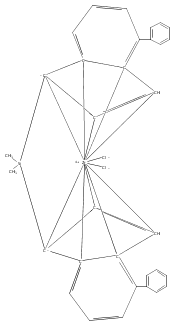
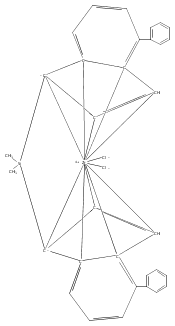
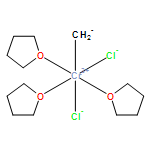
Co-reporter:Fabian F. Karbach, Tibor Macko, and Robbert Duchateau
Macromolecules 2016 Volume 49(Issue 4) pp:1229-1241
Publication Date(Web):February 10, 2016
DOI:10.1021/acs.macromol.5b02430
A silica-supported tandem catalyst system, capable of producing ethylene/1-hexene copolymers from ethylene being the single monomer, has been investigated. As tandem couple a phenoxyimine titanium catalyst for ethylene trimerization was combined with a metallocene catalyst for α-olefin polymerization. Two different approaches were pursued to combine the two catalysts as silica-supported tandem partners. The co-immobilization of the catalysts on the same support particles led to low polymerization activities and yielded products with low comonomer content due to interference of the two catalysts on the support. Immobilization of the two catalysts on separate supports prevented this interaction and led to high polymerization activities while the comonomer content of the product was controlled by the employed catalyst ratio. The copolymers obtained via the latter method were thoroughly analyzed with respect to their chemical composition distribution (CCD) by DSC-SSA, Crystaf, and HT-HPLC. The obtained data indicate a broad and in some cases bimodal CCD, which was explained by the synergy of composition drift during the polymerization and increasing diffusion limitation within the expanding polymer particle.
Co-reporter:Mark P. F. Pepels, Inge Hermsen, Geert J. Noordzij, and Rob Duchateau
Macromolecules 2016 Volume 49(Issue 3) pp:796-806
Publication Date(Web):January 22, 2016
DOI:10.1021/acs.macromol.5b02391
A kinetic analysis on the aluminum salen-catalyzed ring-opening polymerization (ROP) of (macro)lactones is presented, which focuses on how chain transfer agents and the steric hindrance at the α-methylene groups of both the growing chain and the (macro)lactones affect the polymerization rate. It is shown that for branched macrolactones the choice of initiator does not only influence the initiation rate but surprisingly also the apparent rate of polymerization. The increased polymerization rate, when an unbranched initiator was used, was ascribed to transesterification reactions taking place at the polymer chain end, which effectively results in an unprecedented chain growth at the other end of the polymer chain compared to normal ROP. Furthermore, application of a kinetic model including initiation, propagation, and transesterification led to the ability to accurately quantify the effect of steric hindrance for various alkoxide initiators by determining individual rate constants for initiation. It appeared that secondary alcohols not only have a lower reactivity but also are less likely to be bonded to the metal center than primary alcohols when an excess of the two types of alcohols (i.e., chain transfer agents) is used. For the strained seven-membered lactone it was shown that branching mainly has a retarding effect when present at the metal-bonded alkoxide and not on the monomer, which was ascribed to the cisoid conformation of the ester group of the monomer. On the other hand, the transoid conformation of the ester group in macrolactones resulted in a similar decrease in reactivity for branching on the metal-bonded alkoxide and the monomer. Next to this, it was shown that an excess of alcohol chain transfer agent had a retarding effect on the reaction, which was ascribed to coordination of the alcohol to the metal center.
Co-reporter:Mark P.F. Pepels, Rob G. Kleijnen, Johannes G.P. Goossens, Anne B. Spoelstra, Renate Tandler, Hans Martens, Maria Soliman, Rob Duchateau
Polymer 2016 Volume 88() pp:63-70
Publication Date(Web):6 April 2016
DOI:10.1016/j.polymer.2016.01.035
•Blends of PPDL and HDPE are phase separated but show epitaxial growth of PPDL from the HDPE substrate.•Films of blends of PPDL and LDPE are highly transparent and have good optical properties.•PPDL rich films show a shish-kebab type of crystal morphology, in which PPDL kebabs are grown from LDPE shishes.This work describes the phase behavior of blends of ‘polyethylene-like’ polypentadecalactone (PPDL) and polyethylene. Blends of high-density polyethylene (HDPE) and PPDL were shown to be immiscible at the onset of crystallization of polyethylene, resulting in phase-separated morphologies. However, epitaxial crystallization of PPDL onto the HDPE crystals was observed by transmission electron microscopy (TEM), resulting in lamellae penetrating through the interface of the two polymers. Furthermore, PPDL/low-density polyethylene (LDPE) blends were produced and used for film extrusion, yielding clear films with good optical properties, despite the presence of fully phase-separated morphology. For PPDL-rich blends, TEM analysis revealed the formation of highly elongated crystalline domains of LDPE, from which the PPDL domains were epitaxially crystallized yielding a shish-kebab type of morphology. In these structures, the extended LDPE domains formed shishes with LDPE micro-kebabs, onto which PPDL macro-kebabs crystallized. The shish-kebab morphology was furthermore confirmed using x-ray analysis. The high aspect ratio of the LDPE domains is caused by the long relaxation times of LDPE in combination with the low interfacial tension between LDPE and PPDL. As a consequence of the lower relaxation time of PPDL (due to the linear chain architecture), the PPDL domains in the LDPE-rich blends have a lower aspect ratio. The strong epitaxial crystallization in combination with anisotropy in the morphology has a positive effect on the optical properties of the films.Download high-res image (319KB)Download full-size image
Co-reporter:Fabian F. Karbach, John R. Severn, and Robbert Duchateau
ACS Catalysis 2015 Volume 5(Issue 9) pp:5068
Publication Date(Web):July 21, 2015
DOI:10.1021/acscatal.5b01359
A phenoxy-imine titanium catalyst (FI-catalyst) for selective ethylene trimerization was immobilized on methyl aluminoxane (MAO) pretreated silica and its activity and selectivity was compared with that of the corresponding homogeneous catalyst system. The homogeneous and heterogeneous ethylene oligomerization was conducted in the presence of different aluminum alkyls, commonly used as scavengers during olefin polymerization to remove residual oxygen and moisture from the reaction medium. Both the homogeneous and heterogeneous catalysts were strongly affected by the presence of scavenger in the reaction medium. Upon activation with R3Al/MAO (R= Et, nOct, iBu), the homogeneous catalyst switches selectivity from ethylene trimerization to polymerization. NMR spectroscopic investigations indicate that this change of selectivity can be attributed to ligand exchange between the precatalyst and the aluminum alkyl and reduction of the titanium species. The thereby formed ligand-free and/or reduced titanium alkyls act as polymerization catalysts and are responsible for the increasing polymer formation. Using the heterogeneous catalyst, the scavenger employed during ethylene trimerization was found to be of crucial influence regarding the activity of the catalyst and the occurrence of reactor fouling. Employing aluminum alkyls like iBu3Al and nOct3Al resulted in catalyst leaching and homogeneous polymer formation. The latter was prevented using Me3Al or Et3Al as scavengers; however, in general the supported catalyst was poisoned by aluminum alkyls, resulting in a low overall activity. It was found to be beneficial for the heterogeneous trimerization system to employ silica-supported scavengers. By physical separation of the catalyst and the scavenger this poisoning effect was effectively prevented, resulting in a highly active heterogeneous catalyst.Keywords: aluminum alkyls; phenoxy-imine titanium catalyst; selective ethylene oligomerization; silica; supported catalyst
Co-reporter:Aleksandra Wroblewska, Arkadiusz Zych, Shanmugam Thiyagarajan, Dmytro Dudenko, Daan van Es, Michael Ryan Hansen, Cor Koning, Rob Duchateau and Lidia Jasinska-Walc
Polymer Chemistry 2015 vol. 6(Issue 22) pp:4133-4143
Publication Date(Web):20 Apr 2015
DOI:10.1039/C5PY00521C
As part of our ongoing study investigating isohexide-based polyamides, we have synthesized isosorbide(bis(propan-1-amine)) (DAPIS) and studied its reactivity in the polymerization towards fully biobased polyamides. Polycondensation of nylon salts with various contributions of DAPIS afforded a family of homo- and copolyamides, which were characterized using complementary spectroscopic techniques. The chemical structure of the materials was determined by FT-IR, 1D and 2D liquid-state NMR spectroscopy, whilst the supramolecular arrangement, conformational changes upon heating, and molecular mobility of the polymers were investigated by solid-state 13C{1H} Cross-Polarization/Magic-Angle Spinning (CP/MAS) NMR and 13C{1H} Insensitive Nuclei Enhanced by Polarization Transfer (INEPT) experiments. The abundance of the different DAPIS conformers was determined by DFT-D computational methods. The thermal properties of the polyamides were tested for polymers with different amounts of isohexide units in the backbone by DSC and TGA, demonstrating that the increasing amounts of isohexide diamines efficiently decrease their melting points and slightly decrease their thermal stability. The relaxation processes of the isohexide-derived polyamides were studied by DMTA.
Co-reporter:C. Descour, T. Macko, I. Schreur-Piet, M. P. F. Pepels and R. Duchateau
RSC Advances 2015 vol. 5(Issue 13) pp:9658-9666
Publication Date(Web):06 Jan 2015
DOI:10.1039/C4RA11056K
Several compatibilised polyolefin-based blends have been obtained via rather simple and robust chemistry: olefin cross metathesis using Grubbs' second-generation catalyst (G2) of alkenyl-terminated macromolecules of different nature. The viability of the concept was first demonstrated for low molecular weight polyolefin macromolecules before being extended to higher molecular weight polymers, including polar ones such as poly(ε-caprolactone) (PCL), poly(pentadecalactone) (PPDL) and poly(methylmethacrylate) (PMMA). When taking all the possible cross metathesis reactions into account, a statistical distribution of homopolymers and diblock copolymers is likely to be formed. While clear macrophase separation is visible in the uncompatibilised blends of macromolecules, it is absent for the in situ compatibilised products, as was confirmed by optical microscopy. It was demonstrated that even small amounts of diblock copolymers can effectively compatibilise the two phases. All materials were analysed by HT SEC, DSC, HT HPLC and optical microscopy. Such a proof of principle indicates that using cross metathesis on a large library of macromolecules might be a versatile “synthetic handle” to reach a variety of in situ compatibilised blends.
Co-reporter:Mark P. F. Pepels, Ronald A. C. Koeken, Sjoerd J. J. van der Linden, Andreas Heise, and Rob Duchateau
Macromolecules 2015 Volume 48(Issue 14) pp:4779-4792
Publication Date(Web):July 14, 2015
DOI:10.1021/acs.macromol.5b00820
This paper presents a new approach toward the introduction of both short- (SCB) and long-chain branching (LCB) in polyethylene-like polyesters via the ring-opening polymerization of macrolactones. Macrolactones containing an alkyl (S1) or alcohol (S2) branch were obtained using radical thiol–ene chemistry of ambrettolide (Amb). Kinetic studies revealed the need for an excess of thiol to achieve a high conversion of the double bond. Even though homopolymerization of the three monomers Amb, S1, and S2 revealed comparable reactivities, the molecular weight buildup during polymerization of S2 differs drastically from that of Amb and S1. Instead of the linear increase of Mn with conversion observed for Amb and S1, the molecular weight buildup for the ring-opening polymerization of S2 resembles that of a step-growth polymerization—slow buildup at low and moderate conversion followed by a rapid increase in molecular weight at high conversions. This disparity was attributed to the possibility of S2 to function as both an initiator and a monomer, leading to oligomers during the first part of the reaction that are subsequently connected to each other at the final stage of the reaction. Copolymerization of pentadecalactone (PDL) with various ratios of Amb, S1, and S2 in bulk led to the associated random copolymers containing double bonds, short-chain branches, and long-chain branches. The trans-double bonds in poly(PDL-co-Amb) are included in the crystal lattice, leading to a slight decrease in the melting temperature, melting enthalpy and yield stress, while up to 20 double bonds/1000 backbone atoms the crystallinity and lamellar thickness remain similar to those of polypentadecalactone. In contrast, SCBs are fully excluded from the crystal lattice, leading to a more significant decrease in melting temperature and enthalpy as well as crystallinity and lamellar thickness with increasing branching density. The stiffness of these SCB-copolymers exponentially decreases as a function of branching content, effectively changing the mechanical behavior from semicrystalline to elastomeric. The LCB-containing polymers show an even larger linear decrease in melting temperature with increasing branching density than their SCB equivalents, likely due to the particular topology of the polymers consisting of a brush to a hyperbranched structure. However, a rapid decrease of molecular weight as was observed upon increasing the S2 content is also likely to play a role. The observed low molecular weight can be ascribed to both the fact that (macrocycles of) S2 can function as initiator, effectively increasing the amount of polymer chains, and the change of molecular weight buildup.
Co-reporter:Mark P. F. Pepels, Wilma P. Hofman, Rob Kleijnen, Anne B. Spoelstra, Cor E. Koning, Han Goossens, and Rob Duchateau
Macromolecules 2015 Volume 48(Issue 19) pp:6909-6921
Publication Date(Web):September 24, 2015
DOI:10.1021/acs.macromol.5b01620
Block copolymers consisting of a polyethylene block and a polar polymer block are interesting structures for the compatibilization of polyethylene/polar polymer blends or polyethylene-based composites. Since the synthesis of polyethylene-based block copolymers is an elaborate process, diblock copolymers consisting of “polyethylene-like” poly(pentadecalactone) (PPDL) and poly(l-lactide) (PLLA) were synthesized using a one-pot, sequential-feed ring-opening polymerization of pentadecalactone (PDL) and l-lactide (LLA). The peculiar activity of the used aluminum salen catalysts yielded a block copolymer consisting of two blocks with both a high dispersity, as a result of intrablock transesterification. Interestingly, interblock transesterification was effectively suppressed. The obtained poly(PDL-block-LLA) of various block lengths showed coincidental crystallization of the two blocks with an associated microphase-separated morphology, in which PLLA spheres with a high dispersity are distributed within the PPDL matrix. The complex morphologies is believed to arise from the presence of a whole range of block sizes as a consequence of the large dispersity of both blocks. The application of these block copolymers as compatibilizers for high density polyethylene (HDPE)/PLLA blends led to a clear change in blend morphology and a steep decrease in particle size of the dispersed phase. Furthermore, addition of the block copolymers to blends of linear low density polyethylene (LLDPE) and PLLA led to a significant increase in adhesion between the two phases. For both HDPE/PLLA and LLDPE/PLLA blends, the compatibilization efficiency of the poly(PDL-block-LLA) increased when the length of the PPDL block was increased. The presented results clearly show that PPDL can function as a substituent for various types of polyethylene, which opens up a new method for compatibilizing polyethylene with polar polymers using easy attainable “PE-like” block copolymers.
Co-reporter:Shaneesh Vadake Kulangara, Daniel Haveman, Bala Vidjayacoumar, Ilia Korobkov, Sandro Gambarotta, and Rob Duchateau
Organometallics 2015 Volume 34(Issue 7) pp:1203-1210
Publication Date(Web):March 18, 2015
DOI:10.1021/om501013m
Ethylene oligomerization activities of chromium catalysts stabilized by different dipyrrole-based ancillary ligands, [(Ph2C(C4H4N)2)] (2), [Ph2C(C4H4N)(C5H6N)] (3), [(Et)2C(C4H4N)2] (4), and [(C6H5)(C5H4N)C(C4H4N)(C5H6N)] (5), have been investigated using different activation methods, and the results have been compared with the commercial Chevron–Phillips ethylene trimerization system. Upon activation with triethylaluminum (TEA), chromium catalysts stabilized by dipyrrole-based ligands 2–5 showed a lower activity and selectivity compared to the Chevron–Phillips trimerization system based on 2,5-dimethylpyrrole (1) as the ancillary ligand. However, unprecedented increases in both activity and selectivity have been observed by carrying out the oligomerization in methylcyclohexane using depleted-methylaluminoxane (DMAO) along with triisobutylaluminum (TIBA) (1:2 ratio) as cocatalyst system under mild conditions, even for the Chevron–Phillips system itself. Well-defined chromium complexes, [(Ph2C(C4H3N)2)Cr(Cl)(THF)3] (6) and {[Ph2C(C4H3N)(C5H6N]Cr(THF)(μ-Cl)}2 (7), have been synthesized and fully characterized. Upon activating with MAO, catalyst 7 produced a statistical distribution of oligomers, whereas under identical oligomerization conditions catalyst 6/MAO was found to be inactive. The use of MeAlCl2 as cocatalyst to activate 7 resulted in the switching of the catalyst’s behavior from producing a statistical distribution of LAOs to the selective trimerization of ethylene to 1-hexene. The addition of dialkylzinc along with MAO resulted in an unprecedented activity increase.
Co-reporter:Camille Descour;Tibor Macko;Dario Cavallo;Matthew Parkinson;Gerhard Hubner;Anne Spoelstra;Maurizio Villani
Journal of Polymer Science Part A: Polymer Chemistry 2014 Volume 52( Issue 10) pp:1422-1434
Publication Date(Web):
DOI:10.1002/pola.27127
ABSTRACT
Stereoblock polypropylenes comprising of iPP and sPP segments are synthesized by polymerization of the following binary system of metallocenes: the Cs-symmetric [2,7-t-Bu2(Flu)2Ph2C(Cp)ZrCl2] and the C2-symmetric rac-Me2Si(2-Me-4-Ph-Ind)2ZrCl2. Blends of samples made either by each catalyst individually (solution blend) with materials obtained with the mixed catalyst system (reactor blend) are compared. The simultaneous presence of MAO and DEZ, enhancing fast and reversible transfer of the growing chains between the two active centers, leads to the formation of a stereoblock microstructure. In this case, low molecular weight polymers are obtained. The junction between the blocks is qualitatively observed in 13C NMR. When made in toluene, the stereoblock material consists of a majority of syndiotactic sequences, whereas the ratio is more equilibrated when the polymerization was conducted in the more polar chlorobenzene. This is confirmed by the results obtained with 13C NMR, CRYSTAF, HT HPLC, DSC, SSA, WAXD, and optical microscopy. © 2014 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 1422–1434
Co-reporter:Joanna Gurnham, Sandro Gambarotta, Ilia Korobkov, Lidia Jasinska-Walc, and Robbert Duchateau
Organometallics 2014 Volume 33(Issue 17) pp:4401-4409
Publication Date(Web):August 20, 2014
DOI:10.1021/om5005644
Iminopyrrole, aminopyrrole, and aminophosphine ligands were complexed with various chromium sources, producing eight complexes that were tested for their catalytic behavior toward epoxide–CO2 copolymerization. As elucidated by MALDI-TOF-MS, copolymerizations afforded polycarbonates and poly(ether-carbonates) exhibiting linear or cyclic topologies.
Co-reporter:Yun Yang, Joanna Gurnham, Boping Liu, Robbert Duchateau, Sandro Gambarotta, and Ilia Korobkov
Organometallics 2014 Volume 33(Issue 20) pp:5749-5757
Publication Date(Web):July 17, 2014
DOI:10.1021/om5003683
Chromium complexes bearing a series of pyridine–phosphine ligands have been synthesized and examined for their catalytic behavior in ethylene oligomerization. The choice of solvent, toluene versus methylcyclohexane, shows a pronounced influence on the catalytic activity for all these complexes. Variations of the ligand system have been introduced by modifying the phosphine substituents affecting ligand bite angles and flexibility. It has been demonstrated that minor differences in the ligand structure can result in remarkable changes not only in catalytic activity but also in selectivity toward α-olefins versus polyethylene and distribution of oligomeric products. Ligand PyCH2N(Me)PiPr2, in combination with CrCl3(THF)3 afforded selective ethylene tri- and tetramerization, giving 1-hexene and 1-octene with good overall selectivity and high purity, albeit with the presence of small amounts of PE.
Co-reporter:Mark P. F. Pepels, Paul Souljé, Ron Peters, and Rob Duchateau
Macromolecules 2014 Volume 47(Issue 16) pp:5542-5550
Publication Date(Web):August 15, 2014
DOI:10.1021/ma5015353
Polyesters obtained by the catalytic ring-opening polymerization of macrolactones in many aspects resemble the properties of polyethylene. However, the molecular weight distribution is intrinsically different and equals the molecular weight distribution observed for a step-growth process even though macrolactone ring-opening polymerization follows a chain-growth mechanism. The concurrent occurrence of transesterification reactions leading to the formation of cyclic polymers is responsible for the deviation from the molecular weight distribution characteristic for chain-growth polymerization. To explain and extent on the theoretical principles forming the basis of this peculiar molecular weight distribution, in this work the cyclization process during the polymerization of the 17-membered macrolactone ambrettolide has been analyzed. Liquid chromatography under critical conditions has been applied to semiquantitatively analyze the fractions of cyclic and linear products. In addition, low molecular weight size exclusion chromatography has been used to independently quantify the fractions of the smallest cyclics. Using the combination of these techniques it has been shown that cyclics are present during the whole polymerization process. Furthermore, the thermodynamics of the polymerization reaction were determined. The negligible ΔH⊖p = 0.9 ± 1.9 kJ·mol–1 and a positive ΔS⊖p = 38.5 ± 6.5 J·mol–1·K–1 clearly demonstrate the absence of significant ring strain and proofs that the polymerization is driven by entropy. Individual equilibrium concentrations of the cyclics, from monomer to pentamer, were determined and these values were used in combination with the Jacobson and Stockmayer theory to calculate the effective molarity of the cyclic monomer, B = 0.087 M. This value subsequently yields a critical monomer concentration of 0.155 M, for which it was also experimentally determined that polymerizations having a monomer concentration below this value only yield cyclic polymers. Finally, B was used in combination with the monomer and initiator concentration to successfully predict the molecular weight distribution, which shows that real Mn’s are far lower and dispersities far higher than predicted from often-applied theories.
Co-reporter:Miloud Bouyahyi and Rob Duchateau
Macromolecules 2014 Volume 47(Issue 2) pp:517-524
Publication Date(Web):January 14, 2014
DOI:10.1021/ma402072t
This contribution describes our recent results regarding the metal-catalyzed ring-opening polymerization of pentadecalactone and its copolymerization with ε-caprolactone involving single-site metal complexes based on aluminum, zinc, and calcium. Under the right conditions (i.e., monomer concentration, catalyst type, catalyst/initiator ratio, reaction time, etc.), high molecular weight polypentadecalactone with Mn up to 130 000 g mol–1 could be obtained. The copolymerization of a mixture of ε-caprolactone and pentadecalactone yielded random copolymers. Zinc and calcium-catalyzed copolymerization using a sequential feed of pentadecalactone followed by ε-caprolactone afforded perfect block copolymers. The blocky structure was retained even for prolonged times at 100 °C after full conversion of the monomers, indicating that transesterification is negligible. On the other hand, in the presence of the aluminum catalyst, the initially formed block copolymers gradually randomized as a result of intra- and intermolecular transesterification reactions. The formation of homopolymers and copolymers with different architectures has been evidenced by HT-SEC chromatography, NMR, DSC and MALDI-ToF-MS.
Co-reporter:Yun Yang, Zhen Liu, Boping Liu, and Robbert Duchateau
ACS Catalysis 2013 Volume 3(Issue 10) pp:2353
Publication Date(Web):September 3, 2013
DOI:10.1021/cs4004968
A series of N,P-based ancillary ligands have been synthesized, and the corresponding catalysts, formed in situ by mixing one of the N,P-ligands, Cr(acac)3 and MAO, have been tested for ethylene oligomerization. Under standard ethylene oligomerization conditions (30 bar ethylene, 60 °C, methylcyclohexane solvent), all of the in situ-formed complexes show catalytic activity, producing oligomers together with varying amounts of polyethylene (PE). Of all these combinations, only the catalyst formed by mixing N-pyrrolyldiphenylphosphine with Cr(acac)3 and MAO is capable of selectively oligomerizing ethylene, producing a mixture of 1-hexene and 1-octene in varying ratios alongside a small amount of PE. Further investigations on this catalyst system revealed that the presence of a low concentration of toluene favors the production of 1-octene. However, in pure toluene as the solvent, the selectivity toward 1-hexene/1-octene is lost and a statistic mixture of α-olefins is produced. Moreover, the choice of the cocatalyst is found to dramatically influence the composition of the liquid products. By careful adjustment of the reaction conditions (temperature, ethylene pressure, catalyst loading, and ligand/Cr ratio), the 1-hexene/1-octene molar ratio can be tuned from 0.3 to 20 and a selectivity for 1-octene formation of up to 74% can be achieved.Keywords: chromium catalyst; DFT calculation; ethylene tetramerization; ligand design; selective ethylene oligomerization; solvent effect
Co-reporter:Camille Descour, Timo J. J. Sciarone, Dario Cavallo, Tibor Macko, Mauritz Kelchtermans, Ilia Korobkov and Robbert Duchateau
Polymer Chemistry 2013 vol. 4(Issue 17) pp:4718-4729
Publication Date(Web):06 Jun 2013
DOI:10.1039/C3PY00506B
The interactions of a sterically hindered phenol [2,6-(t-Bu)2-4-Me-C6H2OH] (BHT) with the scavenger MAO (AlR3) and ZnR2 during Hf/Zr-based chain shuttling polymerization in a semi-batch reactor have been investigated. NMR model studies indicated preferential binding of BHT to aluminum under these conditions. Subsequently, reproducible polymerization runs gave rise to copolymers that were thoroughly characterized by HT-SEC, HT-HPLC, DSC, thermal fractionation (SSA), 13C NMR, density measurements, CRYSTAF and optical microscopy to unravel their complex microstructures. The obtained materials differ from a simple solution blend of materials, separately produced by single catalysts, but also from multi-block copolymers as obtained by DOW's continuous process, although a blocky structure can be rationalized.
Co-reporter:M. P. F. Pepels, M. Bouyahyi, A. Heise, and R. Duchateau
Macromolecules 2013 Volume 46(Issue 11) pp:4324-4334
Publication Date(Web):May 24, 2013
DOI:10.1021/ma400731c
The kinetic behavior of the catalytic ring-opening polymerization (cROP) of a range of macrolactones, including ω-pentadecalaconte (PDL), ambrettolide (Amb), and butylene adipate (BA), and small-ring lactones, including l-lactide (LLA), ε-caprolactone (ε-CL), ε-decalactone (ε-DL), and β-butyrolactone (B-BL), using various aluminum salen complexes was investigated. The cROP rates were shown to be first order both in catalyst and in monomer. The activation energies of the polymerization of PDL and LLA in combination with aluminum salen complexes, with and without tert-butyl groups, were determined, showing that the increase in steric hindrance is negatively affecting the polymerization rate of LLA more than of PDL. Interestingly, an increase of the salen diimine bridge from ethylene to 2,2-dimethyl propylene leads to a dramatic increase in rate for the polymerization of small-ring lactones, while it leaves the rate of polymerization of macrolactones practically unchanged. In order to exploit this difference in reactivity, the synthesis of block-copolymers of ε-CL and PDL was attempted using kinetic resolution. However, all the polymers obtained over time were found to be fully random, which appeared to be the result of fast transesterification. Poly(PDL-b-CL) block copolymers were successfully synthesized applying the high reactivity of ε-CL in a sequential feed strategy. However, these block copolymers rapidly transform into fully random copolymers as a result of transesterification, which was shown to have a similar rate constant as the rate constant of the polymerization of PDL. By carefully tuning the reaction time polymers with block, gradient or random topology can be obtained.
Co-reporter:Elham Hosseini Nejad, Anita Paoniasari, Carlo G. W. van Melis, Cor E. Koning, and Rob Duchateau
Macromolecules 2013 Volume 46(Issue 3) pp:631-637
Publication Date(Web):January 16, 2013
DOI:10.1021/ma301904y
Catalytic ring-opening copolymerization of limonene oxide with phthalic anhydride was performed applying metal t-Bu–salophen complexes (t-Bu–salophen)MX; M = Cr, X = Cl (1), M = Al, X = Cl (2), M = Co, X = OAc (3), M = Mn, X = Cl (4), t-Bu–salophen = N,N-bis(3,5-di-tert-butylsalicylidene)diimine. The chromium and aluminum catalysts performed best, and catalyst 1 was selected for further studies. Investigating the effect of different cocatalysts in the copolymerization of limonene oxide and phthalic anhydride revealed that the onium salt PPN+Cl– showed the best activity while phosphines and N-heterocyclic-based amines showed a somewhat lower activity. 1H NMR and MALDI-ToF-MS spectra of the copolymers formed confirmed the alternating microstructure. Applying various mono-, di-, and trifunctional chain transfer agents (CTA)s such as water, (poly)alcohols, acids, and diamines in all cases resulted in a decrease in molecular weight while the PDI remained low, characteristic for an immortal system. Catalyst 1 proved to be robust in the presence of these CTAs not showing any drop of catalytic activity, which allows the formation of polyesters with tunable molecular weights and narrow PDIs and varying numbers of functionalities.
Co-reporter:Mark P. F. Pepels, Michael Ryan Hansen, Han Goossens, and Rob Duchateau
Macromolecules 2013 Volume 46(Issue 19) pp:7668-7677
Publication Date(Web):September 25, 2013
DOI:10.1021/ma401403x
Aliphatic long-chain polyesters (ALCPEs) were synthesized using the ring-opening metathesis copolymerization of ambrettolide and cis-cyclooctene followed by exhaustive hydrogenation, yielding saturated ALCPEs with methylene-to-ester ratios (M/E) varying from 15 to 223 and ∞ (polyethylene), of which the ester groups were pseudo randomly distributed over the backbone of the polymer. The melting temperature of these ALCPEs showed an inverse proportional trend with respect to the amount of ester groups in the polymer structure, ranging from 132.1 °C (M/E = ∞) to 91.5 °C (M/E = 15), of which the former is comparable to the melting temperature of high-density polyethylene. The crystallinity and the orthorhombic unit cell of the polymers did not significantly change with increasing M/E. Solid-state NMR was used to show the uniform partitioning of the ester groups over the crystalline and amorphous phase. Even though the lamellar thickness showed a decrease with increasing amount of ester groups, this did only partly explain the decrease in melting temperature. The main factor determining the decrease in melting temperature is the inclusion of ester groups in the crystal lattice, which causes the crystal lattice to be less stable.
Co-reporter:Elham Hosseini Nejad, Anita Paoniasari, Cor E. Koning and Rob Duchateau
Polymer Chemistry 2012 vol. 3(Issue 5) pp:1308-1313
Publication Date(Web):23 Mar 2012
DOI:10.1039/C2PY20026K
Ring-opening copolymerisation of styrene oxide with alicyclic anhydrides containing different ring strains (succinic anhydride, maleic anhydride, citraconic anhydride, cyclopropane-1,2-dicarboxylic acid anhydride, cyclopentane-1,2-dicarboxylic acid anhydride and phthalic anhydride) was performed applying metal salen and tetraphenyl porphyrin complexes where for (salen)MX, M = Cr, X = Cl (1), M = Al, X = Cl (2), M = Mn, X = Cl (3), M = Co, X = OAc (4) and salen = N,N-bis(3,5-di-tert-butylsalicylidene)-diimine and for porphyrin complex, M = Cr, X = Cl (5), M = Mn, X = Cl (6), M = Co, X = OAc (7). The chromium catalysts performed best and therefore 1 was chosen as the selected catalyst for further studies. Investigation of the effect of different cocatalysts on the copolymerisation of styrene oxide and phthalic anhydride revealed that phosphines and onium salt showed quite similar activities whereas N-heterocyclic based amines showed somewhat lower activity. 1H NMR and MALDI-ToF-MS spectra of the copolymers formed confirmed the alternating microstructures. Increasing the monomer to catalyst ratio resulted in the isomerisation of styrene oxide to phenyl ethanal. The aldehyde functions as a chain transfer agent influencing the molecular weight of the polymers. Copolymerisation of styrene oxide with anhydrides bearing a double bond in their structure, such as maleic anhydride and citraconic anhydride, was shown to be highly dependent on temperature, time, type of cocatalyst and solvent used in the copolymerisation reaction.
Co-reporter:Elham Hosseini Nejad, Carlo G. W. van Melis, Tim J. Vermeer, Cor E. Koning, and Rob Duchateau
Macromolecules 2012 Volume 45(Issue 4) pp:1770-1776
Publication Date(Web):February 7, 2012
DOI:10.1021/ma2025804
Ring-opening copolymerization of cyclohexene oxide with alicyclic anhydrides containing different ring strain (succinic anhydride, cyclopropane-1,2-dicarboxylic acid anhydride, and phthalic anhydride) was performed applying metal salen chloride complexes, (salen)MCl (M = Al, Cr, Co; salen = N,N-bis(3,5-di-tert-butylsalicylidene)diimine) with different metals and ligand–diimine backbones. While some of the bulk copolymerizations afforded poly(ester-co-ether)s, all solution polymerizations produced perfect alternating copolymers. The chromium catalysts performed best while the aluminum catalysts were the least active ones. For each metal, the salophen complexes yielded the best performing catalyst. A variety of cocatalysts have been employed: bis(triphenylphosphoranylidene)ammonium chloride, N-heterocyclic nucleophiles including 4-(dimethylamino)pyridine, N-methylimidazole, and 1,5,7-triazabicyclododecene and the phosphines trimesitylphosphine, tris(2,4,6-trimethoxyphenyl)phosphine, tricyclohexylphosphine to triphenylphsophine. Of all cocatalysts, bis(triphenylphosphoranylidene)ammonium chloride was found to be the most efficient cocatalyst in combination with salophenCrCl for the copolymerization of cyclohexene oxide with phthalic anhydride, and 1 equiv was enough to reach optimum activity. N-Heterocyclic nucleophiles showed the lowest activity. Of the three anhydrides used, phthalic anhydride is the most reactive giving the highest conversions and the highest molecular weight products.
Co-reporter:Camille Descour, Tamara Meijer-Vissers, Tibor Macko, Matthew Parkinson, Dario Cavallo, Martin van Drongelen, Gerhard Hubner, Han Goossens, Robbert Duchateau
Polymer 2012 Volume 53(Issue 15) pp:3096-3106
Publication Date(Web):6 July 2012
DOI:10.1016/j.polymer.2012.05.030
The zirconium acetamidinate catalyst {Cp*Zr(Me)2[N(Et)C(Me)N(tBu)]} (Cp* = ŋ5-C5Me5) was used to synthesize both random and block copolymers based on 4-methyl-1-pentene (4M1P) and 1-pentene. The polymers have been characterized by NMR spectroscopy, SEC, DSC, high temperature HPLC and CRYSTAF. Unexpectedly, the yields and molecular weights decreased with increasing amounts of 1-pentene. The reason for this behavior is that 1-pentene occasionally undergoes 2,1-misinsertions trapping the catalyst in a dormant state. These 2,1-misinsertions do not seem to occur with the bulky 4M1P (branched α-olefin). Adding a small amount of ethylene reactivates the catalyst. Unlike most semi-crystalline polymers, the density of the crystalline phase of isotactic P4M1P can be lower than of the amorphous phase, when crystallized under very high pressures. To characterize this peculiar behavior of 4M1P-based polymers, various samples have been subjected to Pressure-Volume-Temperature (PVT) measurements. While the P4M1P homopolymers and block copolymers show the expected decrease in specific volume upon crystallization, the 4M1P-rich random copolymers proved not to vary in specific volume under the same conditions.Graphical abstract
Co-reporter:Shaneesh Vadake Kulangara, Amir Jabri, Yun Yang, Ilia Korobkov, Sandro Gambarotta, and Rob Duchateau
Organometallics 2012 Volume 31(Issue 17) pp:6085-6094
Publication Date(Web):August 14, 2012
DOI:10.1021/om300453a
The synthesis, structural characterization, and ethylene polymerization performance of heterobimetallic aluminum-pyrrolyl complexes of group IV metals are described. The combination of MCl4 (M = Ti, Zr, Hf), aluminum alkyls and pyrroles leads, depending on stoichiometry, to mono- and bis(aluminum-pyrrolyl) complexes that are remiscent of the corresponding mono- and bis- cyclopentadienyl systems. The bis(aluminum-pyrrolyl) complexes (η5-2,5-Me2C4H2NAlClMe2)2TiMe2 (1), zirconium (η5-2,5-Me2C4H2NAlClMe2)2ZrClMe (2), (η5-2,5-Me2C4H2NAlCl2Et)2ZrCl2 (3), and hafnium (η5-2,5-Me2C4H2NAlClMe2)2HfClMe (4) were found to be inactive toward ethylene polymerization. By contrast, the electron-deficient mono(aluminum-pyrrolyl) piano stool complexes (η5-2,5-Me2C4H2NAlCl2Me)TiCl2Me (5), (η5-2,3-Me2C8H4NAlCl2Me)TiCl2Me (6), and (η5-3,4,5,6-C12H12NAlCl2Me)TiClMe2 (7), obtained by treatment of TiCl4 with equimolar amounts of trimethyl aluminum and the corresponding pyrrole ligands, were found to be moderately active for ethylene polymerization with MAO or [Ph3C]+[B(C6F5)4]−, in all cases producing UHMWPE. The NMR scale reaction of 5 with B(C6F5)3 showed the formation of a solvent-separated ion pair, formed by the abstraction by B(C6F5)3 of a methyl group from Al-CH3 rather than from Ti-CH3. 1H and 13C NMR analysis of 6 and 7 revealed that several stable structural isomers exist in solution, with a slow interconversion on the NMR time scale. The dimeric zirconium complexes [(η5,κ1-2,5-Me2C4H2NAlClMe2)ZrMeCl(μ-Cl)]2 (8) and [(η5,κ1-2,5-Me2C4H2NAlCl2Et)ZrCl2(μ-Cl)]2 (9), prepared by the reaction of 2 equivalents of ZrCl4, 1 equivalent of 2,5-dimethylpyrrole, and 1 equivalent of aluminum alkyl, showed structures containing a bridging chloride between aluminum and zirconium, thereby giving structural evidence for the lack of catalytic behavior of these complexes. A possible explanation for the moderate or absence of catalytic activity of the aluminum-pyrrolyl complexes was proposed based on DFT calculations.
Co-reporter:Miloud Bouyahyi, Mark P. F. Pepels, Andreas Heise, and Rob Duchateau
Macromolecules 2012 Volume 45(Issue 8) pp:3356-3366
Publication Date(Web):April 9, 2012
DOI:10.1021/ma3001675
The catalytic behavior of several inexpensive and simple N-heterocyclic organic catalysts in ring-opening polymerization (ROP) of ω-pentadecalactone (PDL) and ε-caprolactone (CL) has been studied. The polymerization reactions, carried out in bulk monomer and in toluene solution at 100 °C, identified 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) in combination with benzyl alcohol (BnOH) as initiator as the only active catalyst for the ring-opening polymerization of PDL and for the copolymerization of PDL and CL. The guanidine N-methyl-TBD (MTBD), 1,2,3-triisopropylguanidine, the amidine 1,8-diazabicycloundec-7-ene (DBU), and other N-heterocyclic organic catalysts such as dialkylaminopyridine (DMAP), imidazole, indoles, and N-heterocyclic carbenes (NHC’s) tested in this study proved to be inactive in the ROP of PDL even for the long reaction times. The polymerization mechanism, kinetic studies, temperature, and monomer concentration effects were investigated both in solution and in bulk monomer. The pseudoliving character of the TBD/BnOH system has been proven by kinetic studies in both toluene solution and bulk monomer. By varying the experimental conditions and the monomer feed composition, highly crystalline poly(PDL-co-CL) random copolymers of various compositions have been prepared using the binary system TBD/ROH as catalyst/initiators. Thermal analysis and 13C NMR spectroscopy show a linear relation of the variation of the random copolymers melting temperatures as a function of comonomer content.
Co-reporter:Camille Descour, Tamara Meijer-Vissers, Tibor Macko, Matthew Parkinson, Dario Cavallo, Martin van Drongelen, Gerhard Hubner, Han Goossens, Robbert Duchateau
Polymer 2012 Volume 53(Issue 18) pp:4018
Publication Date(Web):17 August 2012
DOI:10.1016/j.polymer.2012.06.046
Co-reporter:Yacoob Shaikh;Dr. Khalid Albahily;Matthew Sutcliffe;Valeria Fomitcheva;Dr. Sro Gambarotta;Dr. Ilia Korobkov;Dr. Robbert Duchateau
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:1366-1369
Publication Date(Web):
DOI:10.1002/anie.201106517
Co-reporter:Camille Descour, Rob Duchateau, Mamoeletsi R. Mosia, Gert-Jan M. Gruter, John R. Severn and Sanjay Rastogi
Polymer Chemistry 2011 vol. 2(Issue 10) pp:2261-2272
Publication Date(Web):15 Aug 2011
DOI:10.1039/C1PY00257K
1-Pentene and 4-methyl-1-pentene (4M1P) have been polymerised using several C2-symmetric ansa-zirconocene catalysts rac-X(2-R1,4-R2-Ind)2ZrCl2 [X = C2H4, R1 = R2 = H (1); X = SiMe2, R1 = R2 = H (2); X = SiMe2, R1 = Me, R2 = H (3); X = SiMe2, R1 = H, R2 = Ph (4); X = SiMe2, R1 = Me, R2 = Ph (5)] with MAO as cocatalyst. The effects of polymerisation conditions as well as substituents on the indenyl ligand were studied. Except for the poly-1-pentenes synthesized with 3 and 5 at low temperatures, low molecular weight isotactic polymers were generally obtained. Compared to their behaviour in propylene polymerisation, the relative activity and selectivity of catalysts 1–5 are considerably different for 1-pentene and 4M1P polymerisation. Of the five catalysts, 1 and 4 showed the highest activities for both 1-pentene and 4M1P polymerisation, while 5 resulted in the lowest activities, especially for 4M1P polymerisation. Subsequently, a Cs- and several C1-symmetric zirconocenes, (R1)2C(3-R2-Cp)(2,7,-R3-Flu)ZrCl2 (R1 = Me, R2 = R3 = H (6); R1 = R2 = Me, R3 = H (7); R1 = Me, R2 = t-Bu, R3 = H (8); R1 = Me, R2 = R3 = t-Bu (9); R1 = Ph, R2 = t-Bu, R3 = H (10); R1 = Ph, R2 = R3 = t-Bu (11), were tested in 1-pentene and 4M1P polymerisation with MAO as cocatalyst. The effect of substituents on the bridge, the cyclopentadienyl (Cp) and fluorenyl (Flu) ligand, was studied relative to the polymerisation temperature and type of monomer. The molecular weights of the polymers were considerably higher than those of the poly-1-pentenes and P4M1Ps obtained with C2-symmetric zirconocenes (1–5). The catalytic activities and polymer molecular weights strongly depend on the fluorenyl substituent and the bridge, while the type of substituent on the Cp ligand has a strong influence on the tacticity of the polymers.
Co-reporter:Khalid Albahily, Zeeshan Ahmed, Sandro Gambarotta, Ece Koç, Robbert Duchateau, and Ilia Korobkov
Organometallics 2011 Volume 30(Issue 21) pp:6022-6027
Publication Date(Web):October 13, 2011
DOI:10.1021/om2008474
The reactions of the deprotonated form of cis-{[(μ-N)(t-Bu)]2PN(H)(o-OMeC6H4)]2} (a) with either CrCl2(THF)2 or CrCl3(THF)3 afforded the corresponding dimeric Cr(II) and monomeric Cr(III) complexes {cis-[(μ-N)(t-Bu)]2[PN(o-MeOC6H4)]2Cr}2 (1a) and cis-[(μ-N)(t-Bu)]2[PN-2-MeOC6H4]2CrCl (2a). By replacing the ligand’s o-OMeC6H4 groups with less-crowded CH2CH2N(i-Pr)2 functionalities and reacting the deprotonated form of cis-{[(μ-N)(t-Bu)]2[PN(H)CH2CH2N(i-Pr)2)]2} (b) with CrCl2(THF)2, the dimetallic, divalent {cis-[(μ-N)(t-Bu)]2[PNCH2CH2N(i-Pr)2)]2Cr}2 (1b) was obtained in crystalline form. Upon activation with MAO, both 1a and 1b afforded highly active, polyethylene-free ethylene oligomerization catalysts. Complex 2a is instead catalytically inactive.
Co-reporter:Inge van der Meulen, Erik Gubbels, Saskia Huijser, Rafaël Sablong, Cor E. Koning, Andreas Heise, and Rob Duchateau
Macromolecules 2011 Volume 44(Issue 11) pp:4301-4305
Publication Date(Web):May 6, 2011
DOI:10.1021/ma200685u
The catalytic ring-opening polymerization of macrolactones to polyethylene-like polyesters was investigated using aluminum–salen complexes as the initiators. Contrary to the common understanding that high molecular weights in these reactions can only be achieved by enzymatic ring-opening polymerization due to the absence of ring tension in macrolactones, the aluminum–salen complexes produces poly(pentadecalactone)s with number-average molecular weights (Mn) of over 150 000 g/mol. Moreover, the same catalyst is also active in catalyzing the ROP of small and medium size lactones, which makes these aluminum–salen complexes highly potential catalysts for the cROP of lactones irrespective of ring size. These results show that it is possible to polymerize macrolactones to high molecular weight polyethylene-like polymers using cheap and robust metal-based catalysts. Even the so-called medium-sized lactones (ring size: 9–12) can be polymerized with a reasonably good activity to high molecular weight products, which is truly exceptional. These results complement the common theory of ring-tension-driven cROP.
Co-reporter:Dr. Sebastiano Licciulli;Khalid Albahily;Valeria Fomitcheva;Dr. Ilia Korobkov;Dr. Sro Gambarotta;Dr. Robbert Duchateau
Angewandte Chemie International Edition 2011 Volume 50( Issue 10) pp:2346-2349
Publication Date(Web):
DOI:10.1002/anie.201006953
Co-reporter:Saskia Huijser, Elham HosseiniNejad, Rafaël Sablong, Chris de Jong, Cor E. Koning, and Rob Duchateau
Macromolecules 2011 Volume 44(Issue 5) pp:1132-1139
Publication Date(Web):February 3, 2011
DOI:10.1021/ma102238u
Copolymerization of cyclohexene oxide (CHO) with alicyclic anhydrides applying chromium tetraphenylprophyrinato (TPPCrCl, 1) and salophen (SalophenCrCl, 2) catalysts resulted in polyesters or poly(ester-co-ether)s, depending on the nature of the catalyst, presence of a cocatalyst, solvent and type of anhydride. The combination of 1 as catalyst and 4-N,N-dimethylamino-pyridine (DMAP) as cocatalyst in the copolymerization of CHO with succinic anhydride (SA), cyclopropane-1,2-dicarboxylic acid anhydride (CPrA), cyclopentane-1,2-dicarboxylic acid anhydride (CPA) or phthalic anhydride (PA) invariably resulted in a completely alternating topology and therefore a pure polyester. Contrarily, 2 in combination with DMAP did not afford pure polyesters for the copolymerization of CHO with SA or CPrA but did render the alternating topology when CPA or PA was used as anhydride comonomer. Water proved to be an efficient bifunctional CTA affording α,ω-hydroxyl-terminated polyesters without loss of catalytic activity. When CO2 was introduced as additional monomer to CHO and the anhydrides, both 1 and 2 in combination with DMAP as cocatalyst afforded perfect poly(ester-co-carbonate)s. The presence of CO2 effectively prevents the undesirable side reaction of oxirane homopolymerization.
Co-reporter:Alexander F. R. Kilpatrick, Shaneesh Vadake Kulangara, Michael G. Cushion, Robbert Duchateau and Philip Mountford
Dalton Transactions 2010 vol. 39(Issue 15) pp:3653-3664
Publication Date(Web):05 Mar 2010
DOI:10.1039/B926333K
Reaction of (Me2pz)2CHSiMe2N(H)R (R = iPr or Ph) or (Me2pz)2CHSiMe2NMe2 with CrCl3(THF)3 or CrCl2(THF)2 gave Cr{(Me2pz)2CHSiMe2NR1R2}Cl3 (R1 = H, R2 = iPr (10) or Ph (11); R1 = R2 = Me (15)) or Cr{(Me2pz)2CHSiMe2NR1R2}Cl2(THF) (R1 = H, R2 = iPr (12) or Ph (13); R1 = R2 = Me (16)), respectively. Compounds 10 and 11 were crystallographically characterized and the magnetic behaviour of all the new compounds was evaluated using SQUID magnetometry. Reaction of CrCl3(THF)3 with Li{C(Me2pz)3}(THF) gave the zwitterionic complex Cr{C(Me2pz)3}Cl2(THF) (17) containing an apical carbanion. Reaction of the analogous phenol-based ligand (Me2pz)2CHArOH (ArO = 2-O-3,5-C6H2tBu2) with CrCl3(THF)3 gave Cr{(Me2pz)2CHArOH}Cl3 (19) whereas the corresponding reaction with CrCl2(THF)2 unexpectedly gave the Cr(III) phenolate derivative Cr{(Me2pz)2CHArO}Cl2(THF) (20) which could also be prepared from CrCl3(THF)3 and the sodiated ligand [Na{(Me2pz)2CHArO}(THF)]2. Reaction of the corresponding ether (Me2pz)2CHArOMe with CrCl3(THF)3 or CrCl2(THF)2 gave Cr{(Me2pz)2CHArOMe}Cl3 (23) and Cr{(Me2pz)2CHArOMe}Cl2(THF) (24), respectively. The catalytic performance in ethylene oligomerisation/polymerisation of all of the new Cr(II) and Cr(III) complexes was evaluated. Most of the complexes showed high activity, but produced a Schultz-Flory distribution of α-olefins. Compound 23 had an exceptionally low α-value of 0.37 and showed a preference for 1-hexene and 1-octene formation. While replacing a secondary amine (10-13) for a tertiary amine (15-16) resulted in loss of catalytic activity, replacing a phenol (19) for an anisole (23) group afforded a more selective and more active catalyst. Changing from MAO to DIBAL-O as cocatalyst induced a switch in selectivity to ethylene polymerisation.
Co-reporter:Dr. Sebastiano Licciulli;Indira Thapa;Khalid Albahily;Dr. Ilia Korobkov;Dr. Sro Gambarotta;Dr. Robbert Duchateau;Dr. Reynald Chevalier;Dr. Katrin Schuhen
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:9225-9228
Publication Date(Web):
DOI:10.1002/anie.201003465
Co-reporter:Hellen E. Dyer, Saskia Huijser, Nicolas Susperregui, Fanny Bonnet, Andrew D. Schwarz, Robbert Duchateau, Laurent Maron and Philip Mountford
Organometallics 2010 Volume 29(Issue 16) pp:3602-3621
Publication Date(Web):July 20, 2010
DOI:10.1021/om100513j
The synthesis and ring-opening polymerization (ROP) capability of bis(phenolate)amine-supported samarium borohydride and amide complexes are reported, together with a DFT study. Reaction of Na2O2NL (L = OMe, NMe2, py, or Pr) with Sm(BH4)3(THF)3 gave the borohydride complexes Sm(O2NL)(BH4)(THF) (L = OMe (2), NMe2 (3), or py (4)) or Sm(O2NPr)(BH4)(THF)2 (5). Compounds 4 and 5 lost THF in vacuo, forming phenolate O-bridged dimers 1 and 6, respectively. Reaction of H2O2NL with Sm{N(SiHMe2)2}3(THF)2 formed monomeric Sm(O2NL){N(SiHMe2)2}(THF) (L = OMe (7), NMe2 (8), or py (9)) with tetradentate O2NL ligands, but dimeric Sm2(μ-O2NPr)2(O2NPr)(THF) (10) with tridentate O2NPr. Reaction of Sm{N(SiMe3)2}3 with H2O2NL (L = OMe or NMe2) led to zwitterionic products Sm(O2NL)(HO2NL). The bulkier amide compounds Sm(O2NL){N(SiMe3)2}(OEt2)n (n = 1, L = OMe (12) or py (13); n = 0, L = NMe2 (14)) were prepared by reaction of Sm(O2NL)(BH4)(THF) with KN(SiMe3)2. The X-ray structures of 2, 5, 6, 7, 10, 13, and 14 were determined. The borohydrides 2−5 were very efficient initiators for the ROP of ε-CL, giving linear dihydroxytelechelic poly(ε-CL). Selected amide initiators were also assessed but gave poorer control, as judged by broad PDI (Mw/Mn) values and significant amounts of cyclic poly(ε-CL)s. Of the borohydrides, only 2−4 were active for the ROP of rac-LA, and activity increased in the order O2NL = O2NOMe ≈ O2Npy < O2NNMe2. The latter ligand also gave the best control of the ROP, as judged by the PDIs and Mn values. All gave heterotactically enriched poly(rac-LA) with Pr values in the range 0.82−0.84. The ROP of rac-LA with the amides 7, 9, and 12 was faster but much less well controlled. Overall, the borohydride initiators were superior for the ROP of both ε-CL and rac-LA when compared to otherwise identical amide initiators. MALDI-ToF MS analysis of the poly(rac-LA) formed with 3 showed both −CH(Me)CHO and −CH(Me)CH2OH end groups originating from the insertion of the first LA monomer into the Sm−BH4 moiety of 3. In contrast, 2 and 4 formed only α,ω-dihydroxy-terminated polyesters with −CH(Me)CH2OH and −CH(Me)OH end groups. DFT calculations on Eu(O2′NNMe2)(BH4) found two mechanisms for the initial ring-opening step of LA by the borohydride group, giving pathways leading to either aldehyde- or alcohol-terminated poly(lactide)s. Of these two pathways, the one giving α,ω-dihydroxy-terminated polymers was the most favored, in agreement with experiment. (Ligand abbreviations: O2NL = RCH2N(CH2-2-O-3,5-C6H2tBu2)2 where R = CH2OMe, CH2NMe2, py, or Et for L = OMe, NMe2, py, or Pr, respectively; O2′NNMe2 = Me2NCH2CH2N(CH2-2-O-C6H4)2.)
Co-reporter:Jan Devroede;Cor E. Koning;Jan Meuldijk
Journal of Applied Polymer Science 2009 Volume 114( Issue 4) pp:2435-2444
Publication Date(Web):
DOI:10.1002/app.30782
Abstract
An in depth study is performed on the origin of and influences on the formation of tetrahydrofuran (THF) during the first stage of the terephthalic acid (TPA) based synthesis of poly(butylene terephthalate) (PBT). Although many improvements on the synthesis process of PBT have been reported in literature to suppress this undesired side reaction, only few studies reported on the actual mechanism of the THF formation, which is not completely understood. Low molecular weight compounds have been used to model the side reactions occurring during the polymerization reaction. It could be concluded that, in contrast to previous reportage, only the THF formation from the monomer, 1,4-butanediol, is directly influenced by the use of TPA as a starting material for the production of PBT. © 2009 Wiley Periodicals, Inc. J Appl Polym Sci, 2009
Co-reporter:Jan Devroede;Cor E. Koning
Journal of Applied Polymer Science 2009 Volume 114( Issue 4) pp:2427-2434
Publication Date(Web):
DOI:10.1002/app.30781
Abstract
The production of poly(butylene terephthalate) (PBT) struggles with the formation of substantial amounts of tetrahydrofuran (THF). When PBT is synthesized from terephthalic acid (TPA) instead of dimethyl terephthalate (DMT), even more THF is formed, mainly during the first stage of the melt polymerization process. Although a lot of literature reports on the existence of this side reaction in both processes, to the best of our knowledge, a comparison, which reveals the importance of the acidity and insolubility of TPA on the THF formation, was never described. Finally, an interesting study was performed on the THF formation during the synthesis of PBT from mixtures of DMT and TPA as well as from the completely soluble monomethyl terephthalate (MMT). © 2009 Wiley Periodicals, Inc. J Appl Polym Sci, 2009
Co-reporter:Amir Jabri;ChrisB. Mason;Yan Sim;Sro Gambarotta Dr.;TaraJ. Burchell Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 50) pp:9717-9721
Publication Date(Web):
DOI:10.1002/anie.200803434
Co-reporter:Khalid Albahily;Ece Koç;Danya Al-Baldawi;Didier Savard;Sro Gambarotta Dr.;TaraJ. Burchell Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 31) pp:5816-5819
Publication Date(Web):
DOI:10.1002/anie.200800845
Co-reporter:Rafaël Sablong, Robbert Duchateau, Cor E. Koning, Gert de Wit, Daan van Es, Roelof Koelewijn and Jacco van Haveren
Biomacromolecules 2008 Volume 9(Issue 11) pp:
Publication Date(Web):October 7, 2008
DOI:10.1021/bm800627d
The biomass-based monomer isosorbide was incorporated into poly(butylene terephthalate) (PBT) by solid-state polymerization (SSP) using the macrodiol monomer BTITB-(OH)2, which consists of isosorbide (I), terephthalic acid (T), and 1,4-butandiol (B) residues. This macromonomer can be synthesized by a simple one-pot, two-step reaction. Polymers with number-average molecular weights up to 100 000 g·mol−1 were readily synthesized from various ratios of PBT/BTITB-(OH)2. Their molecular weights, thermal properties, and colors were compared with corresponding copolyesters that were obtained by melt polycondensation. We found that Tm, Tc, and especially Tg were superior for materials that were obtained by SSP. This is ascribed to differences in the microstructures of both types of copolyesters; the SSP products exhibit a more blocky structure than do the more random melt-polymerized counterparts. The SSP method resulted in much higher molecular weights and much less colored polymers, and it seems to be the preferred route for incorporating biobased monomers that exhibit limited thermal stability into engineering plastics.
Co-reporter:Adrianus J. M. van Dijk Dr. Dr.;Emiel J. M. Hensen Dr.;Jan Meuldijk Dr.;Cor E. Koning Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 27) pp:
Publication Date(Web):26 JUN 2007
DOI:10.1002/chem.200601898
To test the potential of heterogeneous catalysts for the nylon-6 synthesis from 6-aminocapronitrile, a number of zeolites, aluminum silicate, and metal oxides were tested as catalysts for the model reaction of pentanenitrile with water and hexylamine to N-hexylpentanamide. All zeolitic and aluminum silicate systems showed an insufficient performance, while the metal oxides (TiO2, ZrO2, Nb2O5) showed very promising results. The kinetic behavior of the metal oxides was further investigated. First the nitrile was catalytically hydrolyzed to the terminal amide and subsequently the amidation of the hexylamine occurred. To polymerize 6-aminocapronitrile into nylon-6, more than 99 % nitrile conversion was required to obtain a high-molecular-weight polymer. Pentanenitrile conversions larger than 99 % can be obtained within six hours, at 230 °C, by using ZrO2 as the catalyst. A kinetic study (by using IR spectroscopy) on the behavior of the metal oxides demonstrated that the adsorbed nitrile was catalytically hydrolyzed at the surface, but remained tightly bound to the surface. Zirconia-catalyzed polymerizations of 6-amino-capronitrile demonstrated that high-molecular-weight nylon-6 is feasible by using this route.
Co-reporter:Adrianus J. M. van Dijk Dr.;Tom Heyligen Dr.;Jan Meuldijk Dr.;Cor E. Koning
Chemistry - A European Journal 2007 Volume 13(Issue 27) pp:
Publication Date(Web):26 JUN 2007
DOI:10.1002/chem.200601897
The synthesis of N-hexylpentanamide from a stoichiometric amount of pentanenitrile and hexylamine has been studied as a model reaction for the synthesis of nylon-6 from 6-aminocapronitrile. The reaction was carried out under mild hydrothermal conditions and in the presence of a homogeneous ruthenium catalyst. For the mild hydrothermal conditions the presence of hexylamine distinctively increases the nitrile hydrolysis compared to the nitrile hydrolysis in the absence of hexylamine. Amine-catalyzed nitrile hydrolysis mainly produces the N-substituted amide. A clear product development is observed, consisting of first the terminal amide formation and second the accumulation of N-hexylpentanamide. With a maximum conversion of only 80 % after 18 h, the nitrile hydrolysis rate at 230 °C is still much too low for nylon-6 synthesis. Ruthenium dihydride phosphine was therefore used as a homogeneous catalyst, which significantly increases the nitrile hydrolysis rate. At a temperature of 140 °C and with only 0.5 mol % [RuH2(PPh3)4] a 60 % nitrile conversion is already reached within 2 h. Initially the terminal amide is the sole product, which is gradually converted into N-hexylpentanamide. The reaction has a high initial rate, however, for higher conversions a strong decrease in hydrolysis rate is observed. This is ascribed to product inhibition, which results from the equilibrium nature of the reaction.
Co-reporter:Amir Jabri;Ilia Korobkov;Sro Gambarotta Dr. and Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 32) pp:
Publication Date(Web):12 JUL 2007
DOI:10.1002/anie.200701703
On its own: A vanadium(II) bis(pyrrolide) complex with a vanadocene-type structure is a single-component catalyst for the preparation of ultrahigh molecular weight polyethylene (PE; see scheme; Mw=weight-average molecular weight, PDI=polydispersity index).
Co-reporter:Amir Jabri;Ilia Korobkov;Sro Gambarotta Dr. and Dr.
Angewandte Chemie 2007 Volume 119(Issue 32) pp:
Publication Date(Web):12 JUL 2007
DOI:10.1002/ange.200701703
Für sich allein: Ein Vanadium(II)-Bis(pyrrolid)-Komplex mit Vanadocen-Struktur wirkt als Einkomponentenkatalysator bei der Herstellung von ultrahochmolekularem Polyethylen (PE; siehe Schema; Mw=gewichtsgemitteltes Molekulargewicht, PDI=Polydispersitätsindex).
Co-reporter:Saskia Huijser;Bastiaan B. P. Staal;Juan Huang Dr.;Cor E. Koning
Angewandte Chemie International Edition 2006 Volume 45(Issue 25) pp:
Publication Date(Web):26 MAY 2006
DOI:10.1002/anie.200600594
Software developed in house in combination with a recently developed method of MALDI-TOF MS enable not only the elucidation of individual chain structures of poly(lactide-co-glycolide), a polymer used extensively in the medical sector, but a full characterization of this copolymer including even its chemical composition and topology (random, gradient, block (see figure), or alternating).
Co-reporter:Claire Temple Dr.;Amir Jabri;Patrick Crewdson;Sro Gambarotta Dr.;Ilia Korobkov Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 42) pp:
Publication Date(Web):28 SEP 2006
DOI:10.1002/anie.200602240
An unprecedented oxidation of chromium(II) by trimethylaluminum which, despite the reducing environment, forms a cationic organochromium(III) complex (see picture; SNS=RSCH2CH2N(H)CH2CH2SR, Cy=cyclohexyl, X=[Me6Al2Cl]), provides a possible catalyst re-activation pathway.
Co-reporter:Claire Temple Dr.;Amir Jabri;Patrick Crewdson;Sro Gambarotta Dr.;Ilia Korobkov Dr.
Angewandte Chemie 2006 Volume 118(Issue 42) pp:
Publication Date(Web):28 SEP 2006
DOI:10.1002/ange.200602240
Wieder aktiv werden! Die erstmals beobachtete Oxidation von Chrom(II) durch Trimethylaluminium führt ungeachtet der reduzierenden Umgebung zu einem kationischen Organochrom(III)-Komplex (siehe Bild; SNS=CySCH2CH2N(H)CH2CH2SCy, Cy=Cyclohexyl, X=[Me6Al2Cl]). Diese Reaktion bietet einen möglichen Weg zur Reaktivierung des Katalysators.
Co-reporter:Robbert Duchateau, Tessa W. Dijkstra, John R. Severn, Rutger A. van Santen and Ilia V. Korobkov
Dalton Transactions 2004 (Issue 17) pp:2677-2682
Publication Date(Web):09 Aug 2004
DOI:10.1039/B407471H
Tin silicate species have shown good catalytic activity in various oxidation reactions. In an attempt to mimic surface tin species, several tin containing silsesquioxanes have been synthesized. Incompletely condensed silsesquioxanes (
c-C
5H
9)
7Si
7O
9(OH)
3 and (
c-C
5H
9)
7Si
7O
9(OSiMe
3)(OH)
2 were reacted with common tin-precursors, which afforded several silsesquioxane ligated tin compounds. Divalent stannasilsesquioxanes form dimers of the type [(
c-C
5H
9)
7Si
7O
11(OX)Sn]
2
(X = H, SiMe
3) with three-coordinated tin centers. The three-coordinated tin(
II) are hydrolytically unstable whereas the octahedrally surrounded tetravalent stannasilsesquioxanes [(
c-C
5H
9)
7Si
7O
11(OX)]Sn(acac)
2
(X = H, OSiMe
3) are hydrolytically robust. An unprecedented anionic trimeric cluster, {[(
c-C
5H
9)
7Si
7O
12Sn]
3(μ
2-OH)
3(μ
3-OH)}
−{HNEt
3}
+, stabilized by bridging hydroxyl groups was formed when the product formed upon reacting (
c-C
5H
9)
7Si
7O
9(OH)
3 with SnCl
4 was slowly hydrolyzed. The stannasilsesquioxanes showed no catalytic activity in oxidation reactions.
Co-reporter:Robbert Duchateau Dr.;Tessa W. Dijkstra Dr.;Rutger A. van Santen Dr.;Glenn P. A. Yap Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 16) pp:
Publication Date(Web):28 JUN 2004
DOI:10.1002/chem.200400206
Incompletely condensed silsesquioxanes of the type R7Si7O9(O{SiR′2O}n)OH (R = c-C5H9, c-C6H11; R′ = Me, Ph; n = 1–4), containing a siloxane ring of variable size and rigidity and a remaining silanol, are described. Compared with a truly isolated silanol [R7Si8O12(OH)], solution and solid state FT-IR spectra of these compounds show a OH shift of approximately 150 cm−1 to lower frequency, which suggests hydrogen bonding of the silanol with the internal siloxane ring. In agreement with this, the relative ion pair acidities of the silanols in THF, determined by UV/Vis, were lowered by 0.8–1.2 compared with a truly isolated silanol. Density functional theory (DFT) calculations on these systems confirm the presence of intramolecular hydrogen bonding. Possible interaction of the silyl ether functionalities with Lewis acidic metal sites was studied for the neutral gallium-substituted systems and cationic titanium silsesquioxane complexes, models for an immobilized titanium olefin polymerization catalyst. The electron donating capability of the siloxide functionalities in 1, 6, and 7 is not sufficient to satisfy the electron deficiency of the corresponding gallium silsesquioxane species, which form dimeric structures with a bridging siloxide unit rather than Lewis base adducts with coordinated siloxide functionalities. Metallation of 1 and 4 with [Cp′′Ti(CH2Ph)3] (Cp′′ = η5-1,3-C5H3(SiMe3)2) in a 1:1 ratio afforded monomeric titanasilsesquioxanes. To probe the effect of the neighboring siloxane ring on the highly Lewis acidic titanium center, the catalytic activities of the corresponding cationic half-sandwich complexes were tested in 1-hexene polymerization. Compared with the catalyst system based on the isolated silanol [(c-C5H9)7Si8O12OH], the presence of a neighboring siloxane ring causes considerable retardation of the polymerization process but also improves the stability of the catalyst.
Co-reporter:Robbert Duchateau;Roelant J. Harmsen;Hendrikus C. L. Abbenhuis;Rutger A. van Santen;Auke Meetsma;Sven K.-H. Thiele;Mirko Kranenburg
Chemistry - A European Journal 1999 Volume 5(Issue 11) pp:
Publication Date(Web):29 OCT 1999
DOI:10.1002/(SICI)1521-3765(19991105)5:11<3130::AID-CHEM3130>3.0.CO;2-Z
Lewis and Brønsted acidic aluminosilsesquioxanes have been synthesized and utilized as homogeneous models for the corresponding acidic aluminum sites found in aluminosilicates and zeolites.
Co-reporter:Alexander F. R. Kilpatrick, Shaneesh Vadake Kulangara, Michael G. Cushion, Robbert Duchateau and Philip Mountford
Dalton Transactions 2010 - vol. 39(Issue 15) pp:NaN3664-3664
Publication Date(Web):2010/03/05
DOI:10.1039/B926333K
Reaction of (Me2pz)2CHSiMe2N(H)R (R = iPr or Ph) or (Me2pz)2CHSiMe2NMe2 with CrCl3(THF)3 or CrCl2(THF)2 gave Cr{(Me2pz)2CHSiMe2NR1R2}Cl3 (R1 = H, R2 = iPr (10) or Ph (11); R1 = R2 = Me (15)) or Cr{(Me2pz)2CHSiMe2NR1R2}Cl2(THF) (R1 = H, R2 = iPr (12) or Ph (13); R1 = R2 = Me (16)), respectively. Compounds 10 and 11 were crystallographically characterized and the magnetic behaviour of all the new compounds was evaluated using SQUID magnetometry. Reaction of CrCl3(THF)3 with Li{C(Me2pz)3}(THF) gave the zwitterionic complex Cr{C(Me2pz)3}Cl2(THF) (17) containing an apical carbanion. Reaction of the analogous phenol-based ligand (Me2pz)2CHArOH (ArO = 2-O-3,5-C6H2tBu2) with CrCl3(THF)3 gave Cr{(Me2pz)2CHArOH}Cl3 (19) whereas the corresponding reaction with CrCl2(THF)2 unexpectedly gave the Cr(III) phenolate derivative Cr{(Me2pz)2CHArO}Cl2(THF) (20) which could also be prepared from CrCl3(THF)3 and the sodiated ligand [Na{(Me2pz)2CHArO}(THF)]2. Reaction of the corresponding ether (Me2pz)2CHArOMe with CrCl3(THF)3 or CrCl2(THF)2 gave Cr{(Me2pz)2CHArOMe}Cl3 (23) and Cr{(Me2pz)2CHArOMe}Cl2(THF) (24), respectively. The catalytic performance in ethylene oligomerisation/polymerisation of all of the new Cr(II) and Cr(III) complexes was evaluated. Most of the complexes showed high activity, but produced a Schultz-Flory distribution of α-olefins. Compound 23 had an exceptionally low α-value of 0.37 and showed a preference for 1-hexene and 1-octene formation. While replacing a secondary amine (10-13) for a tertiary amine (15-16) resulted in loss of catalytic activity, replacing a phenol (19) for an anisole (23) group afforded a more selective and more active catalyst. Changing from MAO to DIBAL-O as cocatalyst induced a switch in selectivity to ethylene polymerisation.

![Poly[oxy(1-butyl-6-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/159400/159350-72-8.png)
![Poly[oxy(1-butyl-6-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/159400/159350-72-8_b.png)