the structural thermodynamics of protic IL– (PIL−) water systems at low water concentrations still remains unclear. Using the rigorous Kirkwood–Buff theory of solutions, which can quantify the interactions between species in IL–water systems solely from thermodynamic data, we have shown the following: (1) Between analogous protic and aprotic ILs (AILs), the AIL cholinium bis(trifluoromethanesulfonyl)imide ([Ch][NTf2]) shows stronger interactions with water at low water concentrations, with the analogous PIL N,N-dimethylethanolammonium bis(trifluoromethanesulfonyl)imide ([DMEtA][NTf2]) having stronger water–ion interactions at higher water contents, despite water–ion interactions weakening with increasing water content in both systems. (2) Water has little effect on the average ion–ion interactions in both protic and aprotic ILs, aside from the AIL [Ch][NTf2], which shows a strengthening of ion–ion interactions with increasing water content. (3) Self-association of water in both PIL–water systems leading to the presence of large aggregates of water in IL-rich compositions has been inferred. Water–water interactions in [DMEtA][NTf2] were found to be similar to those of dialkylimidazolium AILs, whereas these interactions were much larger in the PIL N,N-dimethylethanolammonium propionate ([DMEtA][Pr]), attributed to the change in anion–water interactions.
Co-reporter:Seishi Shimizu, Richard Stenner, Nobuyuki Matubayasi
Food Hydrocolloids 2017 Volume 62(Volume 62) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.foodhyd.2016.07.022
•Statistical thermodynamics is shown to be useful in food science.•The rigorous Kirkwood-Buff theory is derived from scratch.•How soy protein denaturation and gelation is affected by cosolvents have been clarified.•Cosolvent's exclusion from sol and binding to gel both drive gelation.Sugars, alcohols, or salts, when added to food, affects the heat denaturation of proteins and the sol-gel transition of macromolecules. Such an effect of cosolvents has long been known and exploited; yet understanding how they work at a molecular level has been a matter of scientific debate for decades, because of the lack of a definitive theory which can provide a microscopic explanation. Here we show that a rigorous statistical thermodynamic theory, the Kirkwood-Buff (KB) theory, provides not only a long-awaited microscopic explanation but also a clear guideline on how to analyze experimental data. KB theory synthesizes the classical Wyman-Tanford formula and partial molar volume, and enables the determination of biomolecule-water and biomolecule-cosolvent interactions solely from experimental data. Nothing beyond the materials in introductory physical chemistry or chemical thermodynamics textbooks is necessary to follow the derivations presented in this review.Download high-res image (181KB)Download full-size image
Co-reporter:Seishi Shimizu, Nobuyuki Matubayasi
Biophysical Chemistry 2017 Volume 231(Volume 231) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bpc.2017.02.003
•Preferential solvation theory formulated in the presence of a semi-permeable membrane.•The membrane does not perturb thermodynamics when it is located at the osmolyte dividing surface.•Preferential solvation = membrane expansion work against the osmotic pressure (when low).•“Depletion forces” and “hydration forces” are shown to be inequivalent.How osmolytes enhance the folding, binding, and self-assembly of biological macromolecules at a microscopic scale has long been a matter of debate. Ambiguities persist on the key interpretive concepts, such as the “effective membrane” (which marks the boundary of the volume from which osmolytes are excluded) and the “free energy of exclusion” of osmolytes from biomolecular surfaces. In this paper, we formulate these elusive concepts based upon chemical thermodynamics and rigorous statistical thermodynamics (the Kirkwood-Buff theory). Positioning of the membrane at the osmotic dividing surface is crucial in order not to affect the thermodynamics of solvation. The notion of the free energy (work) of excluding osmolytes is refined to the expansion work against the osmotic pressure, which indeed describes the change of solvation free energy at dilute osmolyte concentrations.Download high-res image (222KB)Download full-size image
Co-reporter:Thomas W. J. Nicol, Nobuyuki Matubayasi and Seishi Shimizu
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 22) pp:15205-15217
Publication Date(Web):20 May 2016
DOI:10.1039/C6CP01582D
The low solubility of drugs, which poses a serious problem in drug development, can in part be overcome by the use of cyclodextrins (CDs) and their derivatives. Here, the key to solubilisation is identified as the formation of inclusion complexes with the drug molecule. If inclusion complexation were the only contribution to drug solubility, it would increase linearly with CD concentration (as per the Higuchi–Connors model); this is because inclusion complexation is a 1:1 stoichiometric process. However, solubility curves often deviate from this linearity, whose mechanism is yet to be understood. Here we aim to clarify the origin of such non-linearity, based on the Kirkwood–Buff and the McMillan–Mayer theories of solutions. The rigorous statistical thermodynamic theory shows that non-linearity of solubilisation can be rationalised by two contributions: CD–drug interaction and the drug-induced change of CD–CD interaction.
Co-reporter:Seishi Shimizu and Steven Abbott
The Journal of Physical Chemistry B 2016 Volume 120(Issue 15) pp:3713-3723
Publication Date(Web):April 11, 2016
DOI:10.1021/acs.jpcb.6b01380
Supercritical carbon dioxide (scCO2) on its own can be a relatively poor solvent. However, the addition at relatively modest concentration of “entrainers”, simple solvent molecules such as ethanol or acetone, can provide a significant boost in solubility, thereby enabling its industrial use. However, how entrainers work is still under debate; without an unambiguous explanation, it is hard to optimize entrainers for any specific solute. This paper demonstrates that a fundamental, assumption-free statistical thermodynamic theory, the Kirkwood–Buff (KB) theory, can provide an unambiguous explanation of the entrainer effect through an analysis of published experimental data. The KB theory shows that a strong solute–entrainer interaction accounts for the solubility enhancement, while CO2 density increase and/or CO2–entrainer interactions, which have been assumed widely in the literature, do not account for solubilization. This conclusion, despite the limited completeness of available data, is demonstrably robust; this can be shown by an order-of-magnitude analysis based upon the theory, and can be demonstrated directly through a public-domain “app”, which has been developed to implement the theory.
Co-reporter:Jonathan J. Booth, Muhiadin Omar, Steven Abbott and Seishi Shimizu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 12) pp:8028-8037
Publication Date(Web):27 Feb 2015
DOI:10.1039/C4CP05414H
Nicotinamide is an effective non-micellar hydrotrope (solubilizer) for drugs with low aqueous solubility. To clarify the molecular basis of nicotinamide’s hydrotropic effectiveness, we present here a rigorous statistical thermodynamic theory, based on the Kirkwood–Buff theory of solutions, and our recent application of it to hydrotropy. We have shown that (i) nicotinamide self-association reduces solubilization efficiency, contrary to the previous hypothesis which claimed that self-association drives solubilization and (ii) the minimum hydrotrope concentration (MHC), namely, the threshold concentration above which solubility suddenly increases, is caused not by the bulk-phase self-association of nicotinamides as has been postulated previously, but by the enhancement of nicotinamide–nicotinamide interaction around the drug molecules. We have thus established a new view of hydrotropy – it is nicotinamide’s non-stoichiometric accumulation around the drug that is the basis of solubility increase above MHC.
Co-reporter:Joshua E. S. J. Reid, Adam J. Walker and Seishi Shimizu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 22) pp:14710-14718
Publication Date(Web):14 May 2015
DOI:10.1039/C5CP01854D
How do residual water molecules in ionic liquids (ILs) interact with themselves, as well as with the ions? This question is crucial in understanding why the physical properties of ILs – and chemical reactions performed in them – are strongly affected by the residual water content. There have been three conflicting hypotheses regarding the structure and behaviour of the residual water: (i) water molecules are separated from one another, while interacting strongly with the ions, and dispersed throughout the medium; (ii) water molecules self-associate or form clusters in the ILs; (iii) residual water weakens ion–ion interactions. A satisfactory resolution of these conflicting suggestions has been hindered by the complexity and long range of the interactions in the water–IL mixture and by the often profound differences in physical structure between various different ILs. Here we present a route to resolve this question through a combination of a statistical thermodynamic theory (Kirkwood–Buff theory) with density and osmotic data from the literature. The structure of water–IL mixtures is shown to be water content dependent; at the lowest measured water concentration, strong water–IL interaction and water–water separation are observed in accordance to (i), whereas water in a more hydrophobic IL environment seems to self-associate at moderately low water concentrations, in accordance with (ii).
Co-reporter:Seishi Shimizu and Nobuyuki Matubayasi
The Journal of Physical Chemistry B 2014 Volume 118(Issue 14) pp:3922-3930
Publication Date(Web):April 1, 2014
DOI:10.1021/jp410567c
How do osmolytes affect the conformation and configuration of supramolecular assembly, such as ion channel opening and actin polymerization? The key to the answer lies in the excess solvation numbers of water and osmolyte molecules; these numbers are determinable solely from experimental data, as guaranteed by the phase rule, as we show through the exact solution theory of Kirkwood and Buff (KB). The osmotic stress technique (OST), in contrast, purposes to yield alternative hydration numbers through the use of the dividing surface borrowed from the adsorption theory. However, we show (i) OST is equivalent, when it becomes exact, to the crowding effect in which the osmolyte exclusion dominates over hydration; (ii) crowding is not the universal driving force of the osmolyte effect (e.g., actin polymerization); (iii) the dividing surface for solvation is useful only for crowding, unlike in the adsorption theory which necessitates its use due to the phase rule. KB thus clarifies the true meaning and limitations of the older perspectives on preferential solvation (such as solvent binding models, crowding, and OST), and enables excess number determination without any further assumptions.
Co-reporter:Seishi Shimizu and Nobuyuki Matubayasi
The Journal of Physical Chemistry B 2014 Volume 118(Issue 35) pp:10515-10524
Publication Date(Web):August 21, 2014
DOI:10.1021/jp505869m
Drug molecules with low aqueous solubility can be solubilized by a class of cosolvents, known as hydrotropes. Their action has often been explained by an analogy with micelle formation, which exhibits critical micelle concentration (CMC). Indeed, hydrotropes also exhibit “minimum hydrotrope concentration” (MHC), a threshold concentration for solubilization. However, MHC is observed even for nonaggregating monomeric hydrotropes (such as urea); this raises questions over the validity of this analogy. Here we clarify the effect of micellization on hydrotropy, as well as the origin of MHC when micellization is not accompanied. On the basis of the rigorous Kirkwood-Buff (KB) theory of solutions, we show that (i) micellar hydrotropy is explained also from preferential drug–hydrotrope interaction; (ii) yet micelle formation reduces solubilization effeciency per hydrotrope molecule; (iii) MHC is caused by hydrotrope–hydrotrope self-association induced by the solute (drug) molecule; and (iv) MHC is prevented by hydrotrope self-aggregation in the bulk solution. We thus need a departure from the traditional view; the structure of hydrotrope-water mixture around the drug molecule, not the structure of the aqueous hydrotrope solutions in the bulk phase, is the true key toward understanding the origin of MHC.
Co-reporter:Seishi Shimizu and Nobuyuki Matubayasi
The Journal of Physical Chemistry B 2014 Volume 118(Issue 46) pp:13210-13216
Publication Date(Web):November 6, 2014
DOI:10.1021/jp509099h
Gelation is enhanced by the addition of sugars and polyols. How, at a microscopic level, do such cosolvents enhance gelation? The following two different hypotheses have been proposed so far to answer this question: (i) enhancement of water structure around the biopolymer induced by cosolvents; (ii) exclusion of cosolvents from biopolymer surfaces. To examine the validity of the above hypotheses, as well as to quantify the driving forces of cosolvent-induced gelation, we have constructed a statistical thermodynamic theory of gelation, by extending our Kirkwood–Buff theory of cosolvency; biopolymer–water and biopolymer–cosolvent interactions can both be determined from thermodynamic data. The exclusion of cosolvents is shown to be the dominant contribution, whereas the hydration change is a minor contribution, which may be important only so far as to mediate the exclusion of cosolvents.
Co-reporter:Seishi Shimizu, Jonathan J. Booth and Steven Abbott
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 47) pp:20625-20632
Publication Date(Web):04 Nov 2013
DOI:10.1039/C3CP53791A
Hydrophobic drugs can often be solubilized by the addition of hydrotropes. We have previously shown that preferential drug–hydrotrope association is one of the major factors of increased solubility (but not “hydrotrope clustering” or changes in “water structure”). How, then, can we understand this drug–hydrotrope interaction at a molecular level? Thermodynamic models based upon stoichiometric solute–water and solute–hydrotrope binding have long been used to understand solubilization microscopically. Such binding models have shown that the solvation numbers or coordination numbers of the water and hydrotrope molecules around the drug solute is the key quantity for solute–water and solute–hydrotrope interaction. However, we show that a rigorous statistical thermodynamic theory (the fluctuation solution theory originated by Kirkwood and Buff) requires the total reconsideration of such a paradigm. Here we show that (i) the excess solvation number (the net increase or decrease, relative to the bulk, of the solvent molecules around the solute), not the coordination number, is the key quantity for describing the solute–hydrotrope interaction; (ii) solute–hydrotrope binding is beyond the reach of the stoichiometric models because long-range solvation structure plays an important role.
Co-reporter:Seishi Shimizu
Chemical Physics Letters 2013 Volume 582() pp:129-133
Publication Date(Web):4 September 2013
DOI:10.1016/j.cplett.2013.07.009
•The first exact statistical thermodynamic study of sucrose–water mixture.•Sucrose hydration and self-association both drive water activity changes.•Water activity coefficient minimum explained from basic interactions.What is the structure of sucrose–water mixture? I employ an exact statistical thermodynamic theory (Kirkwood–Buff theory) to calculate information regarding sucrose–water, water–water and sucrose–sucrose interactions solely from volumetric and osmometric data. We found that (i) Long-ranged hydration structure, beyond the first hydration shell, influences thermodynamics; (ii) The inferred minimum of the activity coefficient of water at the high sucrose concentration is due to the increases in the self association of water. These findings from a rigorous theory are consistent with previous simulation studies.
Co-reporter:Jonathan J. Booth, Steven Abbott, and Seishi Shimizu
The Journal of Physical Chemistry B 2012 Volume 116(Issue 51) pp:14915-14921
Publication Date(Web):December 13, 2012
DOI:10.1021/jp309819r
Drugs that are poorly soluble in water can be solubilized by the addition of hydrotropes. Albeit known for almost a century, how they work at a molecular basis is still controversial due to the lack of a rigorous theoretical basis. To clear up this situation, a combination of experimental data and Fluctuation Theory of Solutions (FTS) has been employed; information on the interactions between all the molecular species present in the solution has been evaluated directly. FTS has identified two major factors of hydrotrope-induced solubilization: preferential hydrotrope–solute interaction and water activity depression. The former is dominated by hydrotrope–solute association, and the latter is enhanced by ionic dissociation and hindered by the self-aggregation of the hydrotropes. Moreover, in stark contrast to previous hypotheses, neither the change of solute hydration nor the water structure accounts for hydrotropy. Indeed, the rigorous FTS poses serious doubts over the other common hypothesis: self-aggregation of the hydrotrope hinders, rather than promotes, solubilization.
Co-reporter:Thomas W. J. Nicol, Nobuyuki Matubayasi and Seishi Shimizu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 22) pp:NaN15217-15217
Publication Date(Web):2016/05/20
DOI:10.1039/C6CP01582D
The low solubility of drugs, which poses a serious problem in drug development, can in part be overcome by the use of cyclodextrins (CDs) and their derivatives. Here, the key to solubilisation is identified as the formation of inclusion complexes with the drug molecule. If inclusion complexation were the only contribution to drug solubility, it would increase linearly with CD concentration (as per the Higuchi–Connors model); this is because inclusion complexation is a 1:1 stoichiometric process. However, solubility curves often deviate from this linearity, whose mechanism is yet to be understood. Here we aim to clarify the origin of such non-linearity, based on the Kirkwood–Buff and the McMillan–Mayer theories of solutions. The rigorous statistical thermodynamic theory shows that non-linearity of solubilisation can be rationalised by two contributions: CD–drug interaction and the drug-induced change of CD–CD interaction.
Co-reporter:Joshua E. S. J. Reid, Adam J. Walker and Seishi Shimizu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 22) pp:NaN14718-14718
Publication Date(Web):2015/05/14
DOI:10.1039/C5CP01854D
How do residual water molecules in ionic liquids (ILs) interact with themselves, as well as with the ions? This question is crucial in understanding why the physical properties of ILs – and chemical reactions performed in them – are strongly affected by the residual water content. There have been three conflicting hypotheses regarding the structure and behaviour of the residual water: (i) water molecules are separated from one another, while interacting strongly with the ions, and dispersed throughout the medium; (ii) water molecules self-associate or form clusters in the ILs; (iii) residual water weakens ion–ion interactions. A satisfactory resolution of these conflicting suggestions has been hindered by the complexity and long range of the interactions in the water–IL mixture and by the often profound differences in physical structure between various different ILs. Here we present a route to resolve this question through a combination of a statistical thermodynamic theory (Kirkwood–Buff theory) with density and osmotic data from the literature. The structure of water–IL mixtures is shown to be water content dependent; at the lowest measured water concentration, strong water–IL interaction and water–water separation are observed in accordance to (i), whereas water in a more hydrophobic IL environment seems to self-associate at moderately low water concentrations, in accordance with (ii).
Co-reporter:Jonathan J. Booth, Muhiadin Omar, Steven Abbott and Seishi Shimizu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 12) pp:NaN8037-8037
Publication Date(Web):2015/02/27
DOI:10.1039/C4CP05414H
Nicotinamide is an effective non-micellar hydrotrope (solubilizer) for drugs with low aqueous solubility. To clarify the molecular basis of nicotinamide’s hydrotropic effectiveness, we present here a rigorous statistical thermodynamic theory, based on the Kirkwood–Buff theory of solutions, and our recent application of it to hydrotropy. We have shown that (i) nicotinamide self-association reduces solubilization efficiency, contrary to the previous hypothesis which claimed that self-association drives solubilization and (ii) the minimum hydrotrope concentration (MHC), namely, the threshold concentration above which solubility suddenly increases, is caused not by the bulk-phase self-association of nicotinamides as has been postulated previously, but by the enhancement of nicotinamide–nicotinamide interaction around the drug molecules. We have thus established a new view of hydrotropy – it is nicotinamide’s non-stoichiometric accumulation around the drug that is the basis of solubility increase above MHC.
Co-reporter:Seishi Shimizu, Jonathan J. Booth and Steven Abbott
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 47) pp:NaN20632-20632
Publication Date(Web):2013/11/04
DOI:10.1039/C3CP53791A
Hydrophobic drugs can often be solubilized by the addition of hydrotropes. We have previously shown that preferential drug–hydrotrope association is one of the major factors of increased solubility (but not “hydrotrope clustering” or changes in “water structure”). How, then, can we understand this drug–hydrotrope interaction at a molecular level? Thermodynamic models based upon stoichiometric solute–water and solute–hydrotrope binding have long been used to understand solubilization microscopically. Such binding models have shown that the solvation numbers or coordination numbers of the water and hydrotrope molecules around the drug solute is the key quantity for solute–water and solute–hydrotrope interaction. However, we show that a rigorous statistical thermodynamic theory (the fluctuation solution theory originated by Kirkwood and Buff) requires the total reconsideration of such a paradigm. Here we show that (i) the excess solvation number (the net increase or decrease, relative to the bulk, of the solvent molecules around the solute), not the coordination number, is the key quantity for describing the solute–hydrotrope interaction; (ii) solute–hydrotrope binding is beyond the reach of the stoichiometric models because long-range solvation structure plays an important role.
Co-reporter:Joshua E. S. J. Reid, Filipe Agapito, Carlos E. S. Bernardes, Filomena Martins, Adam J. Walker, Seishi Shimizu and Manuel E. Minas da Piedade
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 30) pp:NaN19936-19936
Publication Date(Web):2017/07/10
DOI:10.1039/C7CP02230A
How does cation functionality influence the strength of intermolecular interactions in protic ionic liquids (PILs)? Quantifying the energetics of PILs can be an invaluable tool to answer this fundamental question. With this in view, we have determined the standard molar enthalpy of vaporization, , and the standard molar enthalpy of formation, , of three tertiary ammonium acetate PILs with varying cation functionality, and of their corresponding precursor amines, through a combination of Calvet-drop microcalorimetry, solution calorimetry, and ab initio calculations. The obtained results suggest that these PILs vaporize as their neutral acid and base precursors. We also found a strong correlation between of the PILs and of their corresponding amines. This suggests that, within this series of PILs, the influence of cation modification on their cohesive energies follows a group additivity rule. Finally, no correlation between the of PILs and the extent of proton transfer, as estimated from the difference in aqueous pKa between the precursor acid and the conjugate acid of the precursor base, was observed.
Co-reporter:Seishi Shimizu and Nobuyuki Matubayasi
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 36) pp:NaN25628-25628
Publication Date(Web):2016/09/05
DOI:10.1039/C6CP04823D
The signature of hydrotropic solubilisation is the sigmoidal solubility curve; when plotted against hydrotrope concentration, solubility increases suddenly after the minimum hydrotrope concentration (MHC), and reaches a plateau at higher hydrotrope concentrations. This sigmoidal curve is characteristic of cooperative phenomena, yet the true molecular basis of hydrotropic cooperativity has long remained unclear. Here we develop a theory, derived from the first principles of statistical thermodynamics using partially-open ensembles, to identify the origin of hydrophobic cooperativity. Our theory bears a close resemblance to the cooperative binding model used for protein–ligand binding. The cause of cooperativity is the enhancement of the hydrotrope m-body interaction induced by the presence of the solute; m can be estimated from the experimental solubility data.