Co-reporter:Yong Pan and Weihua Zhu
The Journal of Physical Chemistry A November 30, 2017 Volume 121(Issue 47) pp:9163-9163
Publication Date(Web):November 9, 2017
DOI:10.1021/acs.jpca.7b10462
We designed four bicyclic nitramines and three cage nitramines by incorporating −N(NO2)–CH2–N(NO2)–, −N(NO2)–, and −O– linkages based on the HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane) framework. Then, their electronic structure, heats of formation, energetic properties, strain energy, thermal stability, and impact sensitivity were systematically studied using density functional theory (DFT). Compared to the parent compound HMX, all the title compounds have much higher density, better detonation properties, and better oxygen balance. Among them, four compounds have extraordinary high detonation properties (D > 9.70 km/s and P > 44.30 GPa). Moreover, most of the title compounds exhibit better thermal stability and lower impact sensitivity than CL-20 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane) or HNHAA (hexanitrohexaazaadamantane). Thus, all of the seven new nitramine compounds are promising candidates for high energy density compounds. In particular, five compounds exhibit a best combination of better oxygen balance, good thermal stability, excellent detonation properties superior to or comparable to CL-20 or HNHAA, and lower impact sensitivity than CL-20 or HNHAA. The results indicate that our unusual design strategy that constructing bicyclic or cage nitramines based on the HMX framework by incorporating the intramolecular linkages is very useful for developing novel energetic compounds with excellent detonation performance and low sensitivity.
Co-reporter:Dong Xiang, Weihua Zhu, Heming Xiao
Fuel 2017 Volume 202(Volume 202) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.fuel.2017.04.043
•The decomposition of DBTD is triggered by the N–H bond breaking to release H radical.•The initiation mechanisms are independent on both the temperature and pressure.•Subsequent decompositions were very sensitive to high temperatures.•The pressure could decelerate the decompositions of DBTD.We investigated the initiation mechanisms and subsequent decompositions of energetic ionic crystal dihydrazinium 3,3′-dinitro-5,5′-bis-1,2,4-triazole-1,1-diolate (DBTD) at pure high temperatures (3842 K and 2000 K) and at high temperatures coupled with detonation pressure (34.2 GPa) by ab initio molecular dynamics simulations. The decomposition of DBTD is triggered by the N–H bond breaking to release H radical. The initiation mechanisms are independent on both the temperature and pressure. Subsequent decompositions were very sensitive to high temperatures, and moreover, the temperature becomes the foremost factor affecting the decomposition. However, the product formation mechanisms indicate that the pressure could decelerate the decompositions. The formation mechanisms of H2O, N2, polynitrogen fragments, heterocyclic clusters, and long chains were investigated. Our study may provide new insights into the initiation mechanisms and subsequent decomposition of ionic salt explosives at extreme conditions.
Co-reporter:Dong Xiang
RSC Advances (2011-Present) 2017 vol. 7(Issue 14) pp:8347-8356
Publication Date(Web):2017/01/23
DOI:10.1039/C6RA27255J
We performed ab initio molecular dynamics simulations to study the initiation chemical reaction and subsequent decomposition mechanism of a 4,10-dinitro-2,6,8,12-tetraoxa-4,10-diazaisowurtzitane (TEX) crystal at 2160 K. It was found that there are three different initial reactions involved in the decomposition of an isolated TEX molecule. The activation energy barriers for the initial decomposition reactions indicate that among the three initial reaction paths, cleavage of the nitrogen–nitro bond is the easiest path to be triggered. The decomposition of the TEX crystal is triggered by the unimolecular C–H bond breaking to form a hydrogen radical. The generated H radicals can prompt other unreacted TEX molecules to decompose. Moreover, there are many multimolecular reactions during the decomposition of the TEX crystal. Overall, after the decomposition of TEX was triggered, some long chains and complicated carbon-rich heterocyclic rings were formed, and then they split to form small fragments. This study may provide useful information to understand the decomposition mechanism of cage explosives and develop new high-energy explosives.
Co-reporter:Zhichao Liu;Yu Liu;Jinshan Li
RSC Advances (2011-Present) 2017 vol. 7(Issue 87) pp:55482-55488
Publication Date(Web):2017/12/01
DOI:10.1039/C7RA10043D
We report the most stable packings of five HMX/solvent supramolecular assemblies. A series of 1 : 1 supramolecular synthons of both α-form and β-form HMX conformers with solvent molecules were investigated. Both α-form and β-form HMX conformers have similar stability when combining with solvent molecules into supramolecular synthons. AIM analysis was performed to evaluate the properties of the intermolecular hydrogen bonds between the HMX and solvent molecules. The most stable polymorphs among the 10 most common space groups for α-HMX/solvent supramolecular assemblies were predicted and compared with available experimental data.
Co-reporter:Yong Pan, Weihua Zhu, Heming Xiao
Computational and Theoretical Chemistry 2017 Volume 1114(Volume 1114) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.comptc.2017.05.021
•The introduction of the oxygen atom in the cage is not helpful for increasing energetic properties of parent compound CL-20.•All the title compounds present remarkable detonation properties superior to or very close to HMX.•All the title compounds exhibit higher thermal stability than parent compound CL-20.•The introduction of the oxygen atom in the cage effectively decreases the sensitivity of parent compound CL-20.Ten novel azaoxaisowurtzitane cage compounds were designed by introducing the oxygen atoms into the azaisowurtzitane cage to replace the N-NO2 groups. Then, their heats of formation (HOFs), energetic properties, strain energies, thermal stability, and impact sensitivity were studied by using density functional theory. The introduction of the oxygen atom in the cage is not helpful for increasing the HOFs, densities, and energetic properties of parent compound CL-20. But all the title compounds exhibit remarkable detonation properties superior to or very close to HMX. All the azaoxaisowurtzitane cage compounds exhibit higher thermal stability than parent compound CL-20. The introduction of the oxygen atom in the cage effectively decreases the sensitivity of parent compound CL-20. Considered the detonation performance, thermal stability, and impact sensitivity, six compounds can be regarded as the potential candidates of HEDC because these azaoxaisowurtzitane cage compounds not only exhibit excellent energetic properties comparable with CL-20, but also have higher thermal stability and lower sensitivity than CL-20.Download high-res image (132KB)Download full-size image
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 10) pp:7093-7099
Publication Date(Web):05 Feb 2016
DOI:10.1039/C6CP00096G
We performed ab initio molecular dynamics simulations to study the initiation mechanisms and subsequent chemical decomposition reactions of the nitrogen-rich furazan explosive 3,3′-dinitroamino-4,4′-azoxyfurazan (DNAAF) at low temperatures (363–963 K) coupled with different pressures (1–5 GPa). Two different initial decomposition mechanisms which are dependent on the temperature and pressure were found: bimolecular intermolecular hydrogen transfer and unimolecular N–NO2 bond breaking. The subsequent decomposition reactions are sensitive to both the temperature and the pressure. The pressure could accelerate or decelerate the decomposition of DNAAF, while the temperature can change the effect of the pressure on the decomposition. Our study may provide new insights into the initial mechanisms and subsequent decomposition of furazan explosives at low temperatures coupled with different pressures in atomic detail.
Co-reporter:Zhichao Liu, Weihua Zhu, and Heming Xiao
The Journal of Physical Chemistry C 2016 Volume 120(Issue 48) pp:
Publication Date(Web):November 11, 2016
DOI:10.1021/acs.jpcc.6b09795
Surface chemistry plays an prominent part in the behaviors of condensed phase materials and nanoparticles. A combinational strategy based on density-functional theory (DFT) and density-functional tight-binding (DFTB) methods was used to study the surface-induced effect on the energetics, electronic structure, and vibrational properties of a series of β-octatetramethylene tetranitramine nanoparticles (β-HMX NPs). A comparative analysis of the NPs, isolated constituent molecule, and periodic solid-state phase of β-HMX indicates that the NPs possess quite different characteristics from either the constituent molecule or the bulk crystal. The anitsotropy of surface energies, enthalpy of sublimation, and melting point for the NPs are predicted. The surface-induced surface states of the HMX NPs lead to a significant reduction of the energy gap and provide active sites at surfaces. The vibrational properties of the experimentally determined strong modes are compared and discussed among the NPs, gas phase, and solid phase of HMX. The possible role of the surface molecules for the NPs in decreasing the material stability is elucidated. Our results provide basic understandings of the high activity of nanosized energetic materials.
Co-reporter:QIONG WU;DONG XIANG;GUOLIN XIONG;HEMING XIAO
Journal of Chemical Sciences 2016 Volume 128( Issue 5) pp:695-705
Publication Date(Web):2016 May
DOI:10.1007/s12039-016-1068-2
Ab initio molecular dynamics simulations were performed to study the initiation of decomposition and formation of first products of two molecular crystals pentaerythritol tetranitrate (PETN) and 5-nitro-2,4-dihydro-1,2,4-triazole-3-one (NTO) under thermal decomposition temperature (475 K for PETN and 531 K for NTO) coupled with different pressures (1-5 GPa). The pressure effects on the initial decomposition steps and initially generated products on PETN and NTO were very different. PETN was triggered by C-H ⋯O intermolecular hydrogen transfer. The initial decomposition mechanism was independent of the pressure. For NTO, two different initial decomposition mechanisms were found. At 1, 2, and 3 GPa, it was triggered by N-H ⋯O intermolecular hydrogen transfer, while at 4 and 5 GPa, it was triggered by N-H ⋯N intermolecular hydrogen transfer. This indicates that the initial decomposition mechanism was dependent on the pressure. Our study may provide new insights into initial mechanisms and decomposition reactions of molecular crystal explosives under thermal decomposition temperature coupled with different pressures with details at atomic level.
Co-reporter:Qiong Wu, Guolin Xiong, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 35) pp:22823-22831
Publication Date(Web):28 Jul 2015
DOI:10.1039/C5CP03257A
We have performed ab initio molecular dynamics simulations to study coupling effects of temperature (534–873 K) and pressure (1–20 GPa) on the initiation mechanisms and subsequent chemical decompositions of nitramine explosive 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX). A new initiation decomposition mechanism of HMX was found to be the unimolecular C–H bond breaking, and this mechanism was independent of the coupling effects of different temperatures and pressures. The formed hydrogen radicals could promote subsequent decompositions of HMX. Subsequent decompositions were very sensitive to the pressure at low temperatures (534 and 608 K), while the temperature became the foremost factor that affected the decomposition at a high temperature (873 K) instead of the pressure. Our study may provide a new insight into understanding the coupling effects of the temperature and pressure on the initiation decomposition mechanisms of nitramine explosives.
Co-reporter:Zhichao Liu, Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 16) pp:10568-10578
Publication Date(Web):13 Mar 2015
DOI:10.1039/C5CP00637F
Density functional theory with dispersion-correction (DFT-D) was employed to study the effects of vacancy and pressure on the structure and initial decomposition of crystalline 5-nitro-2,4-dihydro-3H-1,2,4-triazol-3-one (β-NTO), a high-energy insensitive explosive. A comparative analysis of the chemical behaviors of NTO in the ideal bulk crystal and vacancy-containing crystals under applied hydrostatic compression was considered. Our calculated formation energy, vacancy interaction energy, electron density difference, and frontier orbitals reveal that the stability of NTO can be effectively manipulated by changing the molecular environment. Bimolecular hydrogen transfer is suggested to be a potential initial chemical reaction in the vacancy-containing NTO solid at 50 GPa, which is prior to the C–NO2 bond dissociation as its initiation decomposition in the gas phase. The vacancy defects introduced into the ideal bulk NTO crystal can produce a localized site, where the initiation decomposition is preferentially accelerated and then promotes further decompositions. Our results may shed some light on the influence of the molecular environments on the initial pathways in molecular explosives.
Co-reporter:Zhichao Liu, Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2015 vol. 5(Issue 43) pp:34216-34225
Publication Date(Web):08 Apr 2015
DOI:10.1039/C5RA01829C
Density functional theory with dispersion corrections (DFT-D) was used to study pressure-induced effects in a novel energetic CL-20:HMX cocrystal and to understand what role its constituents CL-20 and HMX have. The structural, electronic, absorption, and mechanical properties of the cocrystal and its constituents were compared and analyzed in detail. The results indicate that the two constituents produce different effects on the crystal structure of the cocrystal in different directions. This distinct energy distribution in the cocrystal suggests that electron transitions may take place between the HMX and CL-20 molecules. The CL-20 in the cocrystal plays a leading role in the electronic structure of the cocrystal. The cocrystal has quite similar absorption spectra to ε-CL-20 but very different ones from β-HMX. Compared with the pure crystals, the mechanical properties of the cocrystal present a great anisotropy, which not only greatly strengthens the stiffness but also affects the preference of the stiffness towards different directions. Our results may provide fundamental insight into the roles of the two constituents in the cocrystal and may be helpful for developing new cocrystals with high energy and good safety.
Co-reporter:DONG XIANG;QIONG WU;ZHICHAO LIU;HEMING XIAO
Journal of Chemical Sciences 2015 Volume 127( Issue 10) pp:1777-1784
Publication Date(Web):2015 October
DOI:10.1007/s12039-015-0938-3
Periodic density functional theory with dispersion correction (DFT-D) was used to study the structural, electronic, and absorption properties of crystalline 5-nitramino-3, 4-dinitropyrazole (NADNP) under hydrostatic compression of 0-140 GPa. The results indicate that the PBE-G06 is the best functional for studying NADNP. As the pressure increases, the lattice of parameters, band gap, density of states and absorption spectra change regularly except for 126 GPa, where NADNP begins to decompose and form a new bond. An analysis of the band gap and density of states indicates that NADNP becomes more and more sensitive under compression. The absorption spectra show that NADNP has relatively high optical activity with increasing pressure.
Co-reporter:Qiong Wu
The Journal of Physical Chemistry C 2015 Volume 119(Issue 29) pp:16500-16506
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.jpcc.5b05041
We performed ab initio molecular dynamics simulations to investigate the initiation mechanisms and subsequent decompositions of a 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) crystal at initial decomposition temperature coupled with different pressures. The initial decomposition step of TATB was found to be the unimolecular intramolecular hydrogen transfer; moreover, this initiation mechanism is independent of the variation of the pressure. However, the intramolecular hydrogen transfer becomes easier to occur when the pressure increases from 1 to 3 GPa and from 4 to 5 GPa, while the situation is just the opposite with the increment of pressure from 3 to 4 GPa and from 5 to 20 GPa. The pressure accelerates subsequent decomposition of TATB with the increase of pressure from 1 to 3 GPa and from 4 to 5 GPa, while it would decelerate the decomposition when the pressure increases from 3 to 4 GPa and from 5 to 20 GPa. The deceleration of the pressure on the decomposition of TATB may be caused by inhibiting initiation decomposition. Our study may provide new insight into understanding of the initiation mechanisms and decomposition reactions of nitro explosives under decomposition temperature coupled with different pressures in atomic detail.
Co-reporter:Qiong Wu;Heming Xiao
Structural Chemistry 2015 Volume 26( Issue 2) pp:477-484
Publication Date(Web):2015 April
DOI:10.1007/s11224-014-0506-3
Dispersion-corrected DFT calculations have been performed to study the crystal structure, electronic structure, and absorption properties of crystalline 5-nitro-2,4-dihydro-1,2,4-triazole-3-one (NTO) under hydrostatic pressure of 0–160 GPa. Our results show that the lattice parameters b and c are sensitive to van der Waals interactions and the structure is the stiffest in the a direction. At 150 GPa, NTO decomposes by the breaking of N–O bond of nitro group and polymerizes by forming a new N–H covalent bond between one nitrogen atom in the ring and one hydrogen atom linked to the ring in another molecule. An analysis of the density of states of NTO indicates that O atom in NO2, N atom in the ring, and O atom in C=O act as active centers. The absorption spectra show that NTO has relatively high absorption activity with the increasing pressure. When pressure increases from 140 to 150 GPa, its optical activity is enhanced and reduced at visible light region and near ultraviolet region, respectively.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Journal of Materials Chemistry A 2014 vol. 2(Issue 32) pp:13006-13015
Publication Date(Web):17 Jun 2014
DOI:10.1039/C4TA01879F
We report a new strategy to design a novel high-energy low-sensitivity explosive, 1-amino-5-nitrotetrazole-3N-oxide (ANTZO), with outstanding overall performance by combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule. Its detonation performance and stability were estimated using the density functional theory method and compared with some well-known explosives such as 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) and 1-methyl-2,4,6-trinitrobenzene (TNT). Its heat of detonation (7.03 kJ g−1) and detonation velocity (9.87 km s−1) are larger than those of CL-20, while its h50 value (29 J) is higher than that of TNT, indicating that ANTZO has both the high detonation performance of CL-20 and the low sensitivity of TNT, making it valuable and attractive for experiments. Intramolecular hydrogen transfer is the initial decomposition step of ANTZO with the activation energy for this thermal decomposition reaction being about 51.5 kcal mol−1. Our results indicate that our new strategy used for designing ANTZO is practical and may be applied to design and develop other explosives with high energetic properties and low sensitivity.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 39) pp:21620-21628
Publication Date(Web):2014/08/14
DOI:10.1039/C4CP02579B
Ab initio molecular dynamics simulations were performed to study the thermal decomposition of isolated and crystal 3,6-di(azido)-1,2,4,5-tetrazine (DiAT). During unimolecular decomposition, the three different initiation mechanisms were observed to be N–N2 cleavage, ring opening, and isomerization, respectively. The preferential initial decomposition step is the homolysis of the N–N2 bond in the azido group. The release mechanisms of nitrogen gas are found to be very different in the early and later decomposition stages of crystal DiAT. In the early decomposition, DiAT decomposes very fast and drastically without forming any stable long-chains or heterocyclic clusters, and most of the nitrogen gases are released through rapid rupture of nitrogen–nitrogen and carbon–nitrogen bonds. But in the later decomposition stage, the release of nitrogen gas is inhibited due to low mobility, long distance from each other, and strong carbon–nitrogen bonds. To overcome the obstacles, the nitrogen gases are released through slow formation and disintegration of polycyclic networks. Our simulations suggest a new decomposition mechanism for the organic polyazido initial explosive at the atomistic level.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2014 vol. 4(Issue 95) pp:53000-53009
Publication Date(Web):03 Oct 2014
DOI:10.1039/C4RA10548F
We presented a useful strategy to design novel explosives by incorporating N-oxides, nitro groups, and amino groups into s-heptazine. Five new high explosives s-heptazine-1,3,4,6,7,9-hexaoxides (HTO), trinitroheptazine-1,3,4,6,7,9-hexaoxides (TNHTO), aminodinitroheptazine-1,3,4,6,7,9-hexaoxides (ADNHTO), diaminonitroheptazine-1,3,4,6,7,9-hexaoxides (DANHTO), and triaminoheptazine-1,3,4,6,7,9-hexaoxides (TAHTO) were designed. Their energetic properties and sensitivity were estimated by using density functional theory and compared with some famous explosives like CL-20, ONC, HMX, and TNT. All five designed new explosives have much higher detonation performance than s-heptazine and HMX, indicating that symmetrically introducing six N-oxides into s-heptazine is a very effective strategy to improve the explosive performance. DANHTO and TAHTO have comparable detonation performance with CL-20 or ONC and TAHTO has lower sensitivity than TNT, indicating that appropriately incorporating N-oxides, amino groups, and nitro groups into s-heptazine can generate new explosives with excellent performance and low sensitivity. This strategy may be used to design and develop other new energetic materials.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2014 vol. 4(Issue 65) pp:34454-34459
Publication Date(Web):31 Jul 2014
DOI:10.1039/C4RA04981K
DFT-based MD simulations were performed to study the thermal decomposition of crystalline furoxan, an energetic nitrogen-containing model compound. The hydrogen radicals play a catalytic role in the following decomposition of furoxan and can promote the decomposition to a great degree. They not only catalyze the opening of furoxan rings and breaking of N–O bonds, but also capture and transport the oxygen atoms from nitrogen atoms to carbon atoms and promote the release of some small products. The catalytic ability may come from their high mobility that enables the hydrogen to transport fast between different species.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2014 vol. 4(Issue 31) pp:15995-16004
Publication Date(Web):24 Mar 2014
DOI:10.1039/C3RA47747A
The structural transformations, electronic structure, elastic constants, and absorption properties of crystalline furoxan under hydrostatic compression of 0–160 GPa have been studied using density functional theory. The results show that furoxan (S1) undergoes a hydrogen transfer to form a new structure (named S2) with a planar conformation approximately at 114–115 GPa. Then, it goes through another hydrogen transfer to form a second new structure (named S3) with chair conformation at 124 GPa. Finally, it undergoes polymerization to form another new structure (named S4) with a trans-conformation at 136 GPa. An analysis of its band gaps and density of states under compression indicates that it changes from an insulator to a semiconductor in the pressure range 0–113 GPa. S2 and S4 have metallic properties, while S3 is an insulator. The calculated elastic constants show that the three new structures are mechanically stable. Its absorption spectra show that S2 has higher optical activity than S1. S3 has higher optical activity than S2 in the far ultraviolet region, while S4 has weaker optical activity than S3 in this region. S4 and S2 have comparative optical activity.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2014 vol. 4(Issue 8) pp:3789-3797
Publication Date(Web):27 Nov 2013
DOI:10.1039/C3RA46625F
We report new strategies to design two novel and super-high energy cage explosives: dodecanitrohexaprismane (DNH) and hexanitrohexaazaprismane (HNHAH). First, the energetic properties and stability of DNH were estimated by using density functional theory. Second, the six carbon atoms of hexaprismane are replaced by using six nitrogen atoms symmetrically to form 1,3,5,7,9,11-hexaazahexaprismane and then all six hydrogen atoms of 1,3,5,7,9,11-hexaazahexaprismane are substituted by nitro groups to form HNHAH. Next, its performance was evaluated and compared with those of DNH and octanitrocubane (ONC). Finally, the molecular mechanics method was used to predict their molecular packing. The results indicate that DNH has much higher energetic properties than ONC, which may be the most powerful nonnuclear explosive known so far. DNH is more sensitive than ONC but has comparative sensitivity with RDX (1,3,5-trinitro-1,3,5-triazinane) and HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane). Although HNHAH has lower energetic properties than DNH, it has higher energetic properties than ONC slightly. HNHAH is much more insensitive than DNH and is slightly less sensitive than ONC.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
RSC Advances 2014 vol. 4(Issue 95) pp:53149-53156
Publication Date(Web):25 Sep 2014
DOI:10.1039/C4RA09123J
The crystal, molecular, and electronic structures of crystalline 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) in the pressure range of 0–100 GPa under room temperature have been studied by ab initio molecular dynamics (MD). The DFT- and DFT-D-based MD were used to investigate the effects of the vdW correction on the results. The lattice parameters and P–V isotherm of TATB under compression by the DFT-D agree well with the experimental results, whereas the DFT without the vdW correction either underestimates or overestimates the results evidently. The DFT-D results show that TATB is chemically stable in the entire investigated pressure range, in agreement with the experiments, but the DFT without the vdW correction misestimates that TATB decomposes at 50 GPa and polymerizes at 100 GPa. Both the intra- and intermolecular hydrogen bonding are strengthened with the increasing pressure from 5 to 50 GPa, consistent with the experimental results. The DFT without the vdW correction misestimates that TATB turns into a metal system at 100 GPa. The PBE0 with the vdW correction was used to study the band structures of TATB, and the obtained results up to 50 GPa are in agreement with the available experiment results.
Co-reporter:Qiong Wu;Heming Xiao
Structural Chemistry 2014 Volume 25( Issue 2) pp:451-461
Publication Date(Web):2014 April
DOI:10.1007/s11224-013-0306-1
Periodic density functional theory calculations have been performed to study the structural, electronic, absorption, and thermodynamic properties of crystalline α-RDX under hydrostatic compression of 0–50 GPa. As the pressure increases, its lattice parameters, bond lengths, bonds angels, torsion angles, cell volumes, and band structure crystal change regularly except at the pressure of 13 GPa, where a structural transformation occurs. The remarkable changes in the bond lengths and bond angles indicate that there are several possible initiation decomposition mechanisms of RDX under compression. An analysis of density of states shows that the interactions between electrons, especially for the valence electrons, are strengthened under the influence of pressure. The absorption spectra show that the structural transformation makes the absorption coefficient of C–H stretching increase significantly. An analysis of thermodynamic properties indicates that the structural transformation is endothermic and not spontaneous at room temperature. The increasing temperature is not favorable for the structural transformation.
Co-reporter:Fang Xiang, Qiong Wu, Weihua Zhu, and Heming Xiao
Journal of Chemical & Engineering Data 2014 Volume 59(Issue 2) pp:295-306
Publication Date(Web):December 31, 2013
DOI:10.1021/je400844x
Density functional theory and volume-based thermodynamics calculations have been performed to study the crystal densities, heats of formation (HOFs), energetic properties, thermodynamics of formation, and impact sensitivity for the salts composed of heterocycle-functionalized nitraminofurazanate-based anions and triaminoguanidinium cation. The results show that the triaminoguanidinium nitraminofurazanate-based salts have high densities and positive HOFs. The substitution of the oxygen-containing substituent (−NO2 or −C(NO2)3) is helpful for enhancing the densities and detonation properties of the nitraminofurazanate-based salts, and the substitution of −C(NO2)3 exhibits the best performance. Incorporating the conjugated bridge −N═N– or −N═N–N═N– into the heterocycle-functionalized nitraminofurazanate-based salts is favorable for improving the density and detonation properties of its salt. The tetrazole-functionalized salts exhibit the best detonation properties among the three salt series. The calculated h50 values suggest that the introduction of the bridge −NH– or −NH–NH– decreases the sensitivities of the heterocycle-functionalized nitraminofurazanate-based salts.
Co-reporter:Qiong Wu;Heming Xiao
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 7) pp:589-595
Publication Date(Web):
DOI:10.1002/poc.3136
Density functional theory calculations have been performed to study the structural, electronic, absorption, and thermodynamic properties of crystalline 2,4,6-triamino-3,5-dinitropyridine-1-oxide (TANPyo) in the pressure range of 0–50 GPa. The variation trends of the lattice constants, bond lengths, bond angles, intramolecular H-bonds, and dihedral angles under compression show that there are two structural transformations at 17 and 38 GPa, respectively. The remarkable changes in the bond lengths indicate that there are two possible initiation decomposition mechanisms of TANPyo under compression. As the pressure increases, the intramolecular H-bond strengthens. The obvious changes of the dihedral angles show that the planar structure of the TANPyo molecule is damaged under compression. Its absorption spectra show that as the pressure increases, the absorption coefficient of the N–H stretching decreases, while that of the O–H stretching increases. TANPyo has relatively high optical activity at high pressure. An analysis of thermodynamic properties indicates that both two structural transformations are endothermic and not spontaneous at room temperature. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Zhichao Liu;Qiong Wu;Heming Xiao
Journal of Physical Organic Chemistry 2013 Volume 26( Issue 11) pp:939-947
Publication Date(Web):
DOI:10.1002/poc.3197
Density functional theory method was used to study the heats of formation, energetic properties, and thermal stability for a series of trinitromethyl-substituted tetrazole and tetrazine derivatives with different substituents. It is found that the group ―NO2, ―NHNO2, or ―NF2 play a very important role in increasing the heats of formation of the derivatives. The calculated detonation velocities and pressures indicate that the group ―CF2NF2, ―NHNO2, ―1H-tetrazolyl, ―2H-tetrazolyl, or ―1,2,4,5-tetrazinyl is an effective structural unit for enhancing their detonation performance. An analysis of the bond dissociation energies for several relatively weak bonds indicates that incorporating the group ―NHNO2 and ―NH2 into parent ring decreases their thermal stability. Considering the detonation performance and thermal stability, 37 compounds may be considered as the potential high-energy compounds. Their oxygen balances are close to zero. These results provide basic information for the molecular design of novel high-energy compounds. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Yong Pan, Weihua Zhu, Heming Xiao
Computational and Theoretical Chemistry 2013 Volume 1019() pp:116-124
Publication Date(Web):1 September 2013
DOI:10.1016/j.comptc.2013.07.010
•The introduction of trinitromethyl group is useful for increasing its HOF.•The trinitromethyl group is an effective substituent for enhancing its detonation properties.•The substitution of the dinitromethyl group is not favorable for increasing its HOF and detonation properties.•The C–NO2 bond is the weakest one and the ring cleavage may happen in thermal decomposition.The heats of formation (HOFs), energetic properties, strain energies, thermal stability, and impact sensitivity for a series of trinitromethyl- or dinitromethyl-modified RDX and β-HMX derivatives were studied by using density functional theory. It is found that the introduction of trinitromethyl group is an effective structural unit for improving the HOFs and energetic properties of the derivatives. However, incorporating the dinitromethyl group into the parent compound is not favorable for increasing its HOF and detonation properties. The effects of trinitromethyl or dinitromethyl groups on the stability of the parent compound are discussed. An analysis of the bond dissociation energies for several relatively weak bonds suggests that the substitution of the trinitromethyl or dinitromethyl group decreases the thermal stability of the derivatives. On the whole, the C–NO2 bond in the trinitromethyl or dinitromethyl group is the weakest one and the C–NO2 cleavage may happen in thermal decomposition. The introduction of the trinitromethyl or dinitromethyl group increases the impact sensitivities of the derivatives. In addition, the introduction of trinitromethyl group intensifies the strain of the central ring for the title compounds, while the effects of the dinitromethyl groups on the strain of the central ring are coupled to those of the ring structures.Graphical abstract
Co-reporter:Qiong Wu, Weihua Zhu, and Heming Xiao
The Journal of Physical Chemistry C 2013 Volume 117(Issue 33) pp:16830-16839
Publication Date(Web):July 30, 2013
DOI:10.1021/jp405591j
The structural, electronic, and absorption properties of crystalline 7-amino-6-nitrobenzodifuroxan (ANBDF) under hydrostatic compression of 0–110 GPa have been studied by using DFT calculations. Our results show that there three structural transformations occurred at the pressure of 35, 70, and 100 GPa. The first structural transformation makes the positions of the molecules in the crystal rearranged and obviously improves the planarity. The second one forms a new C7–O10 covalent bond and transforms a boat conformation. The last one takes place with the rupture of the C7–O10 bond, forming another new C8–O10 covalent bond and transforming of a boat conformation to a chair. An analysis of the band gap and density of states of ANBDF indicates that its electronic character transforms into a metallic system at 70 GPa, and it becomes more sensitive under compression. The absorption spectra show that ANBDF has relatively high optical activity with the increasing pressure, and the structural transformations that occurred at 70 and 100 GPa can broaden its absorption region in the energy range of below 2 eV and above 20 eV, respectively. This work may provide useful information in understanding how ANBDF behaves under high pressures.
Co-reporter:Qiong Wu;Heming Xiao
Journal of Molecular Modeling 2013 Volume 19( Issue 8) pp:2945-2954
Publication Date(Web):2013 August
DOI:10.1007/s00894-013-1825-9
The heats of formation (HOFs), energetic properties, and thermal stability of a series of 1,7-diamino-1,7-dinitrimino-2,4,6-trinitro-2,4,6-triazaheptane derivatives with different substituents, different numbers of substituents, and different original chains are found by using the DFT-B3LYP method. The results show that -NO2 or -NH2 is an effective substituent for increasing the gas-phase HOFs of the title compounds, especially -NO2 group. As the numbers of substitutents increase, their HOFs enhance obviously. Increasing the length of original chain is helpful for improving their HOFs. The substitution of -NO2 is useful for enhancing their detonation performances and the effects of the length of original chains on detonation properties are coupled with those of the substituents. An analysis of the BDE of the weakest bonds indicates that the substitution of the -NH2 groups and replacing the -NO2 groups of N-NO2 by the -NH2 groups are favorable for improving their thermal stability, while the substitution of -NO2 and increasing the length of original chain decrease their thermal stability. Considering the detonation performance and thermal stability, seven compounds may be considered as the potential candidates of high energy density compounds.
Co-reporter:Yuling Shao, Weihua Zhu, Heming Xiao
Journal of Molecular Graphics and Modelling 2013 40() pp: 54-63
Publication Date(Web):1 March 2013
DOI:10.1016/j.jmgm.2012.12.003
Density functional theory and volume-based thermodynamics calculations have been performed to study the crystal densities, heats of formation (HOFs), energetic properties, and thermodynamics of formation for a series of ionic salts composed of triaminoguanidinium or ammonium cations and tetrazole-based anions. Substitution with NF2, CH2NF2, CF2NF2, or C(NO2)2NF2 groups increased the densities of the salts. The densities of the tetrazole-based salts are affected not only by different substituents but also by different cations. The CN or N3 groups are effective substituents for increasing the HOFs of the salts. The triaminoguanidinium cation is more effective than the ammonium cation for increasing the HOF of the tetrazole-based salts. Substitution with NO2, NF2, or C(NO2)2NF2 groups enhances the explosive properties of the salts. The thermodynamics of formation of the salts reveal that all of the tetrazole-based salts with the triaminoguanidinium or ammonium cation could be synthesized using the proposed reactions. Our calculated methods provide a straightforward and inexpensive route for screening a large number of potentially energetic ionic salts.Graphical abstractDownload high-res image (144KB)Download full-size imageHighlights► NF2, CF2NF2, CF2NF2, or C(NO2)2NF2 is a useful substituent for increasing their ρ. ► The CN or N3 group is an effective substituent for increasing their HOFs. ► The substitution of NO2, NF2, or C(NO2)2NF2 is useful for enhancing their D and P. ► All the tetrazole-based salts here could be synthesized by the proposed reactions.
Co-reporter:Qiong Wu, Weihua Zhu, and Heming Xiao
Journal of Chemical & Engineering Data 2013 Volume 58(Issue 10) pp:2748-2762
Publication Date(Web):August 20, 2013
DOI:10.1021/je4004367
The density functional theory method was used to study the geometrical structures, enthalpies of formation (EOFs), energetic properties, and thermal stability of a series of tetrazole- and tetrazine-based derivatives with oxygen balance equal to zero including different substituents and linkages. The results show that the two heterocycles in most of the tetrazole derivatives and all of the tetrazine derivatives are approximately coplanar. Most of the designed compounds have much higher EOFs than HMX, and over half of them have an extremely high EOF above 800 kJ/mol. Seventy-one compounds have better detonation properties than RDX, and 25 compounds have better detonation properties than HMX, indicating that designing the tetrazole- and tetrazine-based derivatives with oxygen balance equal to zero is a very effective way to obtain potential energetic compounds with outstanding detonation properties. Considering the thermal stability and detonation performance, 57 compounds may be considered as potential candidates of high-density energy compounds.
Co-reporter:Qingli Yan;Aimin Pang;Xuhui Chi;Xijuan Du
Journal of Molecular Modeling 2013 Volume 19( Issue 4) pp:1617-1626
Publication Date(Web):2013 April
DOI:10.1007/s00894-012-1724-5
To improve the understanding of the unimolecular decomposition mechanism of nitroglycerin (NG) in the gas phase, density functional theory calculations were performed to determine various decomposition channels at the B3LYP/6-311G** level. For the unimolecular decomposition mechanism of NG, we find two main mechanisms: (I) homolytic cleavage of O-NO2 to form •NO2 and CH2ONO2CHONO2CH2O•, which subsequently decomposes to form •CHO, •NO2, and 2CH2O; (II) successive HONO eliminations to form HONO and CHO-CO-CHO, which subsequently decomposes to form CH2O + 2CO2 and •CHO + CO. We also find that the former channel has slightly smaller activation energy than the latter one. In addition, the rate constants of the initial process of the two decomposition channels were calculated. The results show that the O-NO2 cleavage pathway occurs more easily than the HONO elimination.
Co-reporter:Yong Pan, Jinshan Li, Bibo Cheng, Weihua Zhu, Heming Xiao
Computational and Theoretical Chemistry 2012 Volume 992() pp:110-119
Publication Date(Web):15 July 2012
DOI:10.1016/j.comptc.2012.05.013
The heats of formation (HOF), energetic properties, and thermal stability for a series of furazano[3,4-b]pyrazine derivatives with different substituents or nitrogen-containing heterocycles were studied by using density functional theory. It is found that –N3 or nitrogen-containing heterocycle is an effective structural unit for improving the HOF values of the derivatives. The calculated detonation velocities and detonation pressures indicate that the substitution of –NO2, –NF2, or NO2-substituted heterocycle is very useful for enhancing their detonation performance. An analysis of the bond dissociation energies for several relatively weak bonds suggests that most of the derivatives have good thermal stability. By and large, the N–O bond in the furazano[3,4-b]pyrazine ring is the weakest one and the ring cleavage may happen in thermal decomposition. Considered the detonation performance and thermal stability, three compounds may be considered as the potential candidates of high energy density materials.Graphical abstractHighlights► The substitution of –N3, heterocycles, or NO2-substituted heterocycles is useful for increasing its HOF. ► –NF2, –ONO2, or –NO2 is an effective substituent for enhancing its detonation properties. ► The N–O bond in the ring is the weakest one and the ring cleavage may happen in thermal decomposition. ► Three compounds may be considered as potential high-energy candidates.
Co-reporter:Tao Wei;Jianzhang Wu;Chenchen Zhang
Journal of Molecular Modeling 2012 Volume 18( Issue 8) pp:3467-3479
Publication Date(Web):2012 August
DOI:10.1007/s00894-012-1357-8
The heats of formation (HOFs), thermal stability, and detonation properties for a series of nitrogen-bridged 1,2,4,5-tetrazine-, furazan-, and 1H-tetrazole-based polyheterocyclic compounds (3,6-bis(1H-1,2,3,4-tetrazole-5-ylamino)-1,2,4,5- tetrazine (TST), 3,6-bis(furazan-5-ylamino)-1,2,4,5-tetrazine (FSF), 3,4-bis(1,2,4,5- tetrazine-3-ylamino)-furazan (SFS), 3,4-bis(1H-1,2,3,4-tetrazole-5-ylamino)-furazan (TFT), 1,5-bis(1,2,4,5-tetrazine-3-ylamino)-1H-1,2,3,4-tetrazole (STS), and 1,5-bis(furazan-3-ylamino)-1H-1,2,3,4-tetrazole (FTF) derivatives) were systematically studied by using density functional theory. The results show that the -N3 or -NHNH2 group plays a very important role in increasing the HOF values of the derivatives. Among these series, the SFS derivatives have lower energy gaps, while the TFT derivatives have higher ones. Incorporation of the -NH2 group into the FSF, SFS, STS, or FTF ring is favorable for enhancing its thermal stability, whereas the substitution of the -NHNH2 group could increase the thermal stability of the TST, SFS, STS, or FTF ring. The calculated detonation properties indicate that the -NO2 or -NF2 is very helpful for enhancing the detonation performance for these derivatives. Considering the detonation performance and thermal stability, six derivatives may be regarded as promising candidates of high-energy density materials (HEDMs). These results provide basic information for the molecular design of novel HEDMs.
Co-reporter:Qiong Wu, Weihua Zhu, Heming Xiao
Computational and Theoretical Chemistry (15 February 2014) Volume 1030() pp:
Publication Date(Web):15 February 2014
DOI:10.1016/j.comptc.2013.12.024
•There occur four structural transformations in APX at 2, 4, 10, and 19 GPa, respectively.•The band splitting and dispersion increase at high pressures.•The impact sensitivity for APX becomes more and more sensitive with the increment of pressure.•APX has relatively high optical activity at high pressure.The structural, electronic, and absorption properties of crystalline 1,7-diamino-1,7-dinitrimino-2,4,6-trinitro-2,4,6-triazaheptane (APX) in the pressure range of 0–20 GPa have been studied by using density functional theory. A comprehensive analysis of the variation trends of the lattice constants, bond lengths, bond angles, and intra-molecular H-bonds under compression indicates that there are four structural transformations in APX at 2, 4, 10, and 19 GPa, respectively. Structural transformations occurred at 2 and 10 GPa weaken the H-bonding. The impact sensitivity for APX becomes more and more sensitive with the increment of pressure. An analysis of density of states suggests that the band splitting and dispersion increase due to the enhanced intermolecular interactions at high pressures. APX has relatively high optical activity at high pressure. The structural transformation at 4 GPa makes the absorption coefficient of the C–H stretching increase significantly, but the transformations at 2 and 19 GPa decrease that slightly.Graphical abstract
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 39) pp:NaN21628-21628
Publication Date(Web):2014/08/14
DOI:10.1039/C4CP02579B
Ab initio molecular dynamics simulations were performed to study the thermal decomposition of isolated and crystal 3,6-di(azido)-1,2,4,5-tetrazine (DiAT). During unimolecular decomposition, the three different initiation mechanisms were observed to be N–N2 cleavage, ring opening, and isomerization, respectively. The preferential initial decomposition step is the homolysis of the N–N2 bond in the azido group. The release mechanisms of nitrogen gas are found to be very different in the early and later decomposition stages of crystal DiAT. In the early decomposition, DiAT decomposes very fast and drastically without forming any stable long-chains or heterocyclic clusters, and most of the nitrogen gases are released through rapid rupture of nitrogen–nitrogen and carbon–nitrogen bonds. But in the later decomposition stage, the release of nitrogen gas is inhibited due to low mobility, long distance from each other, and strong carbon–nitrogen bonds. To overcome the obstacles, the nitrogen gases are released through slow formation and disintegration of polycyclic networks. Our simulations suggest a new decomposition mechanism for the organic polyazido initial explosive at the atomistic level.
Co-reporter:Qiong Wu, Guolin Xiong, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 35) pp:NaN22831-22831
Publication Date(Web):2015/07/28
DOI:10.1039/C5CP03257A
We have performed ab initio molecular dynamics simulations to study coupling effects of temperature (534–873 K) and pressure (1–20 GPa) on the initiation mechanisms and subsequent chemical decompositions of nitramine explosive 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX). A new initiation decomposition mechanism of HMX was found to be the unimolecular C–H bond breaking, and this mechanism was independent of the coupling effects of different temperatures and pressures. The formed hydrogen radicals could promote subsequent decompositions of HMX. Subsequent decompositions were very sensitive to the pressure at low temperatures (534 and 608 K), while the temperature became the foremost factor that affected the decomposition at a high temperature (873 K) instead of the pressure. Our study may provide a new insight into understanding the coupling effects of the temperature and pressure on the initiation decomposition mechanisms of nitramine explosives.
Co-reporter:Zhichao Liu, Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 16) pp:NaN10578-10578
Publication Date(Web):2015/03/13
DOI:10.1039/C5CP00637F
Density functional theory with dispersion-correction (DFT-D) was employed to study the effects of vacancy and pressure on the structure and initial decomposition of crystalline 5-nitro-2,4-dihydro-3H-1,2,4-triazol-3-one (β-NTO), a high-energy insensitive explosive. A comparative analysis of the chemical behaviors of NTO in the ideal bulk crystal and vacancy-containing crystals under applied hydrostatic compression was considered. Our calculated formation energy, vacancy interaction energy, electron density difference, and frontier orbitals reveal that the stability of NTO can be effectively manipulated by changing the molecular environment. Bimolecular hydrogen transfer is suggested to be a potential initial chemical reaction in the vacancy-containing NTO solid at 50 GPa, which is prior to the C–NO2 bond dissociation as its initiation decomposition in the gas phase. The vacancy defects introduced into the ideal bulk NTO crystal can produce a localized site, where the initiation decomposition is preferentially accelerated and then promotes further decompositions. Our results may shed some light on the influence of the molecular environments on the initial pathways in molecular explosives.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 10) pp:NaN7099-7099
Publication Date(Web):2016/02/05
DOI:10.1039/C6CP00096G
We performed ab initio molecular dynamics simulations to study the initiation mechanisms and subsequent chemical decomposition reactions of the nitrogen-rich furazan explosive 3,3′-dinitroamino-4,4′-azoxyfurazan (DNAAF) at low temperatures (363–963 K) coupled with different pressures (1–5 GPa). Two different initial decomposition mechanisms which are dependent on the temperature and pressure were found: bimolecular intermolecular hydrogen transfer and unimolecular N–NO2 bond breaking. The subsequent decomposition reactions are sensitive to both the temperature and the pressure. The pressure could accelerate or decelerate the decomposition of DNAAF, while the temperature can change the effect of the pressure on the decomposition. Our study may provide new insights into the initial mechanisms and subsequent decomposition of furazan explosives at low temperatures coupled with different pressures in atomic detail.
Co-reporter:Qiong Wu, Weihua Zhu and Heming Xiao
Journal of Materials Chemistry A 2014 - vol. 2(Issue 32) pp:NaN13015-13015
Publication Date(Web):2014/06/17
DOI:10.1039/C4TA01879F
We report a new strategy to design a novel high-energy low-sensitivity explosive, 1-amino-5-nitrotetrazole-3N-oxide (ANTZO), with outstanding overall performance by combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule. Its detonation performance and stability were estimated using the density functional theory method and compared with some well-known explosives such as 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) and 1-methyl-2,4,6-trinitrobenzene (TNT). Its heat of detonation (7.03 kJ g−1) and detonation velocity (9.87 km s−1) are larger than those of CL-20, while its h50 value (29 J) is higher than that of TNT, indicating that ANTZO has both the high detonation performance of CL-20 and the low sensitivity of TNT, making it valuable and attractive for experiments. Intramolecular hydrogen transfer is the initial decomposition step of ANTZO with the activation energy for this thermal decomposition reaction being about 51.5 kcal mol−1. Our results indicate that our new strategy used for designing ANTZO is practical and may be applied to design and develop other explosives with high energetic properties and low sensitivity.

![1,3,5,7-TETRAAZASPIRO[3.4]OCTANE, 1,3,5,7-TETRANITRO-](http://img.cochemist.com/ccimg/883300/883244-94-8.png)
![1,3,5,7-TETRAAZASPIRO[3.4]OCTANE, 1,3,5,7-TETRANITRO-](http://img.cochemist.com/ccimg/883300/883244-94-8_b.png)
![1,2,4,6-Tetraazaspiro[2.3]hexane, 1,2,4,6-tetranitro-](http://img.cochemist.com/ccimg/883300/883244-93-7.png)
![1,2,4,6-Tetraazaspiro[2.3]hexane, 1,2,4,6-tetranitro-](http://img.cochemist.com/ccimg/883300/883244-93-7_b.png)


![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7.png)
![1,3,4,6-TETRANITRO-2,3A,5,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE](http://img.cochemist.com/ccimg/473800/473796-67-7_b.png)






![tetrazolo[1,5-b][1,2,4,5]tetrazin-6-amine](http://img.cochemist.com/ccimg/161300/161238-62-6.png)
![tetrazolo[1,5-b][1,2,4,5]tetrazin-6-amine](http://img.cochemist.com/ccimg/161300/161238-62-6_b.png)
![2H-Imidazo[4,5-b]pyrazin-2-one, octahydro-1,3,4,7-tetranitro-](http://img.cochemist.com/ccimg/160100/160013-48-9.png)
![2H-Imidazo[4,5-b]pyrazin-2-one, octahydro-1,3,4,7-tetranitro-](http://img.cochemist.com/ccimg/160100/160013-48-9_b.png)
![2,4,6,8,9,10-Hexaazatricyclo[3.3.1.13,7]decane, 2,4,6,8,9,10-hexanitro-](http://img.cochemist.com/ccimg/157200/157110-14-0.png)
![2,4,6,8,9,10-Hexaazatricyclo[3.3.1.13,7]decane, 2,4,6,8,9,10-hexanitro-](http://img.cochemist.com/ccimg/157200/157110-14-0_b.png)
![1,3,5,7-Tetraazaspiro[3.3]heptane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/155600/155582-25-5.png)
![1,3,5,7-Tetraazaspiro[3.3]heptane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/155600/155582-25-5_b.png)



![4H,8H-bis[1,2,5]oxadiazolo[3,4-b:3',4'-e]pyrazine](http://img.cochemist.com/ccimg/132100/132029-07-3.png)
![4H,8H-bis[1,2,5]oxadiazolo[3,4-b:3',4'-e]pyrazine](http://img.cochemist.com/ccimg/132100/132029-07-3_b.png)
![4,8-dinitro-4H,8H-bis[1,2,5]oxadiazolo[3,4-b:3',4'-e]pyrazine](http://img.cochemist.com/ccimg/132100/132029-05-1.png)
![4,8-dinitro-4H,8H-bis[1,2,5]oxadiazolo[3,4-b:3',4'-e]pyrazine](http://img.cochemist.com/ccimg/132100/132029-05-1_b.png)
![Imidazo[4,5-d]imidazol-2(1H)-one, hexahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/130300/130256-72-3.png)
![Imidazo[4,5-d]imidazol-2(1H)-one, hexahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/130300/130256-72-3_b.png)

![2,4,6,8-Tetraazabicyclo[3.3.1]nonane-3,7-dione, 2,4,6,8-tetranitro-](http://img.cochemist.com/ccimg/115100/115029-36-2.png)
![2,4,6,8-Tetraazabicyclo[3.3.1]nonane-3,7-dione, 2,4,6,8-tetranitro-](http://img.cochemist.com/ccimg/115100/115029-36-2_b.png)

![1,3,4,5,7,8-hexanitrooctahydrodiimidazo[4,5-b:4',5'-e]pyrazine-2,6(1H,3H)-dione](http://img.cochemist.com/ccimg/115100/115029-33-9.png)
![1,3,4,5,7,8-hexanitrooctahydrodiimidazo[4,5-b:4',5'-e]pyrazine-2,6(1H,3H)-dione](http://img.cochemist.com/ccimg/115100/115029-33-9_b.png)







![tricyclo[3.3.1.1~3,7~]decane-1,3,5,7-tetrayl tetranitrate](http://img.cochemist.com/ccimg/94700/94616-58-7.png)
![tricyclo[3.3.1.1~3,7~]decane-1,3,5,7-tetrayl tetranitrate](http://img.cochemist.com/ccimg/94700/94616-58-7_b.png)


![2,4,8,10-Tetraazaspiro[5.5]undecane, 2,4,8,10-tetranitro-](http://img.cochemist.com/ccimg/84700/84606-37-1.png)
![2,4,8,10-Tetraazaspiro[5.5]undecane, 2,4,8,10-tetranitro-](http://img.cochemist.com/ccimg/84700/84606-37-1_b.png)


![PYRAZINO[2,3-B]PYRAZINE, DECAHYDRO-1,4,5,8-TETRANITRO-, TRANS-](http://img.cochemist.com/ccimg/83700/83673-31-8.png)
![PYRAZINO[2,3-B]PYRAZINE, DECAHYDRO-1,4,5,8-TETRANITRO-, TRANS-](http://img.cochemist.com/ccimg/83700/83673-31-8_b.png)
![BENZO[1,2-C:3,4-C']BIS[1,2,5]OXADIAZOL-4-AMINE, 5-NITRO-, 3,8-DIOXIDE](http://img.cochemist.com/ccimg/83500/83454-00-6.png)
![BENZO[1,2-C:3,4-C']BIS[1,2,5]OXADIAZOL-4-AMINE, 5-NITRO-, 3,8-DIOXIDE](http://img.cochemist.com/ccimg/83500/83454-00-6_b.png)


![3,7-Diazabicyclo[3.3.1]nonane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/81400/81340-15-0.png)
![3,7-Diazabicyclo[3.3.1]nonane, 1,3,5,7-tetranitro-](http://img.cochemist.com/ccimg/81400/81340-15-0_b.png)
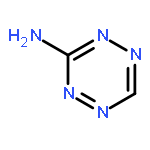
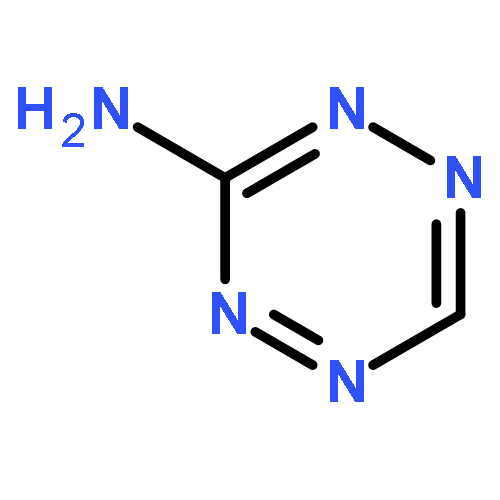
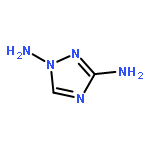

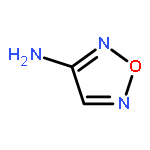
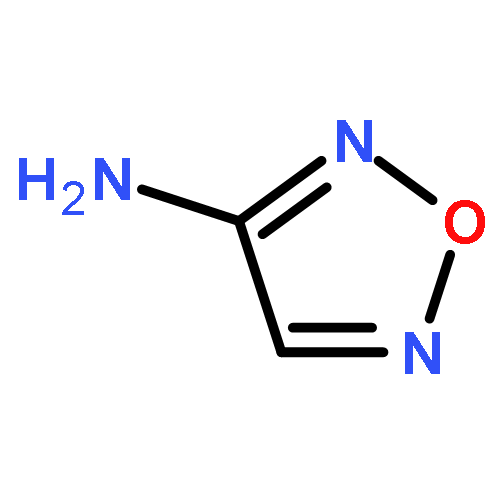
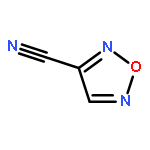
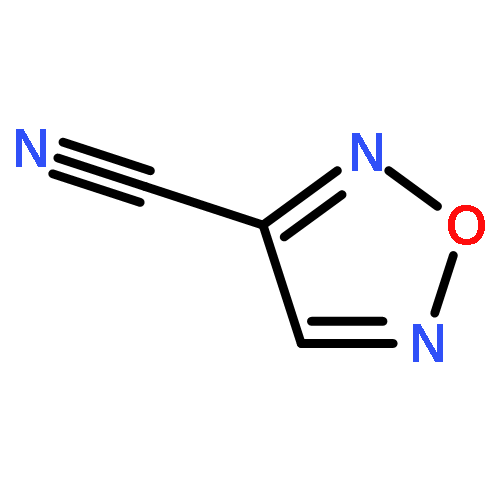


![1,3,7,9-Tetraazaspiro[4.5]decane, 1,3,7,9-tetranitro-](http://img.cochemist.com/ccimg/65500/65479-81-4.png)
![1,3,7,9-Tetraazaspiro[4.5]decane, 1,3,7,9-tetranitro-](http://img.cochemist.com/ccimg/65500/65479-81-4_b.png)
![Tricyclo[3.3.1.13,7]decan-2-ol, nitrate](http://img.cochemist.com/ccimg/64700/64662-51-7.png)
![Tricyclo[3.3.1.13,7]decan-2-ol, nitrate](http://img.cochemist.com/ccimg/64700/64662-51-7_b.png)
![N-(azidomethyl)-N'-{[(azidomethyl)(nitro)amino]methyl}-N,N'-dinitromethanediamine](http://img.cochemist.com/ccimg/62300/62209-57-8.png)
![N-(azidomethyl)-N'-{[(azidomethyl)(nitro)amino]methyl}-N,N'-dinitromethanediamine](http://img.cochemist.com/ccimg/62300/62209-57-8_b.png)
![1,3,5,7-TETRAAZATRICYCLO[3.3.1.13,7]DECANE, 1,3-DIOXIDE](http://img.cochemist.com/ccimg/62200/62190-93-6.png)
![1,3,5,7-TETRAAZATRICYCLO[3.3.1.13,7]DECANE, 1,3-DIOXIDE](http://img.cochemist.com/ccimg/62200/62190-93-6_b.png)
![1,3,5,7-Tetraazatricyclo[3.3.1.13,7]decane, 1-oxide](http://img.cochemist.com/ccimg/62200/62190-92-5.png)
![1,3,5,7-Tetraazatricyclo[3.3.1.13,7]decane, 1-oxide](http://img.cochemist.com/ccimg/62200/62190-92-5_b.png)


![3,6-DINITRO-1,3A,4,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE-2,5-DIONE](http://img.cochemist.com/ccimg/55600/55510-04-8.png)
![3,6-DINITRO-1,3A,4,6A-TETRAHYDROIMIDAZO[4,5-D]IMIDAZOLE-2,5-DIONE](http://img.cochemist.com/ccimg/55600/55510-04-8_b.png)
![Imidazo[4,5-d]imidazole-2,5(1H,3H)-dione,tetrahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/55600/55510-03-7.png)
![Imidazo[4,5-d]imidazole-2,5(1H,3H)-dione,tetrahydro-1,3,4,6-tetranitro-](http://img.cochemist.com/ccimg/55600/55510-03-7_b.png)


![tricyclo[3.3.1.1~3,7~]decane-1,3-diyl dinitrate](http://img.cochemist.com/ccimg/53500/53488-28-1.png)
![tricyclo[3.3.1.1~3,7~]decane-1,3-diyl dinitrate](http://img.cochemist.com/ccimg/53500/53488-28-1_b.png)
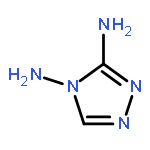
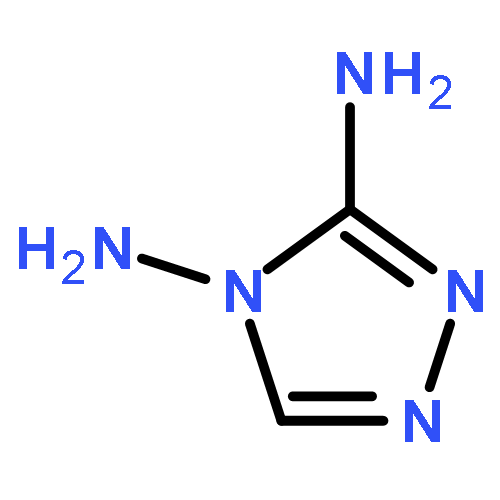

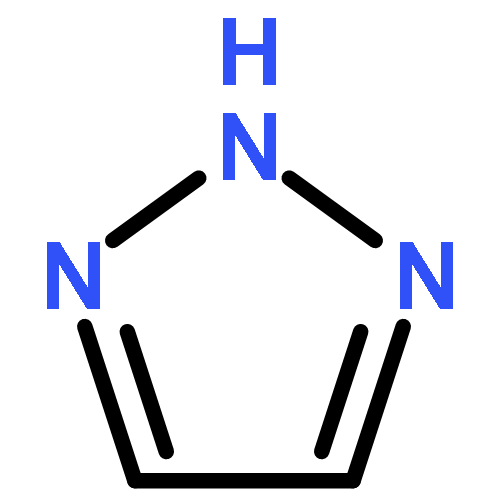


![tricyclo[3.3.1.1~3,7~]dec-1-yl nitrate](http://img.cochemist.com/ccimg/32400/32314-61-7.png)
![tricyclo[3.3.1.1~3,7~]dec-1-yl nitrate](http://img.cochemist.com/ccimg/32400/32314-61-7_b.png)


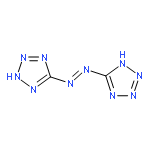

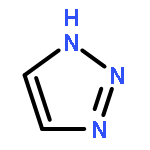




![[1,2,5]Oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/25900/25883-65-2.png)
![[1,2,5]Oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/25900/25883-65-2_b.png)
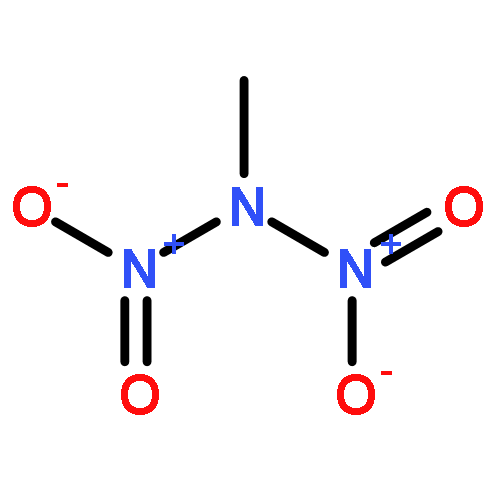
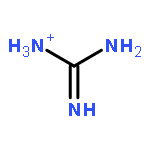

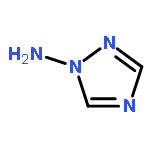
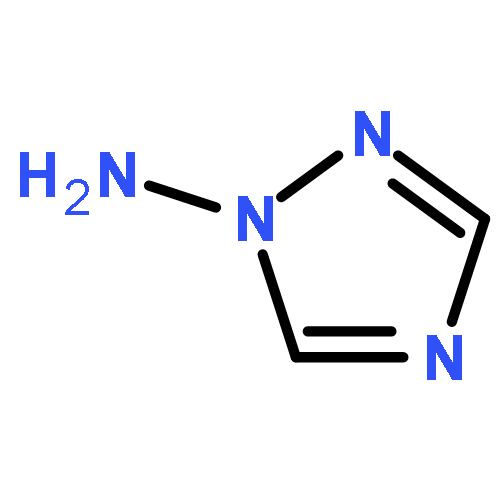

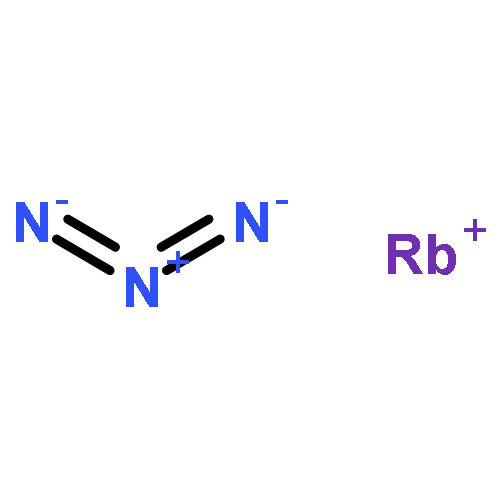
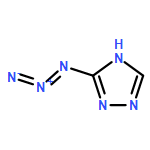
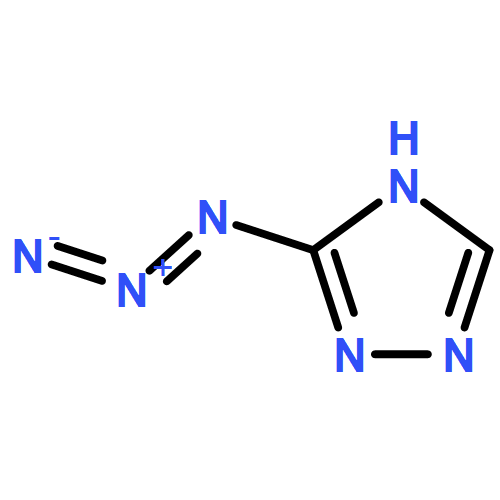
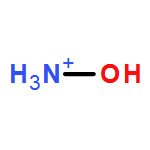









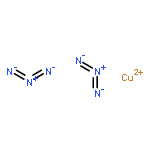
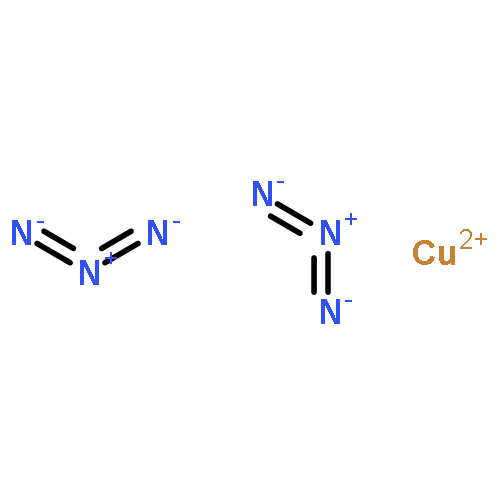
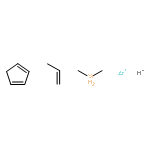




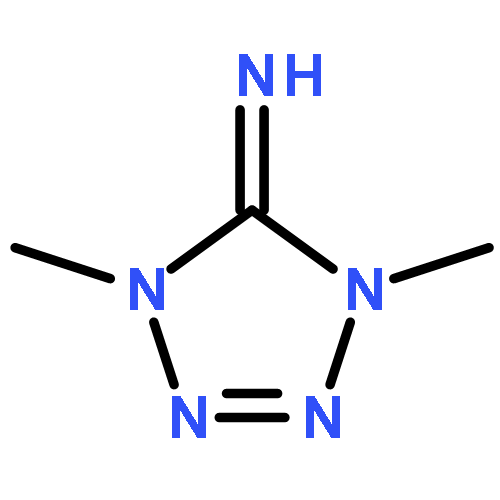

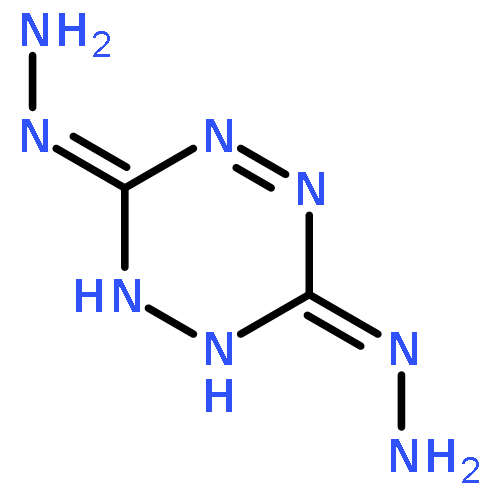




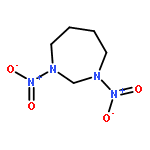







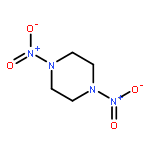

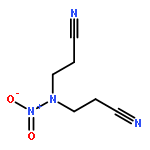





















![1,3-Diazatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/300/281-29-8.png)
![1,3-Diazatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/300/281-29-8_b.png)
![1-Azatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/300/281-27-6.png)
![1-Azatricyclo[3.3.1.13,7]decane](http://img.cochemist.com/ccimg/300/281-27-6_b.png)





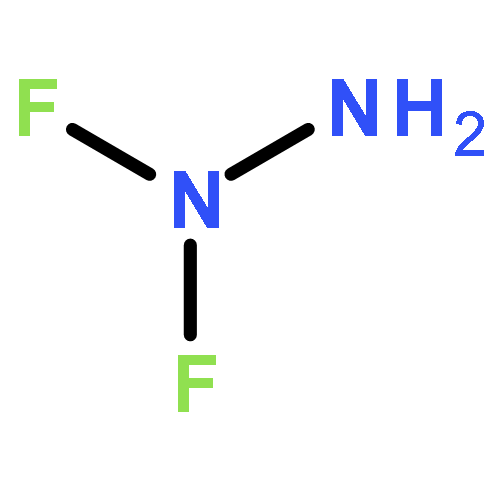
![4,7-dinitro-4,5,6,7-tetrahydro[1,2,5]oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/98800/98778-15-5.png)
![4,7-dinitro-4,5,6,7-tetrahydro[1,2,5]oxadiazolo[3,4-b]pyrazine](http://img.cochemist.com/ccimg/98800/98778-15-5_b.png)


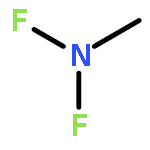
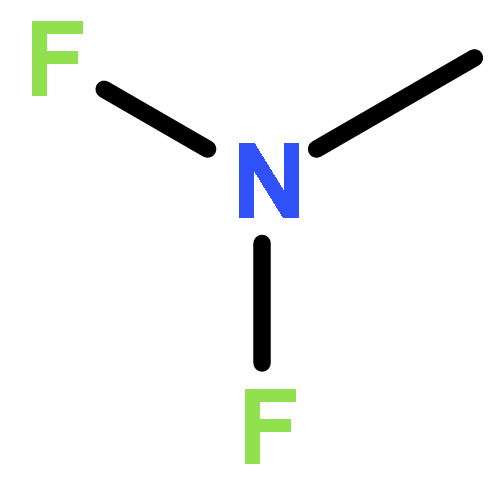


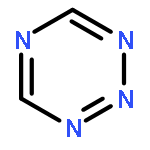



















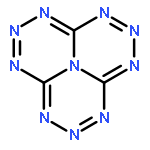
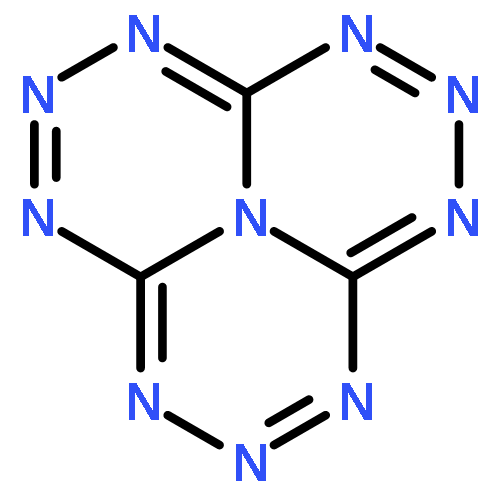
![GUANIDINE, N,N'''-[(NITROIMINO)BIS(METHYLENE)]BIS[N,N'-DINITRO-](http://img.cochemist.com/ccimg/666800/666736-94-3.png)
![GUANIDINE, N,N'''-[(NITROIMINO)BIS(METHYLENE)]BIS[N,N'-DINITRO-](http://img.cochemist.com/ccimg/666800/666736-94-3_b.png)
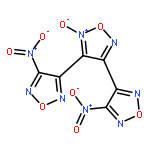
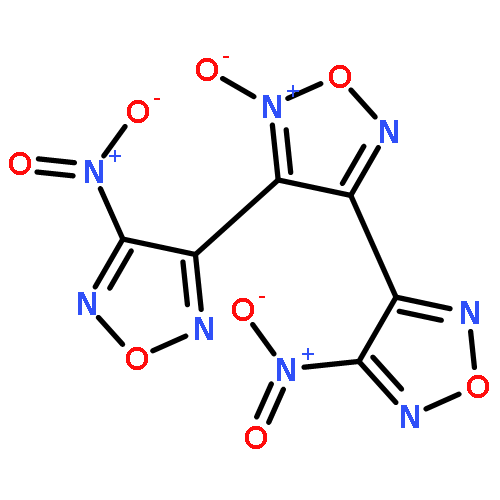
![3-nitro-4-[(4-nitro-1,2,5-oxadiazol-3-yl)-NNO-azoxy]-1,2,5-oxadiazole](http://img.cochemist.com/ccimg/152900/152845-82-4.png)
![3-nitro-4-[(4-nitro-1,2,5-oxadiazol-3-yl)-NNO-azoxy]-1,2,5-oxadiazole](http://img.cochemist.com/ccimg/152900/152845-82-4_b.png)


![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)







![Pentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/300/277-10-1.png)
![Pentacyclo[4.2.0.02,5.03,8.04,7]octane](http://img.cochemist.com/ccimg/300/277-10-1_b.png)