Co-reporter:Alex Panaccione, Yi Zhang, Molly Ryan, Christopher A. Moskaluk, Karen S. Anderson, Wendell G. Yarbrough, Sergey V. Ivanov
Stem Cell Research 2017 Volume 21(Volume 21) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.scr.2017.05.002
•ACC cancer stem cell cultures are authenticated using ACC-intrinsic MYB fusions.•A novel MYB fusion, with a long non-coding RNA from 6q23.4, is identified.•ACC-CSC is marked by expression of CD24 and CD44.•ACC-CSC purification is optimized to establish cell lines for drug screening.•CyTOF is used to explore STAT3 and β-catenin as novel targets for ACC-CSC eradication.Cancer stem cells (CSC) are considered the major cause of aggressive tumor behavior, recurrence, metastases, and resistance to radiation, making them an attractive therapeutic target. However, isolation of CSC from tumor tissue and their characterization are challenging due to uncertainty about their molecular markers and conditions for their propagation. Adenoid cystic carcinoma (ACC), which arises predominantly in the salivary glands, is a slow-growing but relentless tumor that frequently invades nerves and metastasizes. New effective treatment approaches for ACC have not emerged over the last 40 years. Previously, based on a highly conserved SOX10 gene signature that we identified in the majority of ACC tumors, we suggested the existence in ACC of SOX10+ cells with neural stem properties and corroborated this hypothesis via isolation from ACC tissue a novel population of CSC, termed ACC-CSC. These cells activated NOTCH1 signaling and co-expressed SOX10 and other ACC-intrinsic neural crest stem cell markers with CD133, a CSC cell surface marker, suggesting that ACC is driven by a previously uncharacterized population of SOX10+/CD133+ cells with neural stem cell properties. Here, we authenticated ACC identity of our primary cultures by demonstrating that most of them harbor MYB-NFIB fusions, which are found in 86% of ACC. We demonstrated using CyTOF, a novel mass cytometry technology, that these cells express high β-catenin and STAT3 levels and are marked by CD24 and CD44. Finally, to streamline development of ACC cell lines, we developed RT-PCR tests for distinguishing mouse and human cells and used immunomagnetic cell sorting to eliminate mouse cells from long-term cell cultures. Overall, this study describes a new population of CSC that activates signaling pathways associated with poor prognosis, validates their ACC identity, and optimizes approaches that can be used for purification of ACC-CSC and generation of cell lines.
Co-reporter:Won-Gil Lee, Albert H. Chan, Krasimir A. Spasov, Karen S. Anderson, and William L. Jorgensen
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 12) pp:
Publication Date(Web):October 31, 2016
DOI:10.1021/acsmedchemlett.6b00390
Catechol diethers that incorporate a 7-cyano-2-naphthyl substituent are reported as non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs). Many of the compounds have 1–10 nM potencies toward wild-type HIV-1. An interesting conformational effect allows two unique conformers for the naphthyl group in complexes with HIV-RT. X-ray crystal structures for 4a and 4f illustrate the alternatives.Keywords: Anti-HIV agents; NNRTIs; protein crystallography;
Co-reporter:Kathleen M. Frey; David E. Puleo; Krasimir A. Spasov; Mariella Bollini; William L. Jorgensen
Journal of Medicinal Chemistry 2015 Volume 58(Issue 6) pp:2737-2745
Publication Date(Web):February 20, 2015
DOI:10.1021/jm501908a
The development of novel non-nucleoside inhibitors (NNRTIs) with activity against variants of HIV reverse transcriptase (RT) is crucial for overcoming treatment failure. The NNRTIs bind in an allosteric pocket in RT ∼10 Å away from the active site. Earlier analogues of the catechol diether compound series have picomolar activity against HIV strains with wild-type RT but lose potency against variants with single Y181C and double K103N/Y181C mutations. As guided by structure-based and computational studies, removal of the 5-Cl substitution of compound 1 on the catechol aryl ring system led to a new analogue compound 2 that maintains greater potency against Y181C and K103N/Y181C variants and better solubility (510 μg/mL). Crystal structures were determined for wild-type, Y181C, and K103N/Y181C RT in complex with both compounds 1 and 2 to understand the structural basis for these findings. Comparison of the structures reveals that the Y181C mutation destabilizes the binding mode of compound 1 and disrupts the interactions with residues in the pocket. Compound 2 maintains the same conformation in wild-type and mutant structures, in addition to several interactions with the NNRTI binding pocket. Comparison of the six crystal structures will assist in the understanding of compound binding modes and future optimization of the catechol diether series.
Co-reporter:Christal D. Sohl, Molly R. Ryan, BeiBei Luo, Kathleen M. Frey, and Karen S. Anderson
ACS Chemical Biology 2015 Volume 10(Issue 5) pp:1319
Publication Date(Web):February 16, 2015
DOI:10.1021/acschembio.5b00014
Human fibroblast growth factor receptors (FGFRs) 1–4 are a family of receptor tyrosine kinases that can serve as drivers of tumorigenesis. In particular, FGFR1 gene amplification has been implicated in squamous cell lung and breast cancers. Tyrosine kinase inhibitors (TKIs) targeting FGFR1, including AZD4547 and E3810 (Lucitanib), are currently in early phase clinical trials. Unfortunately, drug resistance limits the long-term success of TKIs, with mutations at the “gatekeeper” residue leading to tumor progression. Here we show the first structural and kinetic characterization of the FGFR1 gatekeeper mutation, V561M FGFR1. The V561M mutation confers a 38-fold increase in autophosphorylation achieved at least in part by a network of interacting residues forming a hydrophobic spine to stabilize the active conformation. Moreover, kinetic assays established that the V561M mutation confers significant resistance to E3810, while retaining affinity for AZD4547. Structural analyses of these TKIs with wild type (WT) and gatekeeper mutant forms of FGFR1 offer clues to developing inhibitors that maintain potency against gatekeeper mutations. We show that AZD4547 affinity is preserved by V561M FGFR1 due to a flexible linker that allows multiple inhibitor binding modes. This is the first example of a TKI binding in distinct conformations to WT and gatekeeper mutant forms of FGFR, highlighting adaptable regions in both the inhibitor and binding pocket crucial for drug design. Exploiting inhibitor flexibility to overcome drug resistance has been a successful strategy for combatting diseases such as AIDS and may be an important approach for designing inhibitors effective against kinase gatekeeper mutations.
Co-reporter:William T. Gray, Kathleen M. Frey, Sarah B. Laskey, Andrea C. Mislak, Krasimir A. Spasov, Won-Gil Lee, Mariela Bollini, Robert F. Siliciano, William L. Jorgensen, and Karen S. Anderson
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 10) pp:1075
Publication Date(Web):August 31, 2015
DOI:10.1021/acsmedchemlett.5b00254
Catechol diether compounds have nanomolar antiviral and enzymatic activity against HIV with reverse transcriptase (RT) variants containing K101P, a mutation that confers high-level resistance to FDA-approved non-nucleoside inhibitors efavirenz and rilpivirine. Kinetic data suggests that RT (K101P) variants are as catalytically fit as wild-type and thus can potentially increase in the viral population as more antiviral regimens include efavirenz or rilpivirine. Comparison of wild-type structures and a new crystal structure of RT (K101P) in complex with a leading compound confirms that the K101P mutation is not a liability for the catechol diethers while suggesting that key interactions are lost with efavirenz and rilpivirine.Keywords: HIV; mutations; non-nucleoside reverse transcriptase inhibitors; resistance; reverse transcriptase
Co-reporter:Won-Gil Lee, Kathleen M. Frey, Ricardo Gallardo-Macias, Krasimir A. Spasov, Albert H. Chan, Karen S. Anderson, William L. Jorgensen
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4824-4827
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.06.074
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (HIV-RT) are reported that incorporate a 7-indolizinylamino or 2-naphthylamino substituent on a pyrimidine or 1,3,5-triazine core. The most potent compounds show below 10 nanomolar activity towards wild-type HIV-1 and variants bearing Tyr181Cys and Lys103Asn/Tyr181Cys resistance mutations. The compounds also feature good aqueous solubility. Crystal structures for two complexes enhance the analysis of the structure–activity data.
Co-reporter:Anindita Mukerjee, Pinar Iyidogan, Alejandro Castellanos-Gonzalez, José A. Cisneros, Daniel Czyzyk, Amalendu Prakash Ranjan, William L. Jorgensen, A. Clinton White Jr., Jamboor K. Vishwanatha, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 10) pp:2065-2067
Publication Date(Web):15 May 2015
DOI:10.1016/j.bmcl.2015.03.091
Cryptosporidiosis, a gastrointestinal disease caused by protozoans of the genus Cryptosporidium, is a common cause of diarrheal diseases and often fatal in immunocompromised individuals. Bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) from Cryptosporidium hominis (C. hominis) has been a molecular target for inhibitor design. C. hominis TS-DHFR inhibitors with nM potency at a biochemical level have been developed however drug delivery to achieve comparable antiparasitic activity in Cryptosporidium infected cell culture has been a major hurdle for designing effective therapies. Previous mechanistic and structural studies have identified compound 906 as a nM C. hominis TS-DHFR inhibitor in vitro, having μM antiparasitic activity in cell culture. In this work, proof of concept studies are presented using a nanotherapy approach to improve drug delivery and the antiparasitic activity of 906 in cell culture. We utilized PLGA nanoparticles that were loaded with 906 (NP-906) and conjugated with antibodies to the Cryptosporidium specific protein, CP2, on the nanoparticle surface in order to specifically target the parasite. Our results indicate that CP2 labeled NP-906 (CP2-NP-906) reduces the level of parasites by 200-fold in cell culture, while NP-906 resulted in 4.4-fold decrease. Moreover, the anticryptosporidial potency of 906 improved 15 to 78-fold confirming the utility of the antibody conjugated nanoparticles as an effective drug delivery strategy.
Co-reporter:Y. Whitney Yin;Michal R. Szymanski;Christal D. Sohl;Andrea C. Mislak;Sheida Amiralaei;Christie K. Shumate;Raymond F. Schinazi
PNAS 2015 Volume 112 (Issue 28 ) pp:8596-8601
Publication Date(Web):2015-07-14
DOI:10.1073/pnas.1421733112
Nucleoside analog reverse transcriptase inhibitors (NRTIs) are the essential components of highly active antiretroviral (HAART)
therapy targeting HIV reverse transcriptase (RT). NRTI triphosphates (NRTI-TP), the biologically active forms, act as chain
terminators of viral DNA synthesis. Unfortunately, NRTIs also inhibit human mitochondrial DNA polymerase (Pol γ), causing
unwanted mitochondrial toxicity. Understanding the structural and mechanistic differences between Pol γ and RT in response
to NRTIs will provide invaluable insight to aid in designing more effective drugs with lower toxicity. The NRTIs emtricitabine
[(-)-2,3′-dideoxy-5-fluoro-3′-thiacytidine, (-)-FTC] and lamivudine, [(-)-2,3′-dideoxy-3′-thiacytidine, (-)-3TC] are both
potent RT inhibitors, but Pol γ discriminates against (-)-FTC-TP by two orders of magnitude better than (-)-3TC-TP. Furthermore,
although (-)-FTC-TP is only slightly more potent against HIV RT than its enantiomer (+)-FTC-TP, it is discriminated by human
Pol γ four orders of magnitude more efficiently than (+)-FTC-TP. As a result, (-)-FTC is a much less toxic NRTI. Here, we
present the structural and kinetic basis for this striking difference by identifying the discriminator residues of drug selectivity
in both viral and human enzymes responsible for substrate selection and inhibitor specificity. For the first time, to our
knowledge, this work illuminates the mechanism of (-)-FTC-TP differential selectivity and provides a structural scaffold for
development of novel NRTIs with lower toxicity.
Co-reporter:Kathleen M. Frey;William T. Gray;Krasimir A. Spasov;Mariela Bollini;Ricardo Gallardo-Macias;William L. Jorgensen
Chemical Biology & Drug Design 2014 Volume 83( Issue 5) pp:541-549
Publication Date(Web):
DOI:10.1111/cbdd.12266
Using a computationally driven approach, a class of inhibitors with picomolar potency known as the catechol diethers were developed targeting the non-nucleoside-binding pocket of HIV-1 reverse transcriptase. Computational studies suggested that halogen-bonding interactions between the C5 substituent of the inhibitor and backbone carbonyl of conserved residue Pro95 might be important. While the recently reported crystal structures of the reverse transcriptase complexes confirmed the interactions with the non-nucleoside-binding pocket, they revealed the lack of a halogen-bonding interaction with Pro95. To understand the effects of substituents at the C5 position, we determined additional crystal structures with 5-Br and 5-H derivatives. Using comparative structural analysis, we identified several conformations of the ethoxy uracil dependent on the strength of a van der Waals interaction with the Cγ of Pro95 and the C5 substitution. The 5-Cl and 5-F derivatives position the ethoxy uracil to make more hydrogen bonds, whereas the larger 5-Br and smaller 5-H position the ethoxy uracil to make fewer hydrogen bonds. EC50 values correlate with the trends observed in the crystal structures. The influence of C5 substitutions on the ethoxy uracil conformation may have strategic value, as future derivatives can possibly be modulated to gain additional hydrogen-bonding interactions with resistant variants of reverse transcriptase.
Co-reporter:Won-Gil Lee, Kathleen M. Frey, Ricardo Gallardo-Macias, Krasimir A. Spasov, Mariela Bollini, Karen S. Anderson, and William L. Jorgensen
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 11) pp:1259
Publication Date(Web):October 13, 2014
DOI:10.1021/ml5003713
Catechol diethers that incorporate a 6-cyano-1-naphthyl substituent have been explored as non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs). Promising compounds are reported that show midpicomolar activity against the wild-type virus and sub-20 nM activity against viral variants bearing Tyr181Cys and Lys103Asn mutations in HIV-RT. An X-ray crystal structure at 2.49 Å resolution is also reported for the key compound 6e with HIV-RT.Keywords: Anti-HIV agents; NNRTIs; protein crystallography
Co-reporter:Vidya P. Kumar, Jose A. Cisneros, Kathleen M. Frey, Alejandro Castellanos-Gonzalez, Yiqiang Wang, Aleem Gangjee, A. Clinton White Jr., William L. Jorgensen, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 17) pp:4158-4161
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmcl.2014.07.049
Cryptosporidium is the causative agent of a gastrointestinal disease, cryptosporidiosis, which is often fatal in immunocompromised individuals and children. Thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are essential enzymes in the folate biosynthesis pathway and are well established as drug targets in cancer, bacterial infections, and malaria. Cryptosporidium hominis has a bifunctional thymidylate synthase and dihydrofolate reductase enzyme, compared to separate enzymes in the host. We evaluated lead compound 1 from a novel series of antifolates, 2-amino-4-oxo-5-substituted pyrrolo[2,3-d]pyrimidines as an inhibitor of Cryptosporidium hominis thymidylate synthase with selectivity over the human enzyme. Complementing the enzyme inhibition compound 1 also has anti-cryptosporidial activity in cell culture. A crystal structure with compound 1 bound to the TS active site is discussed in terms of several van der Waals, hydrophobic and hydrogen bond interactions with the protein residues and the substrate analog 5-fluorodeoxyuridine monophosphate (TS), cofactor NADPH and inhibitor methotrexate (DHFR). Another crystal structure in complex with compound 1 bound in both the TS and DHFR active sites is also reported here. The crystal structures provide clues for analog design and for the design of ChTS–DHFR specific inhibitors.
Co-reporter:Hitesh Sharma, Mark J. Landau, Todd J. Sullivan, Vidya P. Kumar, Markus K. Dahlgren, William L. Jorgensen, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1232-1235
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2013.12.039
The parasite Toxoplasma gondii can lead to toxoplasmosis in those who are immunocompromised. To combat the infection, the enzyme responsible for nucleotide synthesis thymidylate synthase–dihydrofolate reductase (TS–DHFR) is a suitable drug target. We have used virtual screening to determine novel allosteric inhibitors at the interface between the two TS domains. Selected compounds from virtual screening inhibited TS activity. Thus, these results show that allosteric inhibition by small drug-like molecules can occur in T. gondii TS–DHFR and pave the way for new and potent species-specific inhibitors.
Co-reporter:Christopher M. Bailey ; Todd J. Sullivan ; Pinar Iyidogan ; Julian Tirado-Rives ; Raymond Chung ; Juliana Ruiz-Caro ; Ebrahim Mohamed ; William Jorgensen ; Roger Hunter
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:3959-3968
Publication Date(Web):May 9, 2013
DOI:10.1021/jm400160s
Human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) is a major target for currently approved anti-HIV drugs. These drugs are divided into two classes: nucleoside and non-nucleoside reverse transcriptase inhibitors (NRTIs and NNRTIs). This study illustrates the synthesis and biochemical evaluation of a novel bifunctional RT inhibitor utilizing d4T (NRTI) and a TMC-derivative (a diarylpyrimidine NNRTI) linked via a poly(ethylene glycol) (PEG) linker. HIV-1 RT successfully incorporates the triphosphate of d4T-4PEG-TMC bifunctional inhibitor in a base-specific manner. Moreover, this inhibitor demonstrates low nanomolar potency that has 4.3-fold and 4300-fold enhancement of polymerization inhibition in vitro relative to the parent TMC-derivative and d4T, respectively. This study serves as a proof-of-concept for the development and optimization of bifunctional RT inhibitors as potent inhibitors of HIV-1 viral replication.
Co-reporter:Hitesh Sharma, Mark J. Landau, Melissa A. Vargo, Krasimir A. Spasov, and Karen S. Anderson
Biochemistry 2013 Volume 52(Issue 41) pp:
Publication Date(Web):September 20, 2013
DOI:10.1021/bi400576t
Most species, such as humans, have monofunctional forms of thymidylate synthase (TS) and dihydrofolate reductase (DHFR) that are key folate metabolism enzymes making critical folate components required for DNA synthesis. In contrast, several parasitic protozoa, including Toxoplasma gondii, contain a unique bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) having the catalytic activities contained on a single polypeptide chain. The prevalence of T. gondii infections across the world, especially for those immunocompromised, underscores the need to understand TS-DHFR enzyme function and to find new avenues to exploit for the design of novel antiparasitic drugs. As a first step, we have solved the first three-dimensional structures of T. gondii TS-DHFR at 3.7 Å and of a loop truncated TS-DHFR, removing several flexible surface loops in the DHFR domain, improving resolution to 2.2 Å. Distinct structural features of the TS-DHFR homodimer include a junctional region containing a kinked crossover helix between the DHFR domains of the two adjacent monomers, a long linker connecting the TS and DHFR domains, and a DHFR domain that is positively charged. The roles of these unique structural features were probed by site-directed mutagenesis coupled with presteady state and steady state kinetics. Mutational analysis of the crossover helix region combined with kinetic characterization established the importance of this region not only in DHFR catalysis but also in modulating the distal TS activity, suggesting a role for TS-DHFR interdomain interactions. Additional kinetic studies revealed that substrate channeling occurs in which dihydrofolate is directly transferred from the TS to DHFR active site without entering bulk solution. The crystal structure suggests that the positively charged DHFR domain governs this electrostatically mediated movement of dihydrofolate, preventing release from the enzyme. Taken together, these structural and kinetic studies reveal unique, functional regions on the T. gondii TS-DHFR enzyme that may be targeted for inhibition, thus paving the way for designing species specific inhibitors.
Co-reporter:Nilesh Zaware, Hitesh Sharma, Jie Yang, Ravi Kumar Vyas Devambatla, Sherry F. Queener, Karen S. Anderson, and Aleem Gangjee
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 12) pp:1148-1151
Publication Date(Web):October 4, 2013
DOI:10.1021/ml400208v
Infection by the parasite Toxoplasma gondii (tg) can lead to toxoplasmosis in immunocompromised patients such as organ transplant, cancer, and HIV/AIDS patients. The bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) enzyme is crucial for nucleotide synthesis in T. gondii and represents a potential target to combat T. gondii infection. While species selectivity with drugs has been attained for DHFR, TS is much more conserved across species, and specificity is significantly more challenging. We discovered novel substituted-9H-pyrimido[4,5-b]indoles 1–3 with single-digit nanomolar Ki for tgTS, two of which, 2 and 3, are 28- and 122-fold selective over human TS (hTS). The synthesis of these compounds, and their structures in complex with tgTS-DHFR are presented along with binding measurements and cell culture data. These results show, for the very first time, that, in spite of the high degree of conservation of active site residues between hTS and the parasite TS, specificity has been accomplished via novel structures and provides a new target (TS) for selective drug development against parasitic infections.Keywords: active-site inhibitors; crystal structure; Opportunistic infection; thymidylate-synthase inhibitors; Toxoplasma gondii;
Co-reporter:Pinar Iyidogan, Todd J. Sullivan, Mahendra D. Chordia, Kathleen M. Frey, and Karen S. Anderson
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 12) pp:1183-1188
Publication Date(Web):October 15, 2013
DOI:10.1021/ml4002979
In a continuing study of potent bifunctional anti-HIV agents, we rationally designed a novel chimeric inhibitor utilizing thymidine (THY) and a TMC derivative (a diarylpyrimidine NNRTI) linked via a polymethylene linker (ALK). The nucleoside, 5′-hydrogen-phosphonate (H-phosphonate), and 5′-triphosphate forms of this chimeric inhibitor (THY-ALK-TMC) were synthesized and the antiviral activity profiles were evaluated at the enzyme and cellular level. The nucleoside triphosphate (11) and the H-phosphonate (10) derivatives inhibited RT polymerization with an IC50 value of 6.0 and 4.3 nM, respectively. Additionally, chimeric nucleoside (9) and H-phosphonate (10) derivatives reduced HIV replication in a cell-based assay with low nanomolar antiviral potencies.Keywords: HIV; NNRTI; nucleoside H-phosphonate; reverse transcriptase; synthesis;
Co-reporter:Mariela Bollini, Kathleen M. Frey, José A. Cisneros, Krasimir A. Spasov, Kalyan Das, Joseph D. Bauman, Eddy Arnold, Karen S. Anderson, William L. Jorgensen
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 18) pp:5209-5212
Publication Date(Web):15 September 2013
DOI:10.1016/j.bmcl.2013.06.093
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (HIV-RT) are reported that feature extension into the entrance channel near Glu138. Complexes of the parent anilinylpyrimidine 1 and the morpholinoethoxy analog 2j with HIV-RT have received crystallographic characterization confirming the designs. Measurement of aqueous solubilities of 2j, 2k, the parent triazene 2a, and other NNRTIs demonstrate profound benefits for addition of the morpholinyl substituent.
Co-reporter:Dongyuan Piao, Aravind Basavapathruni, Pinar Iyidogan, Guangxiu Dai, Wolfgang Hinz, Adrian S. Ray, Eisuke Murakami, Joy Y. Feng, Fei You, Ginger E. Dutschman, David J. Austin, Kathlyn A. Parker, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1511-1518
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.015
The onset of resistance to approved anti-AIDS drugs by HIV necessitates the search for novel inhibitors of HIV-1 reverse transcriptase (RT). Developing single molecular agents concurrently occupying the nucleoside and nonnucleoside binding sites in RT is an intriguing idea but the proof of concept has so far been elusive. As a first step, we describe molecular modeling to guide focused chemical syntheses of conjugates having nucleoside (d4T) and nonnucleoside (TIBO) moieties tethered by a flexible polyethylene glycol (PEG) linker. A triphosphate of d4T–6PEG–TIBO conjugate was successfully synthesized that is recognized as a substrate by HIV-1 RT and incorporated into a double-stranded DNA.
Co-reporter:W. Edward Martucci, Johanna M. Rodriguez, Melissa A. Vargo, Matthew Marr, Andrew D. Hamilton and Karen S. Anderson
MedChemComm 2013 vol. 4(Issue 9) pp:1247-1256
Publication Date(Web):27 Jun 2013
DOI:10.1039/C3MD00141E
The bifunctional enzyme thymidylate synthase–dihydrofolate reductase (TS–DHFR) from the protozoal parasite Cryptosporidium hominis is a potential molecular target for the design of antiparasitic therapies for AIDS-related opportunistic infections. The enzyme exists as a homodimer with each monomer containing a unique swap domain known as a “crossover helix” that binds in a cleft on the adjacent DHFR active site. This crossover helix is absent in species containing monofunctional forms of DHFR such as human. An in-depth understanding of protein–protein interactions between the crossover helix and adjacent DHFR active site that might modulate enzyme integrity or function would allow for insights into rational design of species-specific allosteric inhibitors. Mutational analysis coupled with structural studies and biophysical and kinetic characterization of crossover helix mutants identifies this domain as essential for full enzyme stability and catalytic activity, and pinpoints these effects to distinct faces of the crossover helix important in protein–protein interactions. Moreover, targeting this helical protein interaction with α-helix mimetics of the crossover helix leads to selective inhibition and destabilization of the C. hominis TS–DHFR enzyme, thus validating this region as a new avenue to explore for species-specific inhibitor design.
Co-reporter:Kathleen M. Frey ; Mariela Bollini ; Andrea C. Mislak ; José A. Cisneros ; Ricardo Gallardo-Macias ; William L. Jorgensen
Journal of the American Chemical Society 2012 Volume 134(Issue 48) pp:19501-19503
Publication Date(Web):November 19, 2012
DOI:10.1021/ja3092642
X-ray crystal structures at 2.9 Å resolution are reported for two complexes of catechol diethers with HIV-1 reverse transcriptase. The results help elucidate the structural origins of the extreme antiviral activity of the compounds. The possibility of halogen bonding between the inhibitors and Pro95 is addressed. Structural analysis reveals key interactions with conserved residues P95 and W229 of importance for design of inhibitors with high potency and favorable resistance profiles.
Co-reporter:Youngjoo Kim, Zhimin Li, Mihaela Apetri, BeiBei Luo, Jeffrey E. Settleman, and Karen S. Anderson
Biochemistry 2012 Volume 51(Issue 25) pp:5212-5222
Publication Date(Web):June 1, 2012
DOI:10.1021/bi300476v
Epidermal growth factor receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases (RTK). EGFR overexpression or mutation in many different forms of cancers has highlighted its role as an important therapeutic target. Gefitinib, the first small molecule inhibitor of EGFR kinase function to be approved for the treatment of nonsmall cell lung cancer (NSCLC) by the FDA, demonstrates clinical activity primarily in patients with tumors that harbor somatic kinase domain mutations in EGFR. Here, we compare wild-type EGFR autophosphorylation kinetics to the L834R (also called L858R) EGFR form, one of the most common mutations in lung cancer patients. Using rapid chemical quench, time-resolved electrospray mass spectrometry (ESI-MS), and Western blot analyses, we examined the order of autophosphorylation in wild-type (WT) and L834R EGFR and the effect of gefitinib (Iressa) on the phosphorylation of individual tyrosines. These studies establish that there is a temporal order of autophosphorylation of key tyrosines involved in downstream signaling for WT EGFR and a loss of order for the oncogenic L834R mutant. These studies also reveal unique signature patterns of drug sensitivity for inhibition of tyrosine autophosphorylation by gefitinib: distinct for WT and oncogenic L834R mutant forms of EGFR. Fluorescence studies show that for WT EGFR the binding affinity for gefitinib is weaker for the phosphorylated protein while for the oncogenic mutant, L834R EGFR, the binding affinity of gefitinib is substantially enhanced and likely contributes to the efficacy observed clinically. This mechanistic information is important in understanding the molecular details underpinning clinical observations as well as to aid in the design of more potent and selective EGFR inhibitors.
Co-reporter:Jiae Kim, Ligong Wang, Yongfeng Li, Kimberlynne D. Becnel, Kathleen M. Frey, Scott J. Garforth, Vinayaka R. Prasad, Raymond F. Schinazi, Dennis C. Liotta, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:4064-4067
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.078
Pre-steady state kinetic analysis was utilized for biochemical evaluation of a series of cyclobutyl adenosine nucleotide analogs with HIV-1 RTWT. The phosphonyl-diphosphate form of the cyclobutyl nucleotide, 5, was the most efficiently incorporated of the series. Nucleotide 5 was fourfold more efficiently incorporated than the FDA approved TFV-DP by RTWT. The kinetics of incorporation for 5 using the drug resistant mutant enzyme K65R was also determined. Compound 5 was threefold more efficiently incorporated compared to TFV-DP with RTK65R. These results demonstrate cyclobutyl adenosine analogs can act as substrates for incorporation by HIV-1 RT and be a potential scaffold for HIV inhibitors.
Co-reporter:William L. Jorgensen ; Mariela Bollini ; Vinay V. Thakur ; Robert A. Domaoal ; Krasimir A. Spasov
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15686-15696
Publication Date(Web):August 19, 2011
DOI:10.1021/ja2058583
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) that interfere with the replication of human immunodeficiency virus (HIV) are being pursued with guidance from molecular modeling including free-energy perturbation (FEP) calculations for protein–inhibitor binding affinities. The previously reported pyrimidinylphenylamine 1 and its chloro analogue 2 are potent anti-HIV agents; they inhibit replication of wild-type HIV-1 in infected human T-cells with EC50 values of 2 and 10 nM, respectively. However, they show no activity against viral strains containing the Tyr181Cys (Y181C) mutation in HIV-RT. Modeling indicates that the problem is likely associated with extensive interaction between the dimethylallyloxy substituent and Tyr181. As an alternative, a phenoxy group is computed to be oriented in a manner diminishing the contact with Tyr181. However, this replacement leads to a roughly 1000-fold loss of activity for 3 (2.5 μM). The present report details the efficient, computationally driven evolution of 3 to novel NNRTIs with sub-10 nM potency toward both wild-type HIV-1 and Y181C-containing variants. The critical contributors were FEP substituent scans for the phenoxy and pyrimidine rings and recognition of potential benefits of addition of a cyanovinyl group to the phenoxy ring.
Co-reporter:Alpay Dermenci, Philipp S. Selig, Robert A. Domaoal, Krasimir A. Spasov, Karen S. Anderson and Scott J. Miller
Chemical Science 2011 vol. 2(Issue 8) pp:1568-1572
Publication Date(Web):20 Jun 2011
DOI:10.1039/C1SC00221J
Cysteine plays a number of important functional and structural roles in nature, often in the realm of catalysis. Herein, we present an example of a cysteine-promoted Rauhut-Currier reaction for a potentially biomimetic synthesis of Sch-642305 and related analogs. In this key step of the synthesis we discuss interesting new discoveries and the importance of substrate-catalyst recognition, as well as cysteine's structural features. Also, we investigate the activity of Sch-642305 and four analogs in HIV-infected T-cells.
Co-reporter:Mariela Bollini ; Robert A. Domaoal ; Vinay V. Thakur ; Ricardo Gallardo-Macias ; Krasimir A. Spasov ; Karen S. Anderson ;William L. Jorgensen
Journal of Medicinal Chemistry 2011 Volume 54(Issue 24) pp:8582-8591
Publication Date(Web):November 14, 2011
DOI:10.1021/jm201134m
A 5-μM docking hit has been optimized to an extraordinarily potent (55 pM) non-nucleoside inhibitor of HIV reverse transcriptase. Use of free energy perturbation (FEP) calculations to predict relative free energies of binding aided the optimizations by identifying optimal substitution patterns for phenyl rings and a linker. The most potent resultant catechol diethers feature terminal uracil and cyanovinylphenyl groups. A halogen bond with Pro95 likely contributes to the extreme potency of compound 42. In addition, several examples are provided illustrating failures of attempted grafting of a substructure from a very active compound onto a seemingly related scaffold to improve its activity.
Co-reporter:Cheryl S. Leung, Jacob G. Zeevaart, Robert A. Domaoal, Mariela Bollini, Vinay V. Thakur, Krasimir A. Spasov, Karen S. Anderson, William L. Jorgensen
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 8) pp:2485-2488
Publication Date(Web):15 April 2010
DOI:10.1016/j.bmcl.2010.03.006
Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase is being pursued with the assistance of free energy perturbation (FEP) calculations to predict relative free energies of binding. Extension of azole-containing inhibitors into an ‘eastern’ channel between Phe227 and Pro236 has led to the discovery of potent and structurally novel derivatives.Synthesis, assaying, and computational results are reported for new anti-HIV agents that exhibit high potency and low cytotoxicity. Extension into an eastern channel in the HIV-1 reverse transcriptase binding site is investigated.
Co-reporter:Tina Dasgupta, Penchit Chitnumsub, Sumalee Kamchonwongpaisan, Cherdsak Maneeruttanarungroj, Sara E. Nichols, Theresa M. Lyons, Julian Tirado-Rives, William L. Jorgensen, Yongyuth Yuthavong and Karen S. Anderson
ACS Chemical Biology 2009 Volume 4(Issue 1) pp:29
Publication Date(Web):January 16, 2009
DOI:10.1021/cb8002804
Plasmodium falciparum thymidylate synthase-dihydrofolate reductase (TS-DHFR) is an essential enzyme in folate biosynthesis and a major malarial drug target. This bifunctional enzyme thus presents different design approaches for developing novel inhibitors against drug-resistant mutants. We performed a high-throughput in silico screen of a database of diverse, drug-like molecules against a non-active-site pocket of TS-DHFR. The top compounds from this virtual screen were evaluated by in vitro enzymatic and cellular culture studies. Three compounds active to 20 μM IC50's in both wildtype and antifolate-resistant P. falciparum parasites were identified; moreover, no inhibition of human DHFR enzyme was observed, indicating that the inhibitory effects appeared to be parasite-specific. Notably, all three compounds had a biguanide scaffold. However, relative free energy of binding calculations suggested that the compounds might preferentially interact with the active site over the screened non-active-site region. To resolve the two possible modes of binding, co-crystallization studies of the compounds complexed with TS-DHFR enzyme were performed. Surprisingly, the structural analysis revealed that these novel, biguanide compounds do indeed bind at the active site of DHFR and additionally revealed the molecular basis by which they overcome drug resistance. To our knowledge, these are the first co-crystal structures of novel, biguanide, non-WR99210 compounds that are active against folate-resistant malaria parasites in cell culture.
Co-reporter:Zhili Li, Feng Song, Zhihao Zhuang, Debra Dunaway-Mariano, Karen S. Anderson
Analytical Biochemistry 2009 Volume 394(Issue 2) pp:209-216
Publication Date(Web):15 November 2009
DOI:10.1016/j.ab.2009.07.030
The ability to examine real-time reaction kinetics for multimeric enzymes in their native state may offer unique insights into understanding the catalytic mechanism and its interplay with three-dimensional structure. In this study, we have used a time-resolved electrospray mass spectrometry approach to probe the kinetic mechanism of 4-hydroxybenzoyl–coenzyme A (4-HBA–CoA) thioesterase from Arthrobacter sp. strain SU in the millisecond time domain. Intact tetrameric complexes of 4-HBA–CoA thioesterase with up to four natural substrate (4-HBA–CoA) molecules bound were detected at times as early as 6 ms using an online rapid-mixing device directly coupled to an electrospray ionization time-of-flight mass spectrometer. Species corresponding to the formation of a folded tetramer of the thioesterase at charge states 16+, 17+, 18+, and 19+ around m/z 3800 were observed and assigned as individual tetramers of thioesterase and noncovalent complexes of the tetramers with up to four substrate and/or product molecules. Real-time evaluation of the reaction kinetics was accomplished by monitoring change in peak intensity corresponding to the substrate and product complexes of the tetrameric protein. The mass spectral data suggest that product 4-HBA is released from the active site of the enzyme prior to the release of product CoA following catalytic turnover. This study demonstrates the utility of this technique to provide additional molecular details for an understanding of the individual enzyme states during the thioesterase catalysis and ability to observe real-time interactions between enzyme and substrates and/or products in the millisecond time range.
Co-reporter:W. Edward Martucci, Marina Udier-Blagovic, Chloe Atreya, Oladapo Babatunde, Melissa A. Vargo, William L. Jorgensen, Karen S. Anderson
Bioorganic & Medicinal Chemistry Letters 2009 19(2) pp: 418-423
Publication Date(Web):
DOI:10.1016/j.bmcl.2008.11.054
Co-reporter:W. Edward Martucci, Melissa A. Vargo and Karen S. Anderson
Biochemistry 2008 Volume 47(Issue 34) pp:
Publication Date(Web):August 2, 2008
DOI:10.1021/bi800466z
The essential enzyme TS-DHFR from Cryptosporidium hominis undergoes an unusually rapid rate of catalysis at the conserved TS domain, facilitated by two nonconserved residues, Ala287 and Ser290, in the folate tail-binding region. Mutation of these two residues to their conserved counterparts drastically affects multiple steps of the TS catalytic cycle. We have determined the crystal structures of all three mutants (A287F, S290G, and A287F/S290G) in complex with active site ligands dUMP and CB3717. The structural data show two effects of the mutations: an increased distance between the ligands in the active site and increased flexibility of the folate ligand in the partially open enzyme state that precedes conformational change to the active catalytic state. The latter effect is able to be rescued by the mutants containing the A287F mutation. In addition, the conserved water network of TS is altered in each of the mutants. The structural results point to a role of the folate tail-binding residues in closely positioning ChTS ligands and restricting ligand flexibility in the partially open state to allow for a rapid transition to the active closed state and enhanced rate of catalysis. These results provide an explanation on how folate tail-binding residues at one end of the active site affect long-range interactions throughout the TS active site and validate these residues as targets for species-specific drug design.
Co-reporter:Yagmur Muftuoglu, Christal D. Sohl, Andrea C. Mislak, Hiroaki Mitsuya, Stefan G. Sarafianos, Karen S. Anderson
Antiviral Research (June 2014) Volume 106() pp:1-4
Publication Date(Web):June 2014
DOI:10.1016/j.antiviral.2014.03.001
Co-reporter:Pinar Iyidogan, Karen S. Anderson
Antiviral Research (August 2012) Volume 95(Issue 2) pp:93-103
Publication Date(Web):August 2012
DOI:10.1016/j.antiviral.2012.05.012
Co-reporter:Karen S. Anderson
Methods (August 2010) Volume 51(Issue 4) pp:392-398
Publication Date(Web):1 August 2010
DOI:10.1016/j.ymeth.2010.05.001
Nucleoside analogs play an essential role in treating human immunodeficiency virus (HIV) infection since the beginning of the AIDS epidemic and work by inhibition of HIV-1 reverse transcriptase (RT), a viral polymerase essential for DNA replication. Today, over 90% of all regimens for HIV treatment contain at least one nucleoside. Long-term use of nucleoside analogs has been associated with adverse effects including mitochondrial toxicity due to inhibition of the mitochondrial polymerase, DNA polymerase gamma (mtDNA pol γ). In this review, we describe our efforts to delineate the molecular mechanism of nucleoside inhibition of HIV-1 RT and mtDNA pol γ based upon a transient kinetic approach using rapid chemical quench methodology. Using transient kinetic methods, the maximum rate of polymerization (kpol), the dissociation constant for the ground state binding (Kd), and the incorporation efficiency (kpol/Kd) can be determined for the nucleoside analogs and their natural substrates. This analysis allowed us to develop an understanding of the structure activity relationships that allow correlation between the structural and stereochemical features of the nucleoside analog drugs with their mechanistic behavior toward the viral polymerase, RT, and the host cell polymerase, mtDNA pol γ. An in-depth understanding of the mechanisms of inhibition of these enzymes is imperative in overcoming problems associated with toxicity.
Co-reporter:Scott J. Garforth, Robert A. Domaoal, Chisanga Lwatula, Mark J. Landau, ... Vinayaka R. Prasad
Journal of Molecular Biology (6 August 2010) Volume 401(Issue 1) pp:33-44
Publication Date(Web):6 August 2010
DOI:10.1016/j.jmb.2010.06.001
Lys65 residue, in the fingers domain of human immunodeficiency virus reverse transcriptase (RT), interacts with incoming dNTP in a sequence-independent fashion. We showed previously that a 5-amino-acid deletion spanning Lys65 and a K65A substitution both enhanced the fidelity of dNTP insertion. We hypothesized that the Lys65 residue enhances dNTP misinsertion via interactions with the γ-phosphate of the incoming dNTP. We now examine this hypothesis in pre-steady-state kinetic studies using wild-type human immunodeficiency virus-1 RT and two substitution mutants, K65A and K65R. K65R mutation did not greatly increase misinsertion fidelity, but K65A mutation led to higher incorporation fidelity. For a misinsertion to become a permanent error, it needs to be accompanied by the extension of the mispaired terminus thus formed. Both mutants and the wild-type enzyme discriminated against the mismatched primer at the catalytic step (kpol). Additionally, K65A and K65R mutants displayed a further decrease in mismatch extension efficiency, primarily at the level of dNTP binding. We employed hydroxyl radical footprinting to determine the position of the RT on the primer/template. The wild-type and Lys65-substituted enzymes occupied the same position at the primer terminus; the presence of a mismatched primer terminus caused all three enzymes to be displaced to a − 2 position relative to the primer 3′ end. In the context of an efficiently extended mismatched terminus, the presence of the next complementary nucleotide overcame the displacement, resulting in a complex resembling the matched terminus. The results are consistent with the observed reduction in kpol in mispaired primer extension being due to the position of the enzyme at a mismatched terminus. Our work shows the influence of the stabilizing interactions of Lys65 with the incoming dNTP on two different aspects of polymerase fidelity.
Co-reporter:Jiae Kim, Anne Roberts, Hua Yuan, Yong Xiong, Karen S. Anderson
Journal of Molecular Biology (3 February 2012) Volume 415(Issue 5) pp:866-880
Publication Date(Web):3 February 2012
DOI:10.1016/j.jmb.2011.12.034
Human immunodeficiency virus type 1 (HIV-1) requires reverse transcriptase (RT) and HIV-1 nucleocapsid protein (NCp7) for proper viral replication. HIV-1 NCp7 has been shown to enhance various steps in reverse transcription including tRNA initiation and strand transfer, which may be mediated through interactions with RT as well as RNA and DNA oligonucleotides.With the use of DNA oligonucleotides, we have examined the interaction of NCp7 with RT and the kinetics of reverse transcription during (+)-strand synthesis with an NCp7-facilitated annealed primer–template. Through the use of a pre-steady-state kinetics approach, the NCp7-annealed primer–template has a substantial increase (3- to 7-fold) in the rate of incorporation (kpol) by RT as compared to heat-annealed primer–template with single-nucleotide incorporation. There was also a 2-fold increase in the binding affinity constant (Kd) of the nucleotide. These differences in kpol and Kd were not through direct interactions between HIV-1 RT and NCp7. When extension by RT was examined, the data suggest that the NCp7-annealed primer–template facilitates the formation of a longer product more quickly compared to the heat-annealed primer–template. This enhancement in rate is mediated through interactions with NCp7's zinc fingers and N-terminal domain and nucleic acids. The NCp7-annealed primer–template also enhances the fidelity of RT (3-fold) by slowing the rate of incorporation of an incorrect nucleotide. Taken together, this study elucidates a new role of NCp7 by facilitating DNA-directed DNA synthesis during reverse transcription by HIV-1 RT that may translate into enhanced viral fitness and offers an avenue to exploit for targeted therapeutic intervention against HIV.Download high-res image (153KB)Download full-size imageHighlights► DNA synthesis by HIV RT with an NCp7-faciliated annealed primer–template was studied. ► NCp7 enhances the rate of nucleotide incorporation and fidelity by RT. ► This is mediated through interactions with NCp7's zinc fingers and N-terminal domain. ► As a consequence, the nucleoprotein complex may lead to enhanced viral fitness.
Co-reporter:Alpay Dermenci, Philipp S. Selig, Robert A. Domaoal, Krasimir A. Spasov, Karen S. Anderson and Scott J. Miller
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1572-1572
Publication Date(Web):2011/06/20
DOI:10.1039/C1SC00221J
Cysteine plays a number of important functional and structural roles in nature, often in the realm of catalysis. Herein, we present an example of a cysteine-promoted Rauhut-Currier reaction for a potentially biomimetic synthesis of Sch-642305 and related analogs. In this key step of the synthesis we discuss interesting new discoveries and the importance of substrate-catalyst recognition, as well as cysteine's structural features. Also, we investigate the activity of Sch-642305 and four analogs in HIV-infected T-cells.














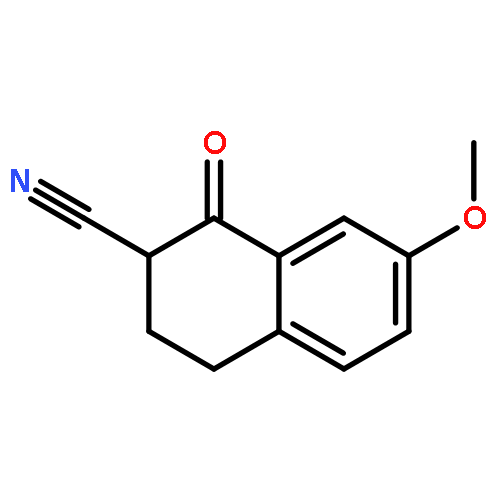
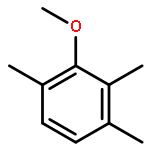
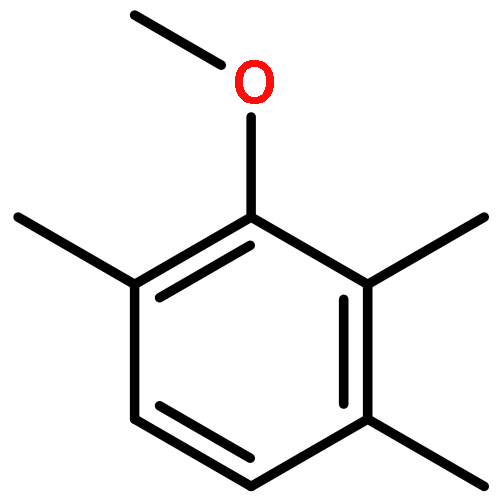

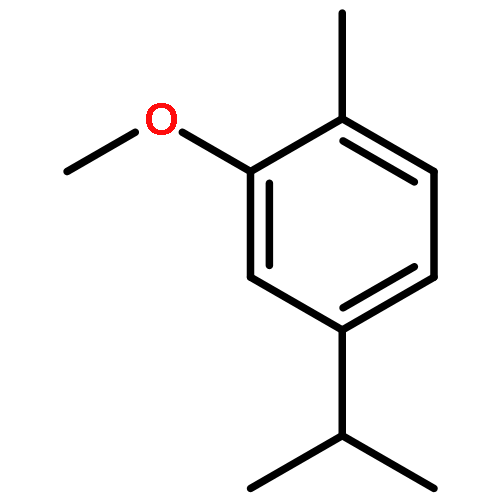




![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-n-methylnaphthalene-1-carboxamide](http://img.cochemist.com/ccimg/1058200/1058137-23-7.png)
![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-n-methylnaphthalene-1-carboxamide](http://img.cochemist.com/ccimg/1058200/1058137-23-7_b.png)
![4-Isoxazolecarboxylic acid, 3-(2,6-dichlorophenyl)-5-methyl-, 2-[2-[[[(2,4-dichlorophenyl)methylene]amino]oxy]acetyl]hydrazide](/data/chemimg/1817700/387353-03-9.png)
![4-Isoxazolecarboxylic acid, 3-(2,6-dichlorophenyl)-5-methyl-, 2-[2-[[[(2,4-dichlorophenyl)methylene]amino]oxy]acetyl]hydrazide](/data/chemimg/1817700/387353-03-9_b.png)
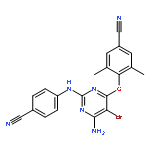

![Benzonitrile,4-[[4-[(2,4,6-trimethylphenyl)amino]-1,3,5-triazin-2-yl]amino]-](http://img.cochemist.com/ccimg/267300/267240-52-8.png)
![Benzonitrile,4-[[4-[(2,4,6-trimethylphenyl)amino]-1,3,5-triazin-2-yl]amino]-](http://img.cochemist.com/ccimg/267300/267240-52-8_b.png)








![4-amino-1-[(2S,5R)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2-one; phosphoric acid](http://img.cochemist.com/ccimg/143200/143188-53-8.png)
![4-amino-1-[(2S,5R)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2-one; phosphoric acid](http://img.cochemist.com/ccimg/143200/143188-53-8_b.png)




![Carbamic acid,[5-(3,4-dihydro-4-oxo-1-phthalazinyl)-1H-benzimidazol-2-yl]-, methyl ester(9CI)](http://img.cochemist.com/ccimg/138600/138525-71-0.png)
![Carbamic acid,[5-(3,4-dihydro-4-oxo-1-phthalazinyl)-1H-benzimidazol-2-yl]-, methyl ester(9CI)](http://img.cochemist.com/ccimg/138600/138525-71-0_b.png)
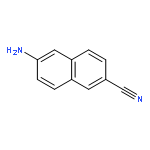
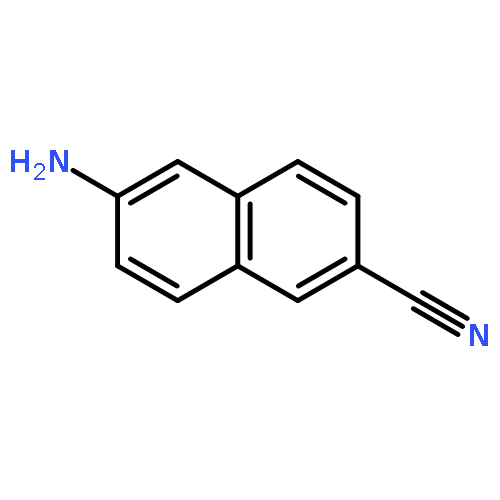


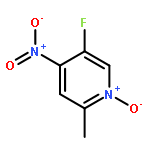


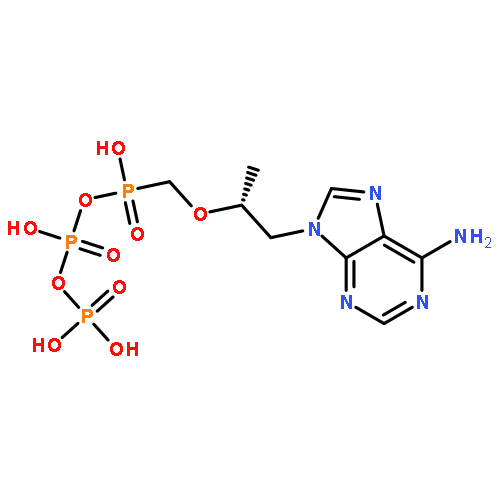
![triphosphoric acid, mono[[(2S,5R)-5-(4-amino-2-oxo-1(2H)-pyrimidinyl)tetrahydro-2-furanyl]methyl] ester](http://img.cochemist.com/ccimg/66100/66004-77-1.png)
![triphosphoric acid, mono[[(2S,5R)-5-(4-amino-2-oxo-1(2H)-pyrimidinyl)tetrahydro-2-furanyl]methyl] ester](http://img.cochemist.com/ccimg/66100/66004-77-1_b.png)








![Triphosphoric acid,P-[[(2R,5S)-5-(4-amino-5-fluoro-2-oxo-1(2H)-pyrimidinyl)-1,3-oxathiolan-2-yl]methyl]ester](http://img.cochemist.com/ccimg/145900/145819-92-7.png)
![Triphosphoric acid,P-[[(2R,5S)-5-(4-amino-5-fluoro-2-oxo-1(2H)-pyrimidinyl)-1,3-oxathiolan-2-yl]methyl]ester](http://img.cochemist.com/ccimg/145900/145819-92-7_b.png)
![4-[[4-[4-[(e)-2-cyanoethenyl]-2,6-dimethylanilino]pyrimidin-2-yl]amino]benzonitrile](http://img.cochemist.com/ccimg/500300/500287-72-9.png)
![4-[[4-[4-[(e)-2-cyanoethenyl]-2,6-dimethylanilino]pyrimidin-2-yl]amino]benzonitrile](http://img.cochemist.com/ccimg/500300/500287-72-9_b.png)