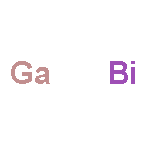Co-reporter:Xiaoming Zhang, Yinong Zhou, Bin Cui, Mingwen Zhao, and Feng Liu
Nano Letters October 11, 2017 Volume 17(Issue 10) pp:6166-6166
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.nanolett.7b02795
Superconductivity is a fascinating quantum phenomenon characterized by zero electrical resistance and the Meissner effect. To date, several distinct families of superconductors (SCs) have been discovered. These include three-dimensional (3D) bulk SCs in both inorganic and organic materials as well as two-dimensional (2D) thin film SCs but only in inorganic materials. Here we predict superconductivity in 2D and 3D organic metal–organic frameworks by using first-principles calculations. We show that the highly conductive and recently synthesized Cu-benzenehexathial (BHT) is a Bardeen–Cooper–Schrieffer SC. Remarkably, the monolayer Cu-BHT has a critical temperature (Tc) of 4.43 K, while Tc of bulk Cu-BHT is 1.58 K. Different from the enhanced Tc in 2D inorganic SCs which is induced by interfacial effects, the Tc enhancement in this 2D organic SC is revealed to be the out-of-plane soft-mode vibrations, analogous to surface mode enhancement originally proposed by Ginzburg. Our findings not only shed new light on better understanding 2D superconductivity but also open a new direction to search for SCs by interface engineering with organic materials.Keywords: enhanced critical temperature; first-principles calculations; metal−organic framework; Superconductivity;
Co-reporter:Mingwen Zhao;Xiangdong Liu;Xuejuan Zhang;Liangmo Mei;Yueyuan Xia;Shishen Yan;Hongyu Zhang;Tao He;Zhenhai Wang;Zexiao Xi
The Journal of Physical Chemistry C August 6, 2009 Volume 113(Issue 31) pp:13610-13615
Publication Date(Web):2017-2-22
DOI:10.1021/jp9032244
We propose stable layered structures and ultrathin tubular configurations of titanium oxide (TiO2) nanomaterials on the basis of first-principles calculations within density functional theory. The thinnest TiO2 nanosheet is characterized by a reconstructed (001) bilayer of rutile TiO2, while ultrathin TiO2 nanotubes can be built by rolling up a TiO2NS. These nanotubes are predicted to have high stability, large Young’s modulus, and tunable electronic properties. A possible synthetic route toward these nanostructures is also presented.
Co-reporter:Yuanyuan Qu, Feng Li, and Mingwen Zhao
The Journal of Physical Chemistry C August 24, 2017 Volume 121(Issue 33) pp:17925-17925
Publication Date(Web):July 28, 2017
DOI:10.1021/acs.jpcc.7b04921
Membrane separation of CO2/N2 in fossil fuel gas is promising for the control of greenhouse gas emission, but challenging due to close kinetic diameters. Here, we propose a generalized model for the design of efficient CO2/N2 separation membranes by taking advantage of the large difference between the electric quadrupole moments of the two molecules. The interaction between the molecular electric quadrupole moment and the built-in electric field of the membrane leads to high CO2/N2 selectivity. We validate this model in five nitrogen-rich membranes, g-C3N4, g-C3N3, C2N-h2D, g-C12N8, and p-BN, and demonstrate via molecular dynamics simulations that highly efficient CO2/N2 separation can be achieved in the theoretically predicted g-C12N8 membrane with a permeance of 2.8 × 105 GPU. This work offers a guidance to improve the separation efficiency of molecules with distinct electric quadrupole moments.
Co-reporter:Aizhu Wang, Xiaoming Zhang, Yuanping Feng, and Mingwen Zhao
The Journal of Physical Chemistry Letters August 17, 2017 Volume 8(Issue 16) pp:3770-3770
Publication Date(Web):July 30, 2017
DOI:10.1021/acs.jpclett.7b01187
Two-dimensional metal–organic frameworks (2D-MOFs) with exotic electronic structures are drawing increasing attention. Here, using first-principles calculations, we demonstrate a spin-gapless MOF, namely, Mn2C6S12, with the coexistence of a spin-polarized Dirac cone and parabolic degenerate points. The Curie temperature evaluated from Monte Carlo simulations implies Mn2C6S12 possessing stable ferromagnetism at room temperature. Taking the spin–orbit coupling into account, the Dirac cone is gapped and the degenerate points are lifted, giving rise to multiple topologically nontrivial states with nonzero Chern number, which imply the possibility of Mn2C6S12 to be a Chern insulator and a Chern half-metal. Our results offer versatile platforms for achieving spin filtering or a quantum anomalous Hall effect with promising application in spintronics devices.
Co-reporter:Junru Wang;Feng Li;Bo Yang;Xiaobiao Liu
Journal of Materials Chemistry A 2017 vol. 5(Issue 40) pp:21486-21490
Publication Date(Web):2017/10/17
DOI:10.1039/C7TA07092F
As a key component of spintronic devices, spin batteries that can generate spin-polarized current are drawing increasing interests. Herein, we propose a simple strategy for spin batteries by introducing a half-metallic anode material in the conventional Li-ion batteries. Using first-principles calculations, we demonstrate a potential half-metallic anode material, TiF3 crystal, which has stable ferromagnetism and half-metallicity under Li insertion. Low Li diffusion barriers (0.16–0.37 eV) and moderate Li storage capacity (256 mA h g−1) are revealed in the TiF3 crystal. The combination of half-metals and Li-ion battery offers a new solution for spin batteries.
Co-reporter:Bo Yang;Xiaoming Zhang
Nanoscale (2009-Present) 2017 vol. 9(Issue 25) pp:8740-8746
Publication Date(Web):2017/06/29
DOI:10.1039/C7NR00411G
As a new type of quantum matter, Dirac node line (DNL) semimetals are currently attracting widespread interest in condensed matter physics and materials science. The DNL, featured by a closed line consisting of linear band crossings in the momentum space, was mostly predicted in three-dimensional materials. Here, we propose a tight-binding (TB) model of pz + px,y or pz + s orbitals defined on the two-dimensional (2D) Lieb lattice for the 2D version of DNL semimetals. The DNL states in these models are caused by the inversion of the bands with different symmetries and thus robust against spin–orbit interaction. By means of first-principles calculations, we demonstrate two candidate materials: Be2C and BeH2 monolayers, which have Fermi circles centred at Γ(0,0) and K(1/2,1/2) points, respectively. Their Fermi velocities are higher than that in graphene. The non-zero Z2 topological invariant accompanied by the edge states is revealed in these materials. This work opens an avenue for the design of 2D DNL semimetals.
Co-reporter:Lin Wei;Xiaoming Zhang;Xiaobiao Liu;Hongcai Zhou;Bo Yang
RSC Advances (2011-Present) 2017 vol. 7(Issue 82) pp:52065-52070
Publication Date(Web):2017/11/07
DOI:10.1039/C7RA10950D
Two-dimensional covalent organic frameworks (2D-COFs) are drawing increasing interest due to the unique configurations and exotic properties. Here, using density-functional theory calculations, we prove the stability of C2N6S3 monolayer by an imagery-frequency-free phonon spectrum, and demonstrate a new ternary 2D-COF: C2N6O3, C2N6Se3 and C2N6Te3 monolayers. The sawtooth-like linkages make the C2N6S3 is soft, and sustain a biaxial tensile strain up to 24% which is as much as graphene. The electronic band structure exhibits linear dispersion near the Fermi level with a flat band right above the Dirac bands, which is unlike the other hexagonal organic monolayers with Dirac cone. The Fermi velocity is comparable to that in graphene and can be tuned by applying biaxial tensile strain. Similar results are also found in its analogs, such as C2N6O3, C2N6Se3 and C2N6Te3 monolayers. This opens an avenue for the design of 2D Dirac materials.
Co-reporter:Junru Wang;Feng Li;Xiaobiao Liu;Hongcai Zhou;Xiaofei Shao;Yuanyuan Qu
Journal of Materials Chemistry A 2017 vol. 5(Issue 18) pp:8762-8768
Publication Date(Web):2017/05/10
DOI:10.1039/C7TA02339A
Electrode materials with low diffusion energy barriers and high storage capacity of lithium are crucial for high performance rechargeable lithium-ion batteries (LIBs). Based on first-principles calculations, we demonstrate a new class of electrode materials. Taking advantage of the large voids in Cu3N crystals, high lithium mobility and storage capacity can be achieved. The diffusion of Li on Cu3N nanosheets experiences an energy barrier of about 0.09 eV, which is much lower than those of presently proposed electrode materials. The maximum Li capacity of Cu3N nanosheets can reach 1008 mA h g−1. In view of a large number of crystals sharing the same lattice structure as Cu3N, this work opens an avenue for developing electrode materials for high performance LIBs.
Co-reporter:Xiaoming Zhang, Lin Wei, Jie Tan, Mingwen Zhao
Carbon 2016 Volume 105() pp:323-329
Publication Date(Web):August 2016
DOI:10.1016/j.carbon.2016.04.058
Searching for the Dirac materials with ultrasoftness is crucial for flexible electronics applications. Based on first-principles calculations, we propose a new carbon allotrope (named as ph-graphene) with a penta-hexagonal framework, which is energetically more favorable than the penta-graphene composing surely of pentagons and some of already-synthesized carbon allotropes. Ph-graphene has an in-plane stiffness of 27.75 GPa·nm, smaller than those of graphene and penta-graphene by one order. The famous isotropic Dirac cones are well preserved in the ultrasoft ph-graphene, exhibiting delocalized feature of pz orbits with the Fermi velocity of 2.8 × 105 m/s. Additionally, surface hydrogenation alters drastically the electronic and mechanical properties of ph-graphene, resulting in electronic spin-polarization and anisotropic negative Poisson’s ratios sequentially with the increase of hydrogenation concentrations.
Co-reporter:Aizhu Wang, Zhenhai Wang, Aijun Du and Mingwen Zhao
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 32) pp:22154-22159
Publication Date(Web):14 Jul 2016
DOI:10.1039/C6CP02617F
To achieve a device application of the quantum spin Hall (QSH) effect, increasing the critical temperature is crucial. A two-dimensional topological insulator (2D-TI) with a sizeable bulk band gap is one of the most promising strategies to reach this goal. Using first-principles calculations, we propose a new 2D-TI, titanium nitride iodide (TiNI) monolayer, which can be exfoliated from a bulk TiNI crystal, thanks to the weak interlayer interaction. We demonstrate that the TiNI monolayer has an inverted band structure accompanied by topologically nontrivial states characterized by a topological invariant of Z2 = 1. The band gap (∼50 meV) opened due to spin–orbit coupling (SOC) is available for achieving the QSH effect at room temperature. The band inversion and topologically nontrivial states are robust under external strain, suggesting that the 2D TiNI monolayer lattice could be a versatile platform for hosting nontrivial topological states with potential applications in 2D spintronics and computer technology.
Co-reporter:Lin Wei, Xiaoming Zhang and Mingwen Zhao
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 11) pp:8059-8064
Publication Date(Web):09 Feb 2016
DOI:10.1039/C6CP00368K
Dirac cones in the band structure make a great contribution to the unique electronic properties of graphene. But the spin-degeneracy of Dirac cones limits the application of graphene in spintronics. Here, using first-principles calculations, we propose a two-dimensional (2D) metal–organic framework (MOF), Ni2C24S6H12, with spin-polarized Dirac cones at the six corners of the Brillouin zone (BZ). Ferromagnetism is quite stable with a high Curie temperature (630 K) as revealed by Monte Carlo simulation within the Ising model. Taking spin–orbit coupling into account, band gaps are opened up at the Dirac point (5.9 meV) and Γ point (10.4 meV) in the BZ, making Ni2C24S6H12 a Chern topological insulator which is implemented for achieving the quantum anomalous Hall effect. These interesting properties enable Ni2C24S6H12 to be a promising candidate material for spintronics device applications.
Co-reporter:Haibin Si, Gang Lian, Aizhu Wang, Deliang Cui, Mingwen Zhao, Qilong Wang, and Ching-Ping Wong
Nano Letters 2015 Volume 15(Issue 12) pp:8122-8128
Publication Date(Web):November 24, 2015
DOI:10.1021/acs.nanolett.5b03569
Investigation of light-element magnetism system is essential in fundamental and practical fields. Here, few-layer (∼3 nm) fluorinated hexagonal boron nitride (F-BN) nanocages with zigzag-edge triangular antidot defects were synthesized via a facile one-step solid-state reaction. They are free of metallic impurities confirmed by X-ray photoelectron spectroscopy, electron energy loss spectroscopy, and inductively coupled plasma atomic emission spectroscopy. Ferromagnetism is obviously observed in the BN nanocages. Saturation magnetization values of them differed by less than 7% between 5 and 300 K, indicating that the Curie temperature (Tc) was much higher than 300 K. By adjusting the concentration of triangular antidot defects and fluorine dopants, the ferromagnetic performance of BN nanocages could be effectively varied, indicating that the observed magnetism originates from triangular antidot defects and fluorination. The corresponding theoretical calculation shows that antidot defects and fluorine doping in BN lattice both favor spontaneous spin polarization and the formation of local magnetic moment, which should be responsible for long-range magnetic ordering in the sp material.
Co-reporter:Linyang Li, Xiaoming Zhang, Xin Chen, and Mingwen Zhao
Nano Letters 2015 Volume 15(Issue 2) pp:1296-1301
Publication Date(Web):January 27, 2015
DOI:10.1021/nl504493d
Quantum spin Hall (QSH) effect is promising for achieving dissipationless transport devices but presently is achieved only at extremely low temperature. Searching for the large-gap QSH insulators with strong spin–orbit coupling (SOC) is the key to increase the operating temperature. We demonstrate theoretically that this can be solved in the chloridized gallium bismuthide (GaBiCl2) monolayer, which has nontrivial gaps of 0.95 eV at the Γ point, and 0.65 eV for bulk, as well as gapless edge states in the nanoribbon structures. The nontrivial gaps due to the band inversion and SOC are robust against external strain. The realization of the GaBiCl2 monolayer will be beneficial for achieving QSH effect and related applications at high temperatures.
Co-reporter:Zhenhai Wang, Xiang-Feng Zhou, Xiaoming Zhang, Qiang Zhu, Huafeng Dong, Mingwen Zhao, and Artem R. Oganov
Nano Letters 2015 Volume 15(Issue 9) pp:6182-6186
Publication Date(Web):August 11, 2015
DOI:10.1021/acs.nanolett.5b02512
Using systematic evolutionary structure searching we propose a new carbon allotrope, phagraphene [fæ’græfi:n], standing for penta-hexa-hepta-graphene, because the structure is composed of 5-6-7 carbon rings. This two-dimensional (2D) carbon structure is lower in energy than most of the predicted 2D carbon allotropes due to its sp2-binding features and density of atomic packing comparable to graphene. More interestingly, the electronic structure of phagraphene has distorted Dirac cones. The direction-dependent cones are further proved to be robust against external strain with tunable Fermi velocities.
Co-reporter:Gang Zhao;Shuo Han;Aizhu Wang;Yongzhong Wu;Zhengping Wang;Xiaopeng Hao
Advanced Functional Materials 2015 Volume 25( Issue 33) pp:5292-5299
Publication Date(Web):
DOI:10.1002/adfm.201501972
2D transition metal dichalcogenides are attracting increased attention because of their excellent electronic and optical properties. Inspired by the natural weathering exfoliation of seaside rocks, a “chemical weathering” concept for fabricating atom-thick 2D materials from their bulk counterparts is proposed. It is experimentally demonstrated that chemical weathering-assisted exfoliation mechanism is a simple and efficient method of preparing atom-thick MoS2 and WS2 monolayers. These monolayers are difficult to prepare using other approaches. Interestingly, the as-prepared MoS2 and WS2 monolayers exhibit excellent saturable absorption and mode-locking properties in all-solid-state lasers because of intermediate states resulting from S-vacancy defects. The obtained passively Q-switched laser operation with 60 ns pulse width and ultrafast mode locking with 8.6 ps pulse width are promising for all-solid-state laser application.
Co-reporter:Bo Yang, Hongcai Zhou, Xiaoming Zhang and Mingwen Zhao
Journal of Materials Chemistry A 2015 vol. 3(Issue 41) pp:10886-10891
Publication Date(Web):16 Sep 2015
DOI:10.1039/C5TC02423D
Graphitic carbon nitrides are attracting increasing interest in many fields such as fuel cells, photocatalytic decomposition of water and spintronic devices. Tailoring electronic band gaps and inducing electron spin-polarization are the keys of these applications. Using first-principles calculations, we demonstrate that these goals can be reached by modifying graphitic carbon nitride via the introduction of additional carbon atoms into its vacancy sites. We found that with the increase of carbon concentration, the band gap of graphitic carbon nitride decreases rapidly and comes to close as the carbon concentration is higher than 2.609%, thus leading to a semiconductor–metal phase transition. More interestingly, local magnetic moments appear in the triangular domain centered by the introduced carbon atom and they interact in a ferromagnetic or “antiferromagnetic” manner depending on their relative positions. The tunable band gap and ferromagnetism revealed in the carbon-modified graphitic carbon nitrides offer a promising approach to achieve applications in hydrogen generation and spintronic devices.
Co-reporter:Xiaoming Zhang, Aizhu Wang, Mingwen Zhao
Carbon 2015 Volume 84() pp:1-8
Publication Date(Web):April 2015
DOI:10.1016/j.carbon.2014.11.049
The Dirac cones in the electronic band structures of graphene cause exotic properties, such as Dirac fermions, but these cones are spin-degenerated. In this study, from first principles, we demonstrate that a honeycomb lattice of modified tri-s-triazine (C7N6) units has spin-polarized Dirac cones in the band structures and exhibits features of spin-gapless semiconductors (SGSs). The hybrid honeycomb lattice of the C7N6 and s-triazine (C3N3) units, however, is a SGS with parabolic energy–momentum dispersion relations near the Fermi level. Ferromagnetic ordering is stable with a Curie temperature (Tc) of 830 and 205 K for the two lattices, as revealed by Monte Carlo simulations within an Ising model. The two honeycomb lattices have topologically nontrivial electronic states with a Chern number of C = −1, implying that the quantum anomalous Hall effect (QAHE) states could be achieved in metal-free materials.
Co-reporter:Aizhu Wang and Mingwen Zhao
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 34) pp:21837-21844
Publication Date(Web):09 Jun 2015
DOI:10.1039/C5CP03060A
Fractals are natural phenomena that exhibit a repeating pattern “exactly the same at every scale or nearly the same at different scales”. Defect-free molecular fractals were assembled successfully in a recent work [Shang et al., Nature Chem., 2015, 7, 389–393]. Here, we adopted the feature of a repeating pattern in searching two-dimensional (2D) materials with intrinsic half-metallicity and high stability that are desirable for spintronics applications. Using first-principles calculations, we demonstrate that the electronic properties of fractal frameworks of carbon nitrides have stable ferromagnetism accompanied by half-metallicity, which are highly dependent on the fractal structure. The ferromagnetism increases gradually with the increase of fractal order. The Curie temperature of these metal-free systems estimated from Monte Carlo simulations is considerably higher than room temperature. The stable ferromagnetism, intrinsic half-metallicity, and fractal characteristics of spin distribution in the carbon nitride frameworks open an avenue for the design of metal-free magnetic materials with exotic properties.
Co-reporter:Xin Chen, Linyang Li and Mingwen Zhao
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 25) pp:16624-16629
Publication Date(Web):21 May 2015
DOI:10.1039/C5CP00046G
A quantum spin Hall (QSH) effect is quite promising for applications in spintronics and quantum computations, but at present, can only be achieved at ultralow temperatures. The determination of large-gap QSH insulators is critical to increase the operating temperature. By using first-principles calculations, we demonstrate that the stable hydrogenated stanene with a dumbbell-like structure (DB stanane) has large topological nontrivial band gaps of 312 meV (Γ point) and 160 meV for the bulk, characterized by a topological invariant of Z2 = 1 because of s–pxy band inversion. Helical gapless edge states appear in the nanoribbon structures with high Fermi velocity comparable to that of graphene. The nontrivial topological states are robust against the substrate effects. The realization of this material is a feasible solution for the applications of QSH effects at room temperature and can be beneficial in the fabrication of high-speed spintronics devices.
Co-reporter:Xin Chen, Linyang Li and Mingwen Zhao
RSC Advances 2015 vol. 5(Issue 89) pp:72462-72468
Publication Date(Web):20 Aug 2015
DOI:10.1039/C5RA10712A
The quantum spin Hall (QSH) effect in two-dimensional topological insulators (2D TIs) is promising for building nanoscaled devices with low energy consumption. The 2D TIs with a bulk band gap larger than the atomic thermal motion energy at room temperature (∼26 meV) are essential for achieving room-temperature QSH effects. We proved from first-principles that this goal may be reached in a hydrogenated germanium–tin (Sn6Ge4H4) dumbbell (DB) structure, where spin–orbit coupling (SOC) opens a bulk band gap of 235 meV. The topological nontriviality is related to the band inversion of s–pxy of Sn atoms induced by surface hydrogenation and can be characterized by a topological invariant of Z2 = 1. This work offers a promising candidate material for achieving long-desired room-temperature QSH effects.
Co-reporter:Xiaoming Zhang and Mingwen Zhao
RSC Advances 2015 vol. 5(Issue 13) pp:9875-9880
Publication Date(Web):06 Jan 2015
DOI:10.1039/C4RA15861J
A quantum anomalous Hall effect (QAHE) has been realized in the Cr-doped magnetic topological insulator at an extremely low temperature (∼30 mK). From first-principles, we predict that QAHE can also be achieved in a well-designed graphene nanomesh without the requirement of transition metal doping. The ferromagnetic ordering arises mainly from the in-plane pxy orbitals, leading to a Curie temperature of 830 K. The bulk band gap due to the intrinsic spin–orbital coupling (SOC) of the pxy orbitals is about 3.7 meV, corresponding to an operating temperature of 43 K. This work reveals a viable approach for realizing QAHE in metal-free graphene nanostructures.
Co-reporter:Aizhu Wang;Aijun Du
Nano Research 2015 Volume 8( Issue 12) pp:3823-3829
Publication Date(Web):2015 December
DOI:10.1007/s12274-015-0882-z
A quantum-spin-Hall (QSH) state was achieved experimentally, albeit at a low critical temperature because of the narrow band gap of the bulk material. Twodimensional topological insulators are critically important for realizing novel topological applications. Using density functional theory (DFT), we demonstrated that hydrogenated GaBi bilayers (HGaBi) form a stable topological insulator with a large nontrivial band gap of 0.320 eV, based on the state-of-the-art hybrid functional method, which is implementable for achieving QSH states at room temperature. The nontrivial topological property of the HGaBi lattice can also be confirmed from the appearance of gapless edge states in the nanoribbon structure. Our results provide a versatile platform for hosting nontrivial topological states usable for important nanoelectronic device applications.
Co-reporter:Shuxian Wang;Haohai Yu;Huaijin Zhang;Aizhu Wang;Yanxue Chen;Liangmo Mei;Jiyang Wang
Advanced Materials 2014 Volume 26( Issue 21) pp:3538-3544
Publication Date(Web):
DOI:10.1002/adma.201306322
Co-reporter:Aizhu Wang, Xiaoming Zhang and Mingwen Zhao
Nanoscale 2014 vol. 6(Issue 19) pp:11157-11162
Publication Date(Web):24 Jul 2014
DOI:10.1039/C4NR02707H
Two-dimensional (2D) graphitic carbon nitride materials have been drawing increasing attentions in energy conversion, environment protection and spintronic devices. Here, based on first-principles calculations, we demonstrate that the already-synthesized honeycomb lattice of s-triazines with a chemical formula of C6N6 (g-C6N6) has topologically nontrivial electronic states characterized by px,y-orbital band structures with a topological invariant of Z2 = 1, and stronger spin-orbital coupling (SOC) than both graphene and silicene. The band gaps opened in the px,y-orbital bands due to SOC are 5.50 meV (K points) and 8.27 eV (Γ point), respectively, implying that the quantum spin Hall effect (QSHE) could be achieved in this 2D graphitic carbon nitride material at a temperature lower than 95 K. This offers a viable approach for searching for 2D Topological Insulators (TIs) in metal-free organic materials.
Co-reporter:Hongxia Bu, Mingwen Zhao, Wenzheng Dong, Shuangwen Lu and Xiaopeng Wang
Journal of Materials Chemistry A 2014 vol. 2(Issue 15) pp:2751-2757
Publication Date(Web):28 Jan 2014
DOI:10.1039/C3TC32083A
Carbon has abundant allotropes with superhardness, but few of them are metallic. From first-principles calculations, we propose a stable metallic carbon allotrope (Hex-C24) phase with superhardness. The Hex-C24 can be thought of as a superlattice of carbon nanotubes and graphene nanoribbons composed of sp2- and sp3-hybridized carbon atoms. A possible synthetic route towards Hex-C24 from graphyne multilayers is evaluated by calculating the transition states between the two phases. Our calculations show that at a uniaxial pressure of around 25 GPa, the energy barrier of this endothermic transition is estimated to be 0.04 eV per atom, while at a pressure of 34 GPa, the transition is barrierless for specific initial configurations. The cohesive energy, elastic constants, and phonon frequencies unambiguously confirm the structural stability. The hardness of the Hex-C24 is estimated to exceed 44.54 GPa, which is 1/2 that of diamond. The Hex-C24 phase is metallic with several bands across the Fermi level. Both mechanical and metallic properties of Hex-C24 are anisotropic.
Co-reporter:Xiujie He, Jinliang Song, Huihao Xia, Jie Tan, Baoliang Zhang, Zhoutong He, Xingtai Zhou, Zhiyong Zhu, Mingwen Zhao, Xiangdong Liu, Li Xu, Shuo Bai
Carbon 2014 Volume 68() pp:95-103
Publication Date(Web):March 2014
DOI:10.1016/j.carbon.2013.10.058
Isotropic pyrolytic carbon (IPyC) prepared at 1300 °C by chemical vapor deposition was implanted with 129Xe26+ ions to obtain a wide range of information and understanding about the coating materials in nuclear energy field. Microstructure of the pristine and ion-implanted IPyC on nuclear graphite substrate was firstly investigated using polarized light microscopy, scanning and transmission electron microscopy, X-ray diffraction, Raman spectroscopy, nanoindentation, and X-ray photoemission spectroscopy. It was demonstrated that the Xe ion irradiation resulted in concurrent changes in both physical and chemical structures of our standard polycrystalline sample. Influences of the thermal annealing temperature on the properties of the implanted IPyC at 500 and 1000 °C were also studied. Ion-irradiation gave rise to the formation of structural deterioration along a and c axis, accompanying with the appearance of widespread clastic morphology among the irradiated zone of IPyC. There was a dose window that could be used to tune the mechanical properties of IPyC: the nanohardness and Young’s modulus increased after an irradiation, but decreased as the amorphization was reached.
Co-reporter:Xiaobiao Liu, Jie Tan, Aizhu Wang, Xiaoming Zhang and Mingwen Zhao
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 42) pp:23286-23291
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4CP03478C
Covalent organic frameworks (COFs) hold great promise in several applications, such as sieves, catalytic supports and gas storage because of their unique structures and electronic properties. However, most of these metal-free COFs are nonmagnetic and cannot be directly used in spintronics. Here, based on first-principles calculations, we predict that substitutional doping of COF-5 with nitrogen and boron atoms can modify the electronic structures, inducing stable electron spin-polarization in the framework. The preferability of the different doping sites is checked. The electronic structures of the doped COF-5 are dependent on the doping sites and doping atoms, which offer high degrees of freedom to tune the electronic properties. Kagome lattices of S = 1/2 spins can be achieved in the COF-5, suggesting a promising candidate for spin-liquid materials.
Co-reporter:Guihua Li, Jie Tan, Xiangdong Liu, Xiaopeng Wang, Feng Li, Mingwen Zhao
Chemical Physics Letters 2014 Volumes 595–596() pp:20-24
Publication Date(Web):18 March 2014
DOI:10.1016/j.cplett.2014.01.042
•A- and Z-silicane nanoribbons (NRs) are studied by DFT.•Structures of both A- and Z-hybrid silicane–silicene-NRs are studied.•Band gaps of hybrid A-system can be separated into three different families.•Gaps of hybrid Z-systems depend on the hydrogenation of Z-chains at the interface.•The electronic and magnetic properties of the hybrid Z-system can be controlled.The intriguing electronic and magnetic properties of silicane and hybrid silicane–silicene nanoribbons are investigated by means of first-principles calculations. Both armchair and zigzag silicane nanoribbons are nonmagnetic semiconductors. Meanwhile, the band gap of armchair hybrid silicane–silicene nanoribbons without spin-split, which is mainly determined by the silicene part, can be separated into three different families. The energy gap of the zigzag counterparts depends on the hydrogenation of the zigzag silicon chain at the silicane–silicene interface. Thus, controlling the hydrogenation ratio along the ribbon width can provide a basis for modulating the electronic and magnetic properties of the zigzag hybrid nanoribbons.Graphical abstract
Co-reporter:Xiujie He, Jinliang Song, Jie Tan, Baoliang Zhang, Huihao Xia, Zhoutong He, Xingtai Zhou, Mingwen Zhao, Xiangdong Liu, Li Xu, Shuo Bai
Journal of Nuclear Materials 2014 Volume 448(1–3) pp:1-3
Publication Date(Web):May 2014
DOI:10.1016/j.jnucmat.2014.01.034
SiC coating is produced on a nuclear graphite (NG) substrate using chemical vapor deposition at 1150 °C to protect it from molten salt diffusion. Infiltration studies, performed in molten FLiNaK salt under an argon atmosphere at 5 atm, show that uncoated NG exhibits significantly higher weight gain than SiC-coated NG. The continuous and compact SiC coating exhibits excellent infiltration resistance in liquid fluoride salt as confirmed by synchrotron radiation X-ray microbeam fluorescence.
Co-reporter:Guihua Li, Feng Li, Xiaopeng Wang, Mingwen Zhao, Xiangdong Liu
Physica E: Low-dimensional Systems and Nanostructures 2014 Volume 59() pp:235-242
Publication Date(Web):May 2014
DOI:10.1016/j.physe.2014.01.014
•Energetic and structural properties of Au/Au-dimer adsorbed on pristine and defective graphene (Gra) and boron nitride monolayer (BN) are investigated using density functional theory.•The binding of Au/Au-dimer to a pristine support is weak, stronger binding could be achieved by introducing a defect in the surface.•BN-NC support Au/Au-dimer well from aspect of adsorption energy.•Bands near the Fermi level of Au/Au-dimer on BN-NC mainly comes from the hybrid of Au-5d, 6s, C-2p, and B-2p.Energetic and structural properties of gold atom (Au) and gold dimer (Au dimer) adsorbed on pristine and defective graphene (Gra) and boron nitride monolayer (BN) are investigated using density functional theory. Substitutional doping models in the neutral charge state are considered by replacing the C site in graphene with B or N atom impurities (Gra-CB and Gra-CN) or by doping the B or N sites in the BN sheet by a C atom (BN-BC and BN-NC). It is shown that while the binding of Au/Au-dimer to a pristine support is weak, stronger binding could be achieved by introducing a defect in the surface indicating that defects can trap metal atoms. It is found that Gra-CB and BN-NC support Au/Au-dimer well and BN-NC is more preferable from aspect of adsorption energy. Interaction between Au/Au-dimer and the BN-NC substrates is explained by assigning appropriate partial charge densities of the valence band maximum (VBM) and conduction band minimum (CBM) at the Г point and projected densities of states (PDOS). The results demonstrate that both pristine and defective BN surfaces can no longer be treated as inert supports for Au/Au-dimer.
Co-reporter:Mingwen Zhao, Aizhu Wang and Xiaoming Zhang
Nanoscale 2013 vol. 5(Issue 21) pp:10404-10408
Publication Date(Web):14 Aug 2013
DOI:10.1039/C3NR03323F
The spin ordering in kagome lattices has long been studied in the search for real materials with a spin-liquid ground state. The synthesis of a nickel bis-dichiolene complex (Ni3C12S12) nanosheet (T. Kambe et al., J. Am. Chem. Soc., 2013, 135, 2462) paved a way for realizing real two-dimensional kagome lattices. Using first-principles calculations, we predicted that a ferromagnetic kagome spin lattice with S = 3/2 on lattice vertices can be achieved in an Mn3C12S12 monolayer formed by substituting Ni with Mn atoms in nonmagnetic Ni3C12S12. Monte Carlo simulations on the basis of the Ising model suggest that it has a Curie temperature of about 212 K. A ferromagnetic Mn3C12S12 monolayer is half metallic with high carrier mobility in one spin channel and a band gap of 1.54 eV in another spin channel, which is quite promising for spintronic device applications. Additionally, a small band gap opens up at the Dirac point of the kagome bands due to the spin–orbital coupling effects, which may be implementable for achieving a quantum anomalous Hall effect.
Co-reporter:Xiaoming Zhang, Mingwen Zhao, Aizhu Wang, Xiaopeng Wang and Aijun Du
Journal of Materials Chemistry A 2013 vol. 1(Issue 39) pp:6265-6270
Publication Date(Web):06 Aug 2013
DOI:10.1039/C3TC31213E
Polymeric graphitic carbon nitride materials have attracted increasing attention in recent years owing to their potential applications in energy conversion, environment protection, and so on. Here, from first-principles calculations, we report the electronic structure modification of graphitic carbon nitride (g-C3N4) in response to carbon doping. We showed that each dopant atom can induce a local magnetic moment of 1.0 μB in non-magnetic g-C3N4. At a doping concentration of 1/14, the local magnetic moments of the most stable doping configuration which has the dopant atom at the center of the heptazine unit prefer to align in a parallel way leading to long-range ferromagnetic (FM) ordering. When the joint N atom is replaced by a C atom, the system favors an antiferromagnetic (AFM) ordering in an unstrained state, but can be tuned to ferromagnetism (FM) by applying biaxial tensile strain. More interestingly, the FM state of the strained system is half-metallic with abundant states at the Fermi level in one spin channel and a band gap of 1.82 eV in another spin channel. The Curie temperature (Tc) was also evaluated using a mean-field theory and Monte Carlo simulations within the Ising model. Such tunable electron spin-polarization and ferromagnetism are quite promising for the applications of graphitic carbon nitride in spintronics.
Co-reporter:Hongxia Bu, Mingwen Zhao, Aizhu Wang, Xiaopeng Wang
Carbon 2013 Volume 65() pp:341-348
Publication Date(Web):December 2013
DOI:10.1016/j.carbon.2013.08.035
Graphdiyne is a recently-synthesized carbon allotrope with a framework of sp- and sp2-hybridized carbon atoms. From first-principles calculations, we propose a possible transition of graphdiyne to a novel carbon allotrope (h-carbon) whose structure is a superlattice of carbon nanotubes and graphene nanoribbons. The energy barrier of this endothermic transition was estimated to be 4.30 kcal/mol at zero pressure, which is much lower than that of the graphite–diamond transition at high pressure. First-principles calculations on the phonon spectrum and the elastic constants of the h-carbon revealed that it is kinetically and mechanically stable. This unique framework of sp2- and sp3-hybridized carbon atoms is energetically neutral versus diamond. The hardness of the h-carbon (35.52 GPa) is 1/3 that of diamond and very close to β-SiC crystal. Accurate electronic structure calculations based on the Heyd, Scuseria, and Ernzerhof approach and GW approximation indicate that the h-carbon is a semiconducting material with a band gap of 2.20–2.56 eV.
Co-reporter:Jie Tan, Weifeng Li, Xiujie He and Mingwen Zhao
RSC Advances 2013 vol. 3(Issue 19) pp:7016-7022
Publication Date(Web):04 Mar 2013
DOI:10.1039/C3RA40502H
Two-dimensional (2D) organic materials with stable electron spin polarization, ferromagnetic ordering and half-metallicity are quite promising for spintronics, due to their long spin coherence length and mechanical flexibility. Here, using porphyrin molecules as building blocks, we propose a novel 2D periodic organic nanomaterial (2D-polyporphyrin) from first-principles calculations. The recent experimental progress on the one-dimensional (1D) Zn-porphyrin arrays hints the plausibility of these 2D polyporphyrin frameworks. We show that electron spin-polarization can be achieved in both metal-free and transition-metal-embedded 2D-polyporphyrins. Cr-polyporphyrin (Cr-PP) in particular has stable ferromagnetic ordering with a Curie temperature (Tc) of about 187 K as indicated by the Monte Carlo simulations based on the 2D Ising model, which is much higher than that reported in the 2D Mn-phthalocyanine framework. The ferromagnetic Cr-PP nanosheet can be tuned to half-metallic by electron doping. The present work opens up an avenue for the development of 2D organic nanostructures with stable ferromagnetism and half-metallicity.
Co-reporter:Hongxia Bu, Xiaopeng Wang, Yan Xi, Xiaoyang Zhao, Mingwen Zhao
Diamond and Related Materials 2013 Volume 37() pp:55-63
Publication Date(Web):August 2013
DOI:10.1016/j.diamond.2013.04.016
•We report a new family of carbon allotropes consisting of sp- and sp3-hybridized atoms.•In contrast to early estimation, yne-diamonds are unlikely superhard materials, but exhibit good ductility.•The hardness, density, and band gaps are tunable as the ratio of acetylenic bonds increases.•The optical parameters can be used to identify these novel carbon allotropes.Yne-diamonds are novel carbon allotropes designed by inserting acetylenic bonds into the framework of diamond. Varying the ratio of acetylenic bonds yields a new family of carbon allotropes consisting of sp- and sp3-hybridized atoms. The study of these novel carbon frameworks is becoming a topic of increasing interest. Here, we report our systematic studies on the stability, mechanical, electronic and optical properties of yne-diamonds. Our calculations indicate that yne-diamonds are mechanically stable, although they are energetically less favorable than diamond due to sp-hybridized carbon atoms in the frameworks. In contrast to early estimation, yne-diamonds are unlikely superhard materials, but exhibit good ductility. The band gaps of yne-diamonds vary from 0.165 eV to 4.850 eV, as the ratio of acetylenic bonds increases. The energetically most favorable yne-diamond has a direct band gap of 2.916 eV. The optical properties of these carbon allotropes are also discussed.
Co-reporter:Xiujie He, Jinliang Song, Li Xu, Jie Tan, Huihao Xia, Baoliang Zhang, Zhoutong He, Lina Gao, Xingtai Zhou, Mingwen Zhao, Zhiyong Zhu, Shuo Bai
Journal of Nuclear Materials 2013 Volume 442(1–3) pp:306-308
Publication Date(Web):November 2013
DOI:10.1016/j.jnucmat.2013.09.015
Infiltration studies were performed on uncoated nuclear graphite and isotropic pyrolytic carbon (PyC) coated graphite in molten FLiNaK salt at 650 °C under argon atmosphere at 1, 3 and 5 atm. Uncoated graphite shows weight gain more obviously than that of PyC coated graphite. Nuclear graphite with PyC coating exhibits excellent infiltration resistance in molten salt due to the small open porosity as conformed from scanning electron microscopy and mercury injection experiments.
Co-reporter:Zhenhai Wang, Mingwen Zhao, Xiaopeng Wang, Yan Xi, Xiujie He, Xiangdong Liu and Shishen Yan
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 45) pp:15693-15698
Publication Date(Web):25 Sep 2012
DOI:10.1039/C2CP42115A
The band alignment in ZnO–GaN and related heterostructures is crucial for uses in solar harvesting technology. Here, we report our density functional calculations of the band alignment and optical properties of ZnO–GaN and ZnO–(Ga1−xZnx)(N1−xOx)–GaN heterostructures using a Heyd–Scuseria–Ernzerhof (HSE) hybrid functional. We found that the conventional GGA functionals underestimate not only the band gap but also the band offset of these heterostructures. Using the hybrid functional calculations, we show that the (Ga1−xZnx)(N1−xOx) solid solution has a direct band gap of about 2.608 eV, in good agreement with the experimental data. More importantly, this solid solution forms type-II band alignment with the host materials. A GaN–(Ga1−xZnx)(N1−xOx)–ZnO core–shell solar cell model is presented to improve the visible light absorption ability and carrier collection efficiency.
Co-reporter:XiuJie He;Jie Tan;HongXia Bu;HongYu Zhang
Science Bulletin 2012 Volume 57( Issue 23) pp:3080-3085
Publication Date(Web):2012/08/01
DOI:10.1007/s11434-012-5300-2
The electronic structures and optical properties of graphyne consisting of sp- and sp2-hybridized carbon atoms are studied using first-principles calculations. A tight-binding model of the 2pz orbitals are proposed to describe the electronic bands near the Fermi level. The results show that the natural band gap of graphyne originates from the inhomogeneous π bindings between differently-hybridized carbon atoms. The interlayer interactions of bulk graphyne narrow the band gap to 0.16 eV and result in redshift of the optical spectral peaks as compared to single-layered graphyne.
Co-reporter:Hongyu Zhang, Xiujie He, Mingwen Zhao, Meng Zhang, Lixia Zhao, Xiaojuan Feng, and Youhua Luo
The Journal of Physical Chemistry C 2012 Volume 116(Issue 31) pp:16634-16638
Publication Date(Web):July 15, 2012
DOI:10.1021/jp304908p
First-principles calculations are carried out to investigate the hydrogen separation characteristics of two-dimensional carbon allotropes consisting of sp- and sp2-hybridized carbon atoms, i.e., graphyne, graphdiyne, and rhombic-graphyne. The selectivities for H2 over several gas molecules, including CO, N2, and CH4, are found to be sensitive to the pore sizes and shapes. The penetration barriers generally decrease exponentially with the pore sizes. Our results reveal that graphyne with small pores is unsuitable for the purpose of hydrogen separation. Graphdiyne, with larger pores, exhibits a high selectivity (109) for hydrogen over large gas molecules such as CH4, but a relatively low selectivity (103) over small molecules such as CO and N2. The large differences in diffusion barriers for molecules penetration through a rhombic-graphyne monolayer, which possesses pore size in between that of graphyne and graphdiyne, lead to a high selectivity (>1016) for hydrogen separation from the others. The results suggest that the abundant pores of different sizes in these carbon allotropes make them ideal molecular sieves for gas separation applications directed toward different separation needs and objectives.
Co-reporter:Hongxia Bu, Mingwen Zhao, Hongyu Zhang, Xiaopeng Wang, Yan Xi, and Zhenhai Wang
The Journal of Physical Chemistry A 2012 Volume 116(Issue 15) pp:3934-3939
Publication Date(Web):March 21, 2012
DOI:10.1021/jp300107d
Graphdiyne, consisting of sp- and sp2-hybridized carbon atoms, is a new member of carbon allotropes which has a natural band gap ∼1.0 eV. Here, we report our first-principles calculations on the stable configurations and electronic structures of graphdiyne doped with boron–nitrogen (BN) units. We show that BN unit prefers to replace the sp-hybridized carbon atoms in the chain at a low doping rate, forming linear BN atomic chains between carbon hexagons. At a high doping rate, BN units replace first the carbon atoms in the hexagons and then those in the chains. A comparison study indicates that these substitution reactions may be easier to occur than those on graphene which composes purely of sp2-hybridized carbon atoms. With the increase of BN component, the band gap increases first gradually and then abruptly, corresponding to the transition between the two substitution motifs. The direct-band gap feature is intact in these BN-doped graphdiyne regardless the doping rate. A simple tight-binding model is proposed to interpret the origin of the band gap opening behaviors. Such wide-range band gap modification in graphdiyne may find applications in nanoscaled electronic devices and solar cells.
Co-reporter:Xuejuan Zhang, Shijie Xie, Yingcai Fan, Zhenhai Wang, Hongyu Zhang, Mingwen Zhao
Physica E: Low-dimensional Systems and Nanostructures 2011 Volume 43(Issue 8) pp:1522-1527
Publication Date(Web):June 2011
DOI:10.1016/j.physe.2011.04.021
We perform first-principles calculations to study the structural, energetic, and electronic properties of axial and core–shell ZnS/ZnO heteronanotubes. The results show that axial heteronanotubes have smooth and defect-free interfaces. The charge redistribution near the interfaces gives rise to a built-in electric field. The band scheme diagram of axial armchair heteronanotube displays the characteristic of type-Ι band alignment, while that of the ZnO@ZnS core–shell heteronanotube has a type-II band alignment. These heteronanotubes may have practical applications as UV optically active materials.Highlights► We model the structural and electronic properties of axial and core–shell ZnS/ZnO heteronanotubes. ► Axial heteronanotubes have smooth and defect-free interfaces. ► Charge redistribution near the interfaces gives rise to a built-in electric field. ► Axial (5,5) heteronanotube has a type-Ι band alignment while the core–shell one has a type-ΙI band alignment. ► They are possible candidates as UV optically active materials.
Co-reporter:Hongyu Zhang ; Mingwen Zhao ; Xiujie He ; Zhenhai Wang ; Xuejuan Zhang ;Xiangdong Liu
The Journal of Physical Chemistry C 2011 Volume 115(Issue 17) pp:8845-8850
Publication Date(Web):April 13, 2011
DOI:10.1021/jp201062m
We have carried out first-principles calculations to explore the energetics and dynamics of Li in graphyne, a novel carbon allotrope consisting of sp–sp2 hybridized carbon atoms, relevant for anode lithium intercalation in rechargeable Li-ion batteries. In contrast to graphite where Li diffusion is confined in the interlayer space (in-plane diffusion), the unique atomic arrangement and electronic structures enable both in-plane and out-plane diffusion of Li ions in graphyne with moderate barriers, 0.53–0.57 eV. The highest Li intercalation density in graphyne can be LiC4, exceeding the up limit of LiC6 in the commonly used graphite. The high lithium mobility and high storage capacity make graphyne a promising candidate for the anode material in battery applications.
Co-reporter:Yan Xi ; Mingwen Zhao ; Xiaopeng Wang ; Shijie Li ; Xiujie He ; Zhenhai Wang ;Hongxia Bu
The Journal of Physical Chemistry C 2011 Volume 115(Issue 36) pp:17743-17749
Publication Date(Web):August 8, 2011
DOI:10.1021/jp2057157
Graphene quantum dots (QDs) hold great promises in spintronics. Here, we report our predictions of honeycomb-patterned QDs beyond graphene, on the basis of first-principles calculations and an extended Hubbard model. Our calculations showed that the electronic structures and spin-polarization of boron nitride (BN) and silicon carbide (SiC) QDs can be well tuned by controlling the shape and size of the QDs. Edge hydrogenation can not only greatly improve the stability but also diminish the spin-polarization of BN-QDs. Triangular SiC-QDs have spin-polarized ground states, and the magnetic moments increase with the increase of QD size. Hexagonal SiC-QDs, however, possess spin-unpolarized ground states whose energy gaps decrease with the increase of QD size. To understand the origins of the composition- and shape-dependent spin-polarization of these honeycomb-patterned QDs, we extended the single-orbital Hubbard model of graphene QDs by taking into account the onsite energy differences of the two sublattices. Our extended Hubbard model reproduces well the results of first-principles calculations and offers a simple model to predict the electronic structures of honeycomb-patterned QDs.
Co-reporter:Weifeng Li, Mingwen Zhao, Xian Zhao, Yueyuan Xia and Yuguang Mu
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 41) pp:13699-13706
Publication Date(Web):20 Sep 2010
DOI:10.1039/C003524F
Density functional theory calculations are performed to explore vacancy-induced magnetism in graphene. The hydrogen saturation not only stabilizes the vacancy structure but also induces distinct magnetic coupling depending on the defect distribution: weak magnetic coupling between defects on different sublattices and strong coupling between defects on the same sublattice. Ferromagnetic ordering has to be accompanied with a semiconducting property. The interaction integral J between defective spins decreases linearly with the increase of the distance between them. Based on the 2D Ising model and Monte Carlo simulations, the possible highest Curie temperature Tc of defective graphene is predicted to be lower than 500 K.
Co-reporter:Hongyu Zhang, Mingwen Zhao, Tao He, Xuejuan Zhang, Zhenhai Wang, Zexiao Xi, Shishen Yan, Xiangdong Liu, Yueyuan Xia and Liangmo Mei
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 41) pp:13674-13680
Publication Date(Web):24 Sep 2010
DOI:10.1039/C002719G
We carried out first-principles calculations to explore the oxidative longitudinal unzipping of single-walled carbon nanotubes (SWCNTs) of different diameters and chiralities. We found that the initial attack leading to nanotube unzipping prefers to occur in the middle region for armchair tubes, and at the tube ends for zigzag tubes. Once the initial attack has taken place, by overcoming an energy barrier whose value decreases with increasing tube diameter, the subsequent breakage of C–C bonds parallel to the ones broken in the former process is barrierless. The energetically preferred unzipping path is parallel to the tube axis for armchair tubes, resulting in straight zigzag-edged graphene nanoribbons. For zigzag tubes, there are two energetically equivalent unzipping directions corresponding to the opening of two types of C–C bonds tilted towards the tube axis, giving rise to helical unzipping paths. This is disadvantageous for the production of straight graphene ribbons. A local curvature modulation procedure is proposed to efficiently control the location of the initial attack and thus the shape of the produced graphene nanoribbons.
Co-reporter:Hongyu Zhang, Mingwen Zhao, Xinmei Yang, Huihao Xia, Xiangdong Liu, Yueyuan Xia
Diamond and Related Materials 2010 Volume 19(Issue 10) pp:1240-1244
Publication Date(Web):October 2010
DOI:10.1016/j.diamond.2010.06.010
The study of dynamics of point defects in graphite is crucial for understanding the evolution of defect-induced ferromagnetism in 12C+ irradiated graphite. In this work, we perform first-principles calculations to explore the diffusion and coalescence of vacancies and interstitials in graphite. Different kinds of point defects, such as monovacancy, divacancy, ‘bridge’ and ‘spiro’ interstitials are considered using non-interacting and interacting models. The energetics, the diffusion paths, and the migration energies of these defects and the energy barriers for the reaction between these defects are predicted. The annealing behaviors of point defects and related ferromagnetism found in 12C+ irradiated graphite are discussed.
Co-reporter:Tao He, Fengchun Pan, Zexiao Xi, Xuejuan Zhang, Hongyu Zhang, Zhenhai Wang, Mingwen Zhao, Shishen Yan and Yueyuan Xia
The Journal of Physical Chemistry C 2010 Volume 114(Issue 20) pp:9234-9238
Publication Date(Web):May 3, 2010
DOI:10.1021/jp912143p
The geometric, electronic, and magnetic properties of titania nanoribbons (TiO2NRs) are investigated with use of first-principles calculations within density-functional theory. The TiO2NRs formed by cutting ultrathin TiO2 nanosheet along armchair and zigzag axes have high energetic stability. Zigzag TiO2NRs are more preferable than armchair ones. The electronic structures of TiO2NRs highly depend on the growth orientation and the ribbon width. Introducing oxygen vacancy defects into the edges of zigzag TiO2NRs under poor oxygen conditions can reduce the band gap and trigger the spin-polarization of edge states. These TiO2NRs with well-defined atomic structures, high stability, and tunable electronic properties are expected to have potential applications in solar cells, spintronic devices, and sensors.
Co-reporter:Hongyu Zhang, Xuejuan Zhang, Mingwen Zhao, Zhenhai Wang
Physica E: Low-dimensional Systems and Nanostructures 2010 Volume 43(Issue 2) pp:610-613
Publication Date(Web):December 2010
DOI:10.1016/j.physe.2010.10.004
Adsorption and diffusion of silicon on the exterior surface of carbon nanotubes are crucial for the growth of silicon carbide nanotubes (SiCNTs) from carbon nanotubes. We have carried out first-principles calculations to explore these processes. We found that a silicon atom prefers to be adsorbed at the bridge site above a C−C bond tilted to the tube axis with binding energy of 1.29−1.61 eV. The adsorbed silicon atoms have high mobility with diffusion energy barriers less than 0.06 eV. The energetically favorable diffusion paths are oriented along the tube axis.Research Highlights► Stable adsorption sites for a single Si atom adsorption on the outer surface of single-walled carbon nanotubes were studied in detail. ► Relative stability of equilibrium configurations above different C−C bonds cannot be judged only by the relative reactivity of the C−C bonds, due to the radial deformation of tube caused by adsorption of Si atom. This is contrary to the case of the adsorption of C and N. ► Energetically favorable diffusion paths were determined, which is crucial for the growth of silicon carbide nanotubes. No studies on this issue have been reported.
Co-reporter:Weifeng Li, Mingwen Zhao, Yueyuan Xia, Ruiqin Zhang and Yuguang Mu
Journal of Materials Chemistry A 2009 vol. 19(Issue 48) pp:9274-9282
Publication Date(Web):26 Oct 2009
DOI:10.1039/B908949G
Magnetism and the mechanism of magnetic coupling in graphene decorated with monovalent and divalent adsorbates were investigated using first-principles calculations based on spin polarized density functional theory. The effects of adsorption concentration and the electronegativity of the adsorbate species on the magnetic and electronic properties were analyzed. For monovalent chemisorptions, the magnetic order originates from the instability of π electrons induced by the adsorption, opening a narrow energy gap and resulting in antiparallel spin directions on adjacent carbon atoms on the graphene sheet. The magnetic order is only possible for the separation between the adsorbing sites less than 10 Å. On the contrary, divalent chemisorptions cause long-range magnetic coupling, which is originated from the exchange interactions between localized nonbonding π electrons (spin-polarized) mediated by the conduction π electrons around the Fermi energy, similar to the s–d interaction in transition metals. We demonstrate that our results are well consistent with recent experimental findings.
Co-reporter:Zhenhai Wang, Mingwen Zhao, Tao He, Hongyu Zhang, Xuejuan Zhang, Zexiao Xi, Shishen Yan, Xiangdong Liu and Yueyuan Xia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 29) pp:12731-12735
Publication Date(Web):June 25, 2009
DOI:10.1021/jp903736v
The energetic stability and electronic properties of hydrogenated silicon carbide nanowires (SiCNWs) with zinc blende (3C) and wurtzite (2H) structures are investigated using first-principles calculations within density functional theory and generalized gradient approximation. The [111]-orientated 3C-SiCNWs are energetically more stable than other kinds of NWs with similar size. In contrast to the indirect band gap features of SiC bulk crystals, all the NWs have direct band gaps except those orientating along the [112] direction. The band gaps of these NWs decrease with the increase of wire size. The direct band gap can be kept for the [111]-orientated 3C-SiCNWs with diameters up to 2.8 nm. The superior stability and electronic structures of the 3C-SiCNWs growing along the [111] direction are in good agreement with the experimental results.
Co-reporter:Zhenhai Wang, Mingwen Zhao, Tao He, Xuejuan Zhang, Zexiao Xi, Shishen Yan, Xiangdong Liu and Yueyuan Xia
The Journal of Physical Chemistry C 2009 Volume 113(Issue 3) pp:856-861
Publication Date(Web):2017-2-22
DOI:10.1021/jp808231s
The energetics and atomic and electronic structures of silicon carbide (SiC) nanowires (NWs) and nanotubes (NTs) with radii ranging from 0.45 to 2.9 nm are investigated using density functional theory in conjunction with an atomistic band-order potential. It is found that the formation energy (Eform) of the NWs decreases with the increase of wire radius, and that of the NTs decreases with the increase of wall thickness, irrespective of the tube radius. NTs with faceted single-crystalline walls are energetically more favorable than the cylindrical single- or multiwalled SiC NTs. Due to the surface states, the faceted NWs and NTs possess indirect band gaps, which are narrower than that of bulk SiC crystal. The highest valence band and the lowest conduction band mainly arise from the undercoordinated C and Si atoms on the facets. The surface states can be passivated by surface hydrogenation, and the hydrogenated SiC NWs and NTs become direct-band gap semiconductors with wider band gaps than that of bulk SiC crystal.
Co-reporter:Zexiao Xi, Mingwen Zhao, Ruiqin Zhang, Shishen Yan, Tao He, Weifeng Li, Xuejuan Zhang, Xiaohang Lin, Zhenhai Wang, Xiangdong Liu and Yueyuan Xia
The Journal of Physical Chemistry C 2008 Volume 112(Issue 44) pp:17071-17075
Publication Date(Web):2017-2-22
DOI:10.1021/jp803668y
We performed first-principles calculations on silica nanorings (NRs) designed via the assembly of two- (2MR), three- (3MR), four- (4MR), and six-membered rings (6MR) in a number of different ways. The stable configurations, energetics, and electronic structures of these NRs are presented. The most stable configurations were found to be size-dependent and to possess different structural features at different size ranges. For small-size silica NRs (SiO2)n with n < 12, the configurations with 2MR−3MR hybrid structures (2−3MR-NRs) were energetically most stable. For 12 < n < 22, the NRs formed from linked 2MRs (2MR-NRs) became most favorable. For n > 22, the configurations composed of uniformly hybrid 2MRs and 4MRs (2−4MR-NRs) were the most stable structures. The 2−4MR-NRs had the narrowest HOMO−LUMO gaps, which decreased with decreasing n.
Co-reporter:Mingwen Zhao, Yueyuan Xia, Zhenyu Tan, Xiangdong Liu, Feng Li, Buda Huang, Yanju Ji, Liangmo Mei
Chemical Physics Letters 2004 Volume 389(1–3) pp:160-164
Publication Date(Web):1 May 2004
DOI:10.1016/j.cplett.2004.03.082
Abstract
We study the strain energy and stability of single-walled aluminum nitride nanotubes (SWAlNNTs) using density functional calculations. We find that SWAlNNTs have strain energy higher than 0.68 eV/atom relative to AlN cubic materials. The energy cost required in order to wrap up an AlN graphitic sheet in to a tube is lower than that required to form BN, GaN and carbon nanotubes with similar diameters from their corresponding graphitic sheets. Our simulations also reveal that SWAlNNTs once synthesized can stably exist at room temperature, and start to melt when temperature is higher than 600 K.
Co-reporter:Bo Yang, Hongcai Zhou, Xiaoming Zhang, Xiaobiao Liu, Mingwen Zhao
Carbon (March 2017) Volume 113() pp:
Publication Date(Web):March 2017
DOI:10.1016/j.carbon.2016.11.028
Graphynes consisting of sp- and sp2-hybridized carbon atoms represent two-dimensional allotropes of the carbon family with intriguing properties. Based on first-principles calculations, we propose a stable three-dimensional (3D) framework (with the space group of I41/amd) of sp- and sp2-hybridized carbon atoms termed super-graphyne. The exotic property of super-graphyne lies in the semi-metallic features with a highly anisotropic band structure in the reciprocal space. Along the Г-X direction, there is a Dirac cone centered at the X point with the Fermi velocity higher than that in graphene, while the bands are nearly dispersionless along the X-P direction, featured by node line Dirac semimetals. Additionally, electron spin-polarization is predicted on the bared (001) surface of super-graphyne, which preserves the semi-metallic properties. These interesting results not only enrich the 3D allotropes of the carbon family but could also find applications in electronic devices.We proposed a new carbon allotrope—super-graphyne, which is a semimetal with highly anisotropic band structure in the reciprocal space.
Co-reporter:Junru Wang, Feng Li, Xiaobiao Liu, Hongcai Zhou, Xiaofei Shao, Yuanyuan Qu and Mingwen Zhao
Journal of Materials Chemistry A 2017 - vol. 5(Issue 18) pp:NaN8768-8768
Publication Date(Web):2017/04/07
DOI:10.1039/C7TA02339A
Electrode materials with low diffusion energy barriers and high storage capacity of lithium are crucial for high performance rechargeable lithium-ion batteries (LIBs). Based on first-principles calculations, we demonstrate a new class of electrode materials. Taking advantage of the large voids in Cu3N crystals, high lithium mobility and storage capacity can be achieved. The diffusion of Li on Cu3N nanosheets experiences an energy barrier of about 0.09 eV, which is much lower than those of presently proposed electrode materials. The maximum Li capacity of Cu3N nanosheets can reach 1008 mA h g−1. In view of a large number of crystals sharing the same lattice structure as Cu3N, this work opens an avenue for developing electrode materials for high performance LIBs.
Co-reporter:Weifeng Li, Mingwen Zhao, Xian Zhao, Yueyuan Xia and Yuguang Mu
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 41) pp:NaN13706-13706
Publication Date(Web):2010/09/20
DOI:10.1039/C003524F
Density functional theory calculations are performed to explore vacancy-induced magnetism in graphene. The hydrogen saturation not only stabilizes the vacancy structure but also induces distinct magnetic coupling depending on the defect distribution: weak magnetic coupling between defects on different sublattices and strong coupling between defects on the same sublattice. Ferromagnetic ordering has to be accompanied with a semiconducting property. The interaction integral J between defective spins decreases linearly with the increase of the distance between them. Based on the 2D Ising model and Monte Carlo simulations, the possible highest Curie temperature Tc of defective graphene is predicted to be lower than 500 K.
Co-reporter:Weifeng Li, Mingwen Zhao, Yueyuan Xia, Ruiqin Zhang and Yuguang Mu
Journal of Materials Chemistry A 2009 - vol. 19(Issue 48) pp:NaN9282-9282
Publication Date(Web):2009/10/26
DOI:10.1039/B908949G
Magnetism and the mechanism of magnetic coupling in graphene decorated with monovalent and divalent adsorbates were investigated using first-principles calculations based on spin polarized density functional theory. The effects of adsorption concentration and the electronegativity of the adsorbate species on the magnetic and electronic properties were analyzed. For monovalent chemisorptions, the magnetic order originates from the instability of π electrons induced by the adsorption, opening a narrow energy gap and resulting in antiparallel spin directions on adjacent carbon atoms on the graphene sheet. The magnetic order is only possible for the separation between the adsorbing sites less than 10 Å. On the contrary, divalent chemisorptions cause long-range magnetic coupling, which is originated from the exchange interactions between localized nonbonding π electrons (spin-polarized) mediated by the conduction π electrons around the Fermi energy, similar to the s–d interaction in transition metals. We demonstrate that our results are well consistent with recent experimental findings.
Co-reporter:Bo Yang, Hongcai Zhou, Xiaoming Zhang and Mingwen Zhao
Journal of Materials Chemistry A 2015 - vol. 3(Issue 41) pp:NaN10891-10891
Publication Date(Web):2015/09/16
DOI:10.1039/C5TC02423D
Graphitic carbon nitrides are attracting increasing interest in many fields such as fuel cells, photocatalytic decomposition of water and spintronic devices. Tailoring electronic band gaps and inducing electron spin-polarization are the keys of these applications. Using first-principles calculations, we demonstrate that these goals can be reached by modifying graphitic carbon nitride via the introduction of additional carbon atoms into its vacancy sites. We found that with the increase of carbon concentration, the band gap of graphitic carbon nitride decreases rapidly and comes to close as the carbon concentration is higher than 2.609%, thus leading to a semiconductor–metal phase transition. More interestingly, local magnetic moments appear in the triangular domain centered by the introduced carbon atom and they interact in a ferromagnetic or “antiferromagnetic” manner depending on their relative positions. The tunable band gap and ferromagnetism revealed in the carbon-modified graphitic carbon nitrides offer a promising approach to achieve applications in hydrogen generation and spintronic devices.
Co-reporter:Xiaobiao Liu, Jie Tan, Aizhu Wang, Xiaoming Zhang and Mingwen Zhao
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 42) pp:NaN23291-23291
Publication Date(Web):2014/09/10
DOI:10.1039/C4CP03478C
Covalent organic frameworks (COFs) hold great promise in several applications, such as sieves, catalytic supports and gas storage because of their unique structures and electronic properties. However, most of these metal-free COFs are nonmagnetic and cannot be directly used in spintronics. Here, based on first-principles calculations, we predict that substitutional doping of COF-5 with nitrogen and boron atoms can modify the electronic structures, inducing stable electron spin-polarization in the framework. The preferability of the different doping sites is checked. The electronic structures of the doped COF-5 are dependent on the doping sites and doping atoms, which offer high degrees of freedom to tune the electronic properties. Kagome lattices of S = 1/2 spins can be achieved in the COF-5, suggesting a promising candidate for spin-liquid materials.
Co-reporter:Zhenhai Wang, Mingwen Zhao, Xiaopeng Wang, Yan Xi, Xiujie He, Xiangdong Liu and Shishen Yan
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 45) pp:NaN15698-15698
Publication Date(Web):2012/09/25
DOI:10.1039/C2CP42115A
The band alignment in ZnO–GaN and related heterostructures is crucial for uses in solar harvesting technology. Here, we report our density functional calculations of the band alignment and optical properties of ZnO–GaN and ZnO–(Ga1−xZnx)(N1−xOx)–GaN heterostructures using a Heyd–Scuseria–Ernzerhof (HSE) hybrid functional. We found that the conventional GGA functionals underestimate not only the band gap but also the band offset of these heterostructures. Using the hybrid functional calculations, we show that the (Ga1−xZnx)(N1−xOx) solid solution has a direct band gap of about 2.608 eV, in good agreement with the experimental data. More importantly, this solid solution forms type-II band alignment with the host materials. A GaN–(Ga1−xZnx)(N1−xOx)–ZnO core–shell solar cell model is presented to improve the visible light absorption ability and carrier collection efficiency.
Co-reporter:Xiaoming Zhang, Mingwen Zhao, Aizhu Wang, Xiaopeng Wang and Aijun Du
Journal of Materials Chemistry A 2013 - vol. 1(Issue 39) pp:NaN6270-6270
Publication Date(Web):2013/08/06
DOI:10.1039/C3TC31213E
Polymeric graphitic carbon nitride materials have attracted increasing attention in recent years owing to their potential applications in energy conversion, environment protection, and so on. Here, from first-principles calculations, we report the electronic structure modification of graphitic carbon nitride (g-C3N4) in response to carbon doping. We showed that each dopant atom can induce a local magnetic moment of 1.0 μB in non-magnetic g-C3N4. At a doping concentration of 1/14, the local magnetic moments of the most stable doping configuration which has the dopant atom at the center of the heptazine unit prefer to align in a parallel way leading to long-range ferromagnetic (FM) ordering. When the joint N atom is replaced by a C atom, the system favors an antiferromagnetic (AFM) ordering in an unstrained state, but can be tuned to ferromagnetism (FM) by applying biaxial tensile strain. More interestingly, the FM state of the strained system is half-metallic with abundant states at the Fermi level in one spin channel and a band gap of 1.82 eV in another spin channel. The Curie temperature (Tc) was also evaluated using a mean-field theory and Monte Carlo simulations within the Ising model. Such tunable electron spin-polarization and ferromagnetism are quite promising for the applications of graphitic carbon nitride in spintronics.
Co-reporter:Aizhu Wang and Mingwen Zhao
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 34) pp:NaN21844-21844
Publication Date(Web):2015/06/09
DOI:10.1039/C5CP03060A
Fractals are natural phenomena that exhibit a repeating pattern “exactly the same at every scale or nearly the same at different scales”. Defect-free molecular fractals were assembled successfully in a recent work [Shang et al., Nature Chem., 2015, 7, 389–393]. Here, we adopted the feature of a repeating pattern in searching two-dimensional (2D) materials with intrinsic half-metallicity and high stability that are desirable for spintronics applications. Using first-principles calculations, we demonstrate that the electronic properties of fractal frameworks of carbon nitrides have stable ferromagnetism accompanied by half-metallicity, which are highly dependent on the fractal structure. The ferromagnetism increases gradually with the increase of fractal order. The Curie temperature of these metal-free systems estimated from Monte Carlo simulations is considerably higher than room temperature. The stable ferromagnetism, intrinsic half-metallicity, and fractal characteristics of spin distribution in the carbon nitride frameworks open an avenue for the design of metal-free magnetic materials with exotic properties.
Co-reporter:Hongyu Zhang, Mingwen Zhao, Tao He, Xuejuan Zhang, Zhenhai Wang, Zexiao Xi, Shishen Yan, Xiangdong Liu, Yueyuan Xia and Liangmo Mei
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 41) pp:NaN13680-13680
Publication Date(Web):2010/09/24
DOI:10.1039/C002719G
We carried out first-principles calculations to explore the oxidative longitudinal unzipping of single-walled carbon nanotubes (SWCNTs) of different diameters and chiralities. We found that the initial attack leading to nanotube unzipping prefers to occur in the middle region for armchair tubes, and at the tube ends for zigzag tubes. Once the initial attack has taken place, by overcoming an energy barrier whose value decreases with increasing tube diameter, the subsequent breakage of C–C bonds parallel to the ones broken in the former process is barrierless. The energetically preferred unzipping path is parallel to the tube axis for armchair tubes, resulting in straight zigzag-edged graphene nanoribbons. For zigzag tubes, there are two energetically equivalent unzipping directions corresponding to the opening of two types of C–C bonds tilted towards the tube axis, giving rise to helical unzipping paths. This is disadvantageous for the production of straight graphene ribbons. A local curvature modulation procedure is proposed to efficiently control the location of the initial attack and thus the shape of the produced graphene nanoribbons.
Co-reporter:Hongxia Bu, Mingwen Zhao, Wenzheng Dong, Shuangwen Lu and Xiaopeng Wang
Journal of Materials Chemistry A 2014 - vol. 2(Issue 15) pp:NaN2757-2757
Publication Date(Web):2014/01/28
DOI:10.1039/C3TC32083A
Carbon has abundant allotropes with superhardness, but few of them are metallic. From first-principles calculations, we propose a stable metallic carbon allotrope (Hex-C24) phase with superhardness. The Hex-C24 can be thought of as a superlattice of carbon nanotubes and graphene nanoribbons composed of sp2- and sp3-hybridized carbon atoms. A possible synthetic route towards Hex-C24 from graphyne multilayers is evaluated by calculating the transition states between the two phases. Our calculations show that at a uniaxial pressure of around 25 GPa, the energy barrier of this endothermic transition is estimated to be 0.04 eV per atom, while at a pressure of 34 GPa, the transition is barrierless for specific initial configurations. The cohesive energy, elastic constants, and phonon frequencies unambiguously confirm the structural stability. The hardness of the Hex-C24 is estimated to exceed 44.54 GPa, which is 1/2 that of diamond. The Hex-C24 phase is metallic with several bands across the Fermi level. Both mechanical and metallic properties of Hex-C24 are anisotropic.
Co-reporter:Lin Wei, Xiaoming Zhang and Mingwen Zhao
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 11) pp:NaN8064-8064
Publication Date(Web):2016/02/09
DOI:10.1039/C6CP00368K
Dirac cones in the band structure make a great contribution to the unique electronic properties of graphene. But the spin-degeneracy of Dirac cones limits the application of graphene in spintronics. Here, using first-principles calculations, we propose a two-dimensional (2D) metal–organic framework (MOF), Ni2C24S6H12, with spin-polarized Dirac cones at the six corners of the Brillouin zone (BZ). Ferromagnetism is quite stable with a high Curie temperature (630 K) as revealed by Monte Carlo simulation within the Ising model. Taking spin–orbit coupling into account, band gaps are opened up at the Dirac point (5.9 meV) and Γ point (10.4 meV) in the BZ, making Ni2C24S6H12 a Chern topological insulator which is implemented for achieving the quantum anomalous Hall effect. These interesting properties enable Ni2C24S6H12 to be a promising candidate material for spintronics device applications.
Co-reporter:Aizhu Wang, Zhenhai Wang, Aijun Du and Mingwen Zhao
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 32) pp:NaN22159-22159
Publication Date(Web):2016/07/14
DOI:10.1039/C6CP02617F
To achieve a device application of the quantum spin Hall (QSH) effect, increasing the critical temperature is crucial. A two-dimensional topological insulator (2D-TI) with a sizeable bulk band gap is one of the most promising strategies to reach this goal. Using first-principles calculations, we propose a new 2D-TI, titanium nitride iodide (TiNI) monolayer, which can be exfoliated from a bulk TiNI crystal, thanks to the weak interlayer interaction. We demonstrate that the TiNI monolayer has an inverted band structure accompanied by topologically nontrivial states characterized by a topological invariant of Z2 = 1. The band gap (∼50 meV) opened due to spin–orbit coupling (SOC) is available for achieving the QSH effect at room temperature. The band inversion and topologically nontrivial states are robust under external strain, suggesting that the 2D TiNI monolayer lattice could be a versatile platform for hosting nontrivial topological states with potential applications in 2D spintronics and computer technology.
Co-reporter:Xin Chen, Linyang Li and Mingwen Zhao
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 25) pp:NaN16629-16629
Publication Date(Web):2015/05/21
DOI:10.1039/C5CP00046G
A quantum spin Hall (QSH) effect is quite promising for applications in spintronics and quantum computations, but at present, can only be achieved at ultralow temperatures. The determination of large-gap QSH insulators is critical to increase the operating temperature. By using first-principles calculations, we demonstrate that the stable hydrogenated stanene with a dumbbell-like structure (DB stanane) has large topological nontrivial band gaps of 312 meV (Γ point) and 160 meV for the bulk, characterized by a topological invariant of Z2 = 1 because of s–pxy band inversion. Helical gapless edge states appear in the nanoribbon structures with high Fermi velocity comparable to that of graphene. The nontrivial topological states are robust against the substrate effects. The realization of this material is a feasible solution for the applications of QSH effects at room temperature and can be beneficial in the fabrication of high-speed spintronics devices.