Co-reporter:Alexander H. Reath, Joseph W. Ziller, Charlene Tsay, Austin J. Ryan, and Jenny Y. Yang
Inorganic Chemistry March 20, 2017 Volume 56(Issue 6) pp:3713-3713
Publication Date(Web):February 27, 2017
DOI:10.1021/acs.inorgchem.6b03098
Redox inactive Lewis acidic cations are thought to facilitate the reactivity of metalloenzymes and their synthetic analogues by tuning the redox potential and electronic structure of the redox active site. To explore and quantify this effect, we report the synthesis and characterization of a series of tetradentate Schiff base ligands appended with a crown-like cavity incorporating a series of alkali and alkaline earth Lewis acidic cations (1M, where M = Na+, K+, Ca2+, Sr2+, and Ba2+) and their corresponding Co(II) complexes (2M). Cyclic voltammetry of the 2M complexes revealed that the Co(II/I) redox potentials are 130 mV more positive for M = Na+ and K+ and 230–270 mV more positive for M = Ca2+, Sr2+, and Ba2+compared to Co(salen–OMe) (salen–OMe = N,N′-bis(3-methoxysalicylidene)-1,2-diaminoethane), which lacks a proximal cation. The Co(II/I) redox potentials for the dicationic compounds also correlate with the ionic size and Lewis acidity of the alkaline metal. Electronic absorption and infrared spectra indicate that the Lewis acid cations have a minor effect on the electronic structure of the Co(II) ion, which suggests the shifts in redox potential are primarily a result of electrostatic effects due to the cationic charge.
Co-reporter:Bianca M. Ceballos;Charlene Tsay
Chemical Communications 2017 vol. 53(Issue 53) pp:7405-7408
Publication Date(Web):2017/06/29
DOI:10.1039/C7CC02511D
The hydricity (ΔGH−) of a newly synthesized nickel hydride was experimentally determined in acetonitrile (50.6 kcal mol−1), dimethyl sulfoxide (47.1 kcal mol−1), and water (22.8 kcal mol−1). The hydricity values indicate hydride transfer from [HNi(TMEPE)2][BF4] (TMEPE = 1,2-bis[di(methoxyethyl)phosphino]ethane) to CO2 is exergonic in water and endergonic in the organic solvents.
Co-reporter:Charlene Tsay and Jenny Y. Yang
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14174-14177
Publication Date(Web):July 14, 2016
DOI:10.1021/jacs.6b05851
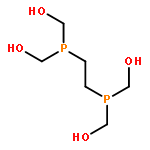
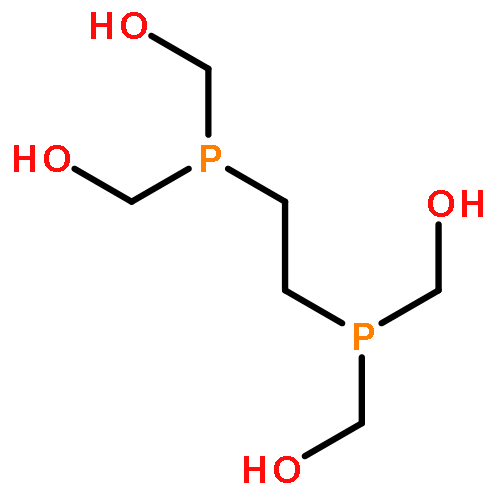
Electrocatalytic activity of a water-soluble nickel complex, [Ni(DHMPE)2]2+ (DHMPE = 2-bis(di(hydroxymethyl)phosphino)ethane), for the hydrogen evolution reaction (HER) at pH 1 is reported. The catalyst functions at a rate of ∼103 s–1 (kobs) with high Faradaic efficiency. Quantification of the complex before and after 18+ hours of electrolysis reveals negligible decomposition under catalytic conditions. Although highly acidic conditions are common in electrolytic cells, this is a rare example of a homogeneous catalyst for HER that functions with high stability at low pH. The stability of the compound and proposed catalytic intermediates enabled detailed mechanistic studies. The thermodynamic parameters governing electron and proton transfer were used to determine the appropriate reductants and acids to access the catalytic cycle in a stepwise fashion, permitting direct spectroscopic identification of intermediates. These studies support a mechanism for proton reduction that proceeds through two-electron reduction of the nickel(II) complex, protonation to generate [HNi(DHMPE)2]+, and further protonation to initiate hydrogen bond formation.
Co-reporter:Zachary Thammavongsy, Ivy M. Kha, Joseph W. Ziller and Jenny Y. Yang
Dalton Transactions 2016 vol. 45(Issue 24) pp:9853-9859
Publication Date(Web):24 Feb 2016
DOI:10.1039/C6DT00326E
The Tolman electronic parameters (TEP) and cone angles were experimentally measured for a series of substituted proazaphosphatrane ligands by synthesizing their respective Ni(LR)(CO)3 complexes, where L = P(RNCH2CH2)3N and R = Me, iPr, iBu and Bz. The complexes Ni(LMe)(CO)3 (1), Ni(LiPr)(CO)3 (2), Ni(LiBu)(CO)3 (3) and Ni(LBz)(CO)3 (4) display CO vibrational frequencies (A1 mode) at 2057.0, 2054.6, 2054.9 and 2059.1 cm−1, respectively. The TEPs for the phosphine ligands in 1–3 are among the lowest measured, with values close to P(tBu)3 the most donating phosphine measured by Tolman. The cone angles of LR measured in 1–4 are 152, 179, 200 and 207° for R = Me, iPr, iBu and Bz, respectively. The substituted proazaphosphatranes have larger cone angles compared to the analogous trialkyl subsituted monophosphines. Our study demonstrates that while the cone angles have a significant dependence on R, all of the substituted proazaphosphatranes are strong electron donors. Additionally, in order to determine the electronic donor strength of our previously reported multidentate ligand, TPAP, Ni(TPAP)(CO)2 (5) (TPAP = tris(2-pyridylmethyl)azaphosphatrane) and Ni(LMe)2(CO)2 (6) were also synthesized and evaluated in a similar fashion.
Co-reporter:Charlene Tsay; Brooke N. Livesay; Samantha Ruelas
Journal of the American Chemical Society 2015 Volume 137(Issue 44) pp:14114-14121
Publication Date(Web):October 14, 2015
DOI:10.1021/jacs.5b07777
The free energy of hydride donation (hydricity) for [HNi(DHMPE)2][BF4] (DHMPE = 1,2-bis(dihydroxymethylphosphino)ethane was experimentally determined versus the heterolytic cleavage energy of hydrogen in acetonitrile, dimethyl sulfoxide, and water to be 57.4, 55.5, and 30.0 kcal/mol, respectively. This work represents the first reported hydricity values for a transition metal hydride donor in three different solvents. A comparison between our values and the hydricity of hydrogen and formate reveals a narrowing in the range of values with increasing solvent polarity. The thermochemical values also reveal solvation effects that impact the overall thermodynamic favorability of hydride generation from hydrogen and transfer to carbon dioxide. The quantitative solvation effects described herein have important consequences to the design and reactivity of catalysts for transformations that have hydride transfer steps throughout synthetic chemistry.
Co-reporter:Zachary Thammavongsy, Juliet F. Khosrowabadi Kotyk, Charlene Tsay, and Jenny Y. Yang
Inorganic Chemistry 2015 Volume 54(Issue 23) pp:11505-11510
Publication Date(Web):November 9, 2015
DOI:10.1021/acs.inorgchem.5b02133
Tripyridylamine (TPA), a tetradentate ligand that forms 5-membered chelate rings upon metal coordination, has demonstrated significant utility in synthetic inorganic chemistry. An analogue with a phosphorus apical donor is a desirable target for tuning electronic structure and enhancing reactivity. However, this congener has been synthetically elusive. Prior attempts have resulted in tridentate coordination to transition metal ions due to a lack of ligand flexibility. Herein, we report the successful synthesis of tris(2-pyridylmethyl)proazaphosphatrane (TPAP), a more accommodating tripyridyl ligand containing an apical phosphorus donor. The TPAP ligand forms 6-membered chelate rings upon coordination and binds in the desired tetradentate fashion to a Co(II) ion. Structural studies elucidate the importance of ligand flexibility in tripodal ligands featuring phosphorus donors. Cyclic voltammetry, UV–vis, and solution magnetic susceptibility experiments of [Co(TPAP)(CH3CN)]2+ are also reported and compared to [Co(TPA)(CH3CN)]2+. Notably, magnetic susceptibility measurements of [Co(TPAP)(CH3CN)]2+ indicate a low spin electronic configuration, in contrast to [Co(TPA)(CH3CN)]2+, which is high spin.
Co-reporter:David W. Shaffer, Samantha I. Johnson, Arnold L. Rheingold, Joseph W. Ziller, William A. Goddard III, Robert J. Nielsen, and Jenny Y. Yang
Inorganic Chemistry 2014 Volume 53(Issue 24) pp:13031-13041
Publication Date(Web):December 3, 2014
DOI:10.1021/ic5021725
The preparation and characterization of a series of isostructural cobalt complexes [Co(t-Bu)2PEPyEP(t-Bu)2(CH3CN)2][BF4]2 (Py = pyridine, E = CH2, NH, O, and X = BF4 (1a–c)) and the corresponding one-electron reduced analogues [Co(t-Bu)2PEPyEP(t-Bu)2(CH3CN)2][BF4]2 (2a–c) are reported. The reactivity of the reduced cobalt complexes with CO2, CO, and H+ to generate intermediates in a CO2 to CO and H2O reduction cycle are described. The reduction of 1a–c and subsequent reactivity with CO2 was investigated by cyclic voltammetry, and for 1a also by infrared spectroelectrochemistry. The corresponding CO complexes of (2a–c) were prepared, and the Co–CO bond strengths were characterized by IR spectroscopy. Quantum mechanical methods (B3LYP-d3 with solvation) were used to characterize the competitive reactivity of the reduced cobalt centers with H+ versus CO2. By investigating a series of isostructural complexes, correlations in reactivity with ligand electron withdrawing effects are made.
Co-reporter:Jenny Y. Yang ; Stuart E. Smith ; Tianbiao Liu ; William G. Dougherty ; Wesley A. Hoffert ; W. Scott Kassel ; M. Rakowski DuBois ; Daniel L. DuBois ;R. Morris Bullock
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9700-9712
Publication Date(Web):April 30, 2013
DOI:10.1021/ja400705a
A nickel bis(diphosphine) complex containing pendant amines in the second coordination sphere, [Ni(PCy2Nt-Bu2)2](BF4)2 (PCy2Nt-Bu2 = 1,5-di(tert-butyl)-3,7-dicyclohexyl-1,5-diaza-3,7-diphosphacyclooctane), is an electrocatalyst for hydrogen oxidation. The addition of hydrogen to the NiII complex gives three isomers of the doubly protonated Ni0 complex [Ni(PCy2Nt-Bu2H)2](BF4)2. Using the pKa values and NiII/I and NiI/0 redox potentials in a thermochemical cycle, the free energy of hydrogen addition to [Ni(PCy2Nt-Bu2)2]2+ was determined to be −7.9 kcal mol–1. The catalytic rate observed in dry acetonitrile for the oxidation of H2 depends on base size, with larger bases (NEt3, t-BuNH2) resulting in much slower catalysis than n-BuNH2. The addition of water accelerates the rate of catalysis by facilitating deprotonation of the hydrogen addition product before oxidation, especially for the larger bases NEt3 and t-BuNH2. This catalytic pathway, where deprotonation occurs prior to oxidation, leads to an overpotential that is 0.38 V lower compared to the pathway where oxidation precedes proton movement. Under the optimal conditions of 1.0 atm H2 using n-BuNH2 as a base and with added water, a turnover frequency of 58 s–1 is observed at 23 °C.
Co-reporter:Zachary Thammavongsy, Ivy M. Kha, Joseph W. Ziller and Jenny Y. Yang
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9859-9859
Publication Date(Web):2016/02/24
DOI:10.1039/C6DT00326E
The Tolman electronic parameters (TEP) and cone angles were experimentally measured for a series of substituted proazaphosphatrane ligands by synthesizing their respective Ni(LR)(CO)3 complexes, where L = P(RNCH2CH2)3N and R = Me, iPr, iBu and Bz. The complexes Ni(LMe)(CO)3 (1), Ni(LiPr)(CO)3 (2), Ni(LiBu)(CO)3 (3) and Ni(LBz)(CO)3 (4) display CO vibrational frequencies (A1 mode) at 2057.0, 2054.6, 2054.9 and 2059.1 cm−1, respectively. The TEPs for the phosphine ligands in 1–3 are among the lowest measured, with values close to P(tBu)3 the most donating phosphine measured by Tolman. The cone angles of LR measured in 1–4 are 152, 179, 200 and 207° for R = Me, iPr, iBu and Bz, respectively. The substituted proazaphosphatranes have larger cone angles compared to the analogous trialkyl subsituted monophosphines. Our study demonstrates that while the cone angles have a significant dependence on R, all of the substituted proazaphosphatranes are strong electron donors. Additionally, in order to determine the electronic donor strength of our previously reported multidentate ligand, TPAP, Ni(TPAP)(CO)2 (5) (TPAP = tris(2-pyridylmethyl)azaphosphatrane) and Ni(LMe)2(CO)2 (6) were also synthesized and evaluated in a similar fashion.
Co-reporter:David W. Shaffer, Indrani Bhowmick, Arnold L. Rheingold, Charlene Tsay, Brooke N. Livesay, Matthew P. Shores and Jenny Y. Yang
Dalton Transactions 2016 - vol. 45(Issue 44) pp:NaN17917-17917
Publication Date(Web):2016/10/13
DOI:10.1039/C6DT03461F
We describe the structural and electronic impacts of modifying the bridging atom in a family of Co(II) pincer complexes with the formula Co(t-Bu)2PEPyEP(t-Bu)2Br2 (Py = pyridine, E = CH2, NH, and O for compounds 1–3, respectively). Structural characterization by single crystal X-ray diffraction indicates that compounds 1 and 3 are 5-coordinate complexes with both bromides bound to the Co(II) ion, while compound 2 is square planar with one bromide in the outer coordination sphere. The reduction potentials of 1–3, characterized by cyclic voltammetry, are consistent with the increasing electron-withdrawing character of the pincer ligand as the linker (E) between the pyridine and phosphine arms becomes more electronegative. Magnetic property studies of compounds 1 and 2 confirm high- and low-spin behavior, respectively, through a broad temperature range. However, complex 3 features an unusual combination of high spin S = 3/2 Co(II) and temperature dependent spin-crossover between S = 3/2 and S = 1/2 states. The different magnetic behaviors observed among the three CoBr2 pincer complexes reflects the importance of small ligand perturbations on overall coordination geometry and resulting spin state properties.
Co-reporter:Bianca M. Ceballos, Charlene Tsay and Jenny Y. Yang
Chemical Communications 2017 - vol. 53(Issue 53) pp:NaN7408-7408
Publication Date(Web):2017/06/16
DOI:10.1039/C7CC02511D
The hydricity (ΔGH−) of a newly synthesized nickel hydride was experimentally determined in acetonitrile (50.6 kcal mol−1), dimethyl sulfoxide (47.1 kcal mol−1), and water (22.8 kcal mol−1). The hydricity values indicate hydride transfer from [HNi(TMEPE)2][BF4] (TMEPE = 1,2-bis[di(methoxyethyl)phosphino]ethane) to CO2 is exergonic in water and endergonic in the organic solvents.
Co-reporter:Brian R. Lydon, Alex Germann and Jenny Y. Yang
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 6) pp:NaN841-841
Publication Date(Web):2016/03/09
DOI:10.1039/C6QI00010J
Chemically modifying electrode surfaces with redox active molecular complexes is an effective route to fabricating tailored functional materials. Surface modification has generally required the installation of reactive functional groups for direct covalent attachment that can present synthetic challenges. An alternative, milder method that utilizes π-interactions to physisorb the molecular complex onto a surface is described herein. Firstly, a gold electrode was modified with pyrene via covalent thiolate bonds. A pyrene-functionalized ferrocene was then physisorbed onto the pyrene-modified gold electrode. X-ray photoelectron spectroscopy, infrared spectroscopy, and cyclic voltammetry were used to demonstrate successful physisorption of the pyrene-functionalized ferrocene onto the pyrene-modified gold surface. Physisorption is attributed to pyrene–pyrene (π) interactions, as the ferrocene compound was not observed after identical treatment of a clean gold electrode surface. Additionally, cyclic voltammetry demonstrates facile electron transfer between the electrode and ferrocene through the non-covalent interactions at the interface. Since this approach of surface modification only requires functionalizing the target molecular complex with the relatively inert pyrene functionality, it broadens the range of experimentally accessible molecular precursors for chemically modified electrodes.
.jpg)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0.png)
![PROPAN-2-YL (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-DIOXOPYRIMIDIN-1-YL)-4-FLUORO-3-HYDROXY-4-METHYLOXOLAN-2-YL]METHOXY-PHENOXYPHOSPHORYL]AMINO]PROPANOATE](http://img.cochemist.com/ccimg/1190400/1190307-88-0_b.png)
![2,5,8,9-Tetraaza-1-phosphabicyclo[3.3.3]undecane,2,8,9-tris(1-methylethyl)-](http://img.cochemist.com/ccimg/175900/175845-21-3.png)
![2,5,8,9-Tetraaza-1-phosphabicyclo[3.3.3]undecane,2,8,9-tris(1-methylethyl)-](http://img.cochemist.com/ccimg/175900/175845-21-3_b.png)


![2,5,8,9-Tetraaza-1-phosphabicyclo[3.3.3]undecane,2,8,9-trimethyl-](http://img.cochemist.com/ccimg/120700/120666-13-9.png)
![2,5,8,9-Tetraaza-1-phosphabicyclo[3.3.3]undecane,2,8,9-trimethyl-](http://img.cochemist.com/ccimg/120700/120666-13-9_b.png)
![3,3'-[1,2-ETHANEDIYLBIS(OXY-2,1-ETHANEDIYLOXY)]BIS[2-HYDROXYBENZALDEHYDE]](http://img.cochemist.com/ccimg/115200/115142-66-0.png)
![3,3'-[1,2-ETHANEDIYLBIS(OXY-2,1-ETHANEDIYLOXY)]BIS[2-HYDROXYBENZALDEHYDE]](http://img.cochemist.com/ccimg/115200/115142-66-0_b.png)