Co-reporter:Binan Zhu, Yuting Lu, Leilin Chen, Binbin Yu, ... Taijun Hang
Journal of Pharmaceutical Analysis 2017 Volume 7, Issue 4(Volume 7, Issue 4) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.jpha.2017.03.008
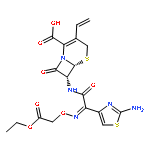
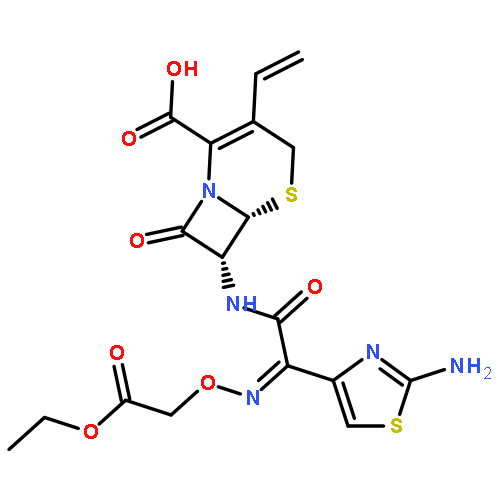
A sensitive and selective method was developed for the separation and characterization of related substances (RSs) in EVT-401 by hyphenated LC–MS techniques. Complete separation of the RSs was achieved with an Inertsil ODS-SP column (250 mm×4.6 mm, 5 µm) by linear gradient elution using a mobile phase consisting of 0.2% formic acid solution, methanol and acetonitrile. EVT-401 was found to be susceptible to acid, alkaline and oxidative stresses, while relatively stable under photolytic and thermal dry stress conditions. Fourteen RSs including six process-related substances and eight degradation products were detected and identified in EVT-401 with positive ESI high-resolution TOF-MS analysis of their parent ions and the corresponding product mass spectra elucidation, and some of them were further verified by chemical synthesis and NMR spectroscopy. The specific LC–MS method developed for separation, identification and characterization of RSs is valuable for EVT-401 manufacturing process optimization and quality control.
Co-reporter:Yuting Lu, Danyi Yang, Xiaoni Song, Sheng Wang, Min Song, Taijun Hang
Journal of Trace Elements in Medicine and Biology 2017 Volume 44(Volume 44) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jtemb.2017.05.006
•CV-AFS method was developed to determine the total and bioaccessible Hg of cinnabar containing TCMs through acid digestion and in vitro extraction by simulated gastrointestinal fluids, respectively.•Bioaccessible and total Hg of 29 cinnabar containing TCMs were measured and compared, and the health risks of Hg exposures were evaluated.•Bioaccessible Hg was found to be a more rational criteria for evaluating the health risks of cinnabar containing TCMs than the total Hg.Cinnabar (α-HgS), has been formulated in Traditional Chinese Medicines (TCMs) for thousands of years. Since the total Hg content was accepted widely as the toxicity criteria, the safety alerts have been issued about the cinnabar containing TCMs for exceeding Hg limits. However, cinnabar is almost insoluble in water, the oral absorption is extremely low. Hence, it is not suitable to use the total Hg content alone to evaluate the toxicity of cinnabar containing TCMs. In instead, the bioaccessible Hg is a much reasonable safety indicator.In this study, bioaccessible Hg contents of 29 cinnabar containing TCMs were determined by cold vapor-atomic fluorescence spectrometry after in vitro extractions with the simulated gastrointestinal fluids, while the total Hg contents were determined after acid digestion. According to the daily dosages, the bioaccessible Hg exposures of these TCMs were evaluated, and most of them were within the permitted daily exposure set by the International Council for Harmonisation, demonstrating that these TCMs are safe when administrated following the instructions. However, the obtained results also suggested that the Hg exposure could also be influenced by the herbal ingredients in TCMs and the bioactivities in gastrointestinal tract, indicating the possible health risks after excessive or long-term medication of cinnabar containing TCMs. Considering the influencing factors of the Hg intakes after oral administration of cinnabar containing TCMs, the bioaccessible Hg exposure should be considered as a more rational criterion for evaluating the health risks than the total Hg content. Furthermore, precautions should also be taken to ensure safe usages of cinnabar containing TCMs from both the cinnabar contents and the processing procedures points of view, as well as the daily dosage regimen, for all of them are directly related with the bioaccessible Hg exposures.Download high-res image (190KB)Download full-size image
Co-reporter:Huai-Song Wang, Min Song, and Tai-Jun Hang
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 5) pp:2881
Publication Date(Web):January 19, 2016
DOI:10.1021/acsami.5b10465
The high-value applications of functional polymers in analytical science generally require well-defined interfaces, including precisely synthesized molecular architectures and compositions. Controlled/living radical polymerization (CRP) has been developed as a versatile and powerful tool for the preparation of polymers with narrow molecular weight distributions and predetermined molecular weights. Among the CRP system, atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) are well-used to develop new materials for analytical science, such as surface-modified core–shell particles, monoliths, MIP micro- or nanospheres, fluorescent nanoparticles, and multifunctional materials. In this review, we summarize the emerging functional interfaces constructed by RAFT and ATRP for applications in analytical science. Various polymers with precisely controlled architectures including homopolymers, block copolymers, molecular imprinted copolymers, and grafted copolymers were synthesized by CRP methods for molecular separation, retention, or sensing. We expect that the CRP methods will become the most popular technique for preparing functional polymers that can be broadly applied in analytical chemistry.Keywords: analytical chemistry; controlled/living radical polymerization; fluorescent or colorimetric sensors; molecularly imprinted polymers; separation materials
Co-reporter:Mingluo Du;Chunyan Pan;Jing Chen;Min Song;Tingting Zhu ;Taijun Hang
Journal of Separation Science 2016 Volume 39( Issue 15) pp:2907-2918
Publication Date(Web):
DOI:10.1002/jssc.201600324
A simple, sensitive, and accurate stability-indicating analytical method has been developed and validated using ultra high performance liquid chromatography. The developed method is used to evaluate the related substances of eplerenone (EP). The degradation behavior of EP under stress conditions was determined, and the major degradants were identified by ultra high performance liquid chromatography with tandem mass spectrometry. The chromatographic conditions were optimized using an impurity-spiked solution, and the samples, generated from forced degradation studies. The resolution of EP, its potential impurities, and its degradation products was performed on a Waters UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm) by linear gradient elution using a mobile phase consisting of 10 mmol/L ammonium acetate adjusted to pH 4.5, methanol and acetonitrile. A photo-diode array detector set at 245 nm was used for detection. The flow rate was set at 0.3 mL/min. The procedure had good specificity, linearity (0.02–3.14 μg/mL), recovery (96.1–103.9%), limit of detection (0.01–0.02 μg/mL), limit of quantitation (0.03–0.05 μg/mL), and robustness. The correction factors of the process-related substances were calculated.
Co-reporter:Yuting Lu, Danyi Yang, Zhiyu Li, Taijun Hang, Min Song
Journal of Pharmaceutical and Biomedical Analysis 2016 Volume 128() pp:253-263
Publication Date(Web):5 September 2016
DOI:10.1016/j.jpba.2016.04.032
•7 related substances were identified, and five of them had not been reported.•LC-QTOF mass spectrometric method was developed for the identification.•Two related substances were synthesized and confirmed by NMR.•The manufacturing processes of alogliptin and its tablets were optimized.A highly specific and efficient LC-QTOF mass spectrometric method was developed for the separation and characterization of process related substances and the major degradation products in alogliptin benzoate and its tablets. The separation was performed on Phenomenex Gemini-NX C18 column (250 mm × 4.6 mm, 5 μm) using 0.2% formic acid-0.2% ammonium acetate in water as mobile phase A, acetonitrile and methanol (60:40, v/v) as mobile phase B in linear gradient elution mode. Forced degradation studies were also conducted under ICH prescribed stress conditions. Alogliptin benzoate and its tablets were tending to degrade under acid, alkaline, oxidative and thermal stresses, while relatively stable to photolytic stress. A total of seven related substances were detected and characterized through liquid chromatography-high resolution QTOF mass spectrometry techniques, including process related substances and degradation products, and two of them were further synthesized and characterized by NMR spectroscopy. Based on the related substances elucidation and the plausible formation mechanisms, efficient approaches were proposed to reduce or eliminate related substances, and in consequence the quality of alogliptin benzoate and its tablets have been promoted obviously. Therefore, the impurity profiles obtained are critical to the quality control and manufacturing processes optimization and monitoring of alogliptin benzoate and its tablets.
Co-reporter:Ping Lu, Lei Wang, Min Song, Tai-jun Hang
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 114() pp:159-167
Publication Date(Web):10 October 2015
DOI:10.1016/j.jpba.2015.05.018
•13 related substances (RSs) were characterized in pomalidomide (PML).•Incorporation of LC–TOF–MS, LC–MS–MS and NMR to characterize these RSs.•The forced degradation study was performed.•Most of these RSs were characterized as the hydroxylation products of PML.The current study dealt with the separation, identification and characterization of related substances in pomalidomide by hyphenated techniques. Complete separation was obtained with an Inertsil ODS-SP column (250 mm × 4.6 mm, 5 μm) by linear gradient elution using a mobile phase consisting of 0.1% formic acid solution and acetonitrile. They were characterized by hyphenated chromatographic techniques with the accurate mass determination using high resolution LC–TOF–MS methods as well as the product MS spectra determination and elucidation. The degradation behaviors of pomalidomide under ICH prescribed stress conditions were also conducted. Pomalidomide was found to be labile to degrade under acid, alkaline, oxidative and thermal stress conditions, while it was relatively stable to photolytic stress. 13 related substances were detected and identified to be 10 degradation products and three process related substances. The hyphenated LC–MS method with high resolution accurate mass determination facilitated the qualitative analysis of the unknown compounds than that of the conventional HPLC–UV. The related compounds identified are valuable for pomalidomide manufacturing process optimization and quality control.
Co-reporter:Fang Yan, Miaomiao Sun, Taijun Hang, Jing Sun, Xia Zhou, Xin Deng, Liang Ge, Hai Qian, Ding Ya, Wenlong Huang
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 102() pp:246-252
Publication Date(Web):5 January 2015
DOI:10.1016/j.jpba.2014.09.017
•A UPLC–MS/MS method was developed to determine HZ08 in rat plasma and tissues.•The total analysis time was 4.0 min.•The method was applied to pharmacokinetic study of HZ08 in rats.•HZ08 had linear pharmacokinetic properties with a rapid elimination phase.•The major distribution tissues of HZ08 in rats were lung, spleen and liver.Overexpression of P-glycoprotein leads to tumor multidrug resistance (MDR). HZ08, a novel tetrahydro-isoquinoline derivate, was discovered to inhibit the MDR in the cancer cell lines of MCF-7/ADM, K562/ADM and KBV in our previous studies. A rapid and sensitive ultra-performance liquid chromatography–tandem mass spectrometric method (UPLC–MS/MS) was developed and validated for determination of HZ08 in rat plasma and tissues after intravenous administration of HZ08 liposome injection at different doses. The analytes were extracted from plasma and tissues using protein precipitation by acetonitrile with clotrimazole as internal standard. The chromatographic separation was performed on a Thermo BDS HYPERSIL C18 column (100 mm × 4.6 mm, 2.4 μm) at a flow rate of 0.7 ml/min using 0.2% ammonium acetate solution (containing 0.1% formic acid) and methanol as mobile phase. The total run time was 4 min. The tandem mass detection was applied with electrospray ionization in positive ion selected reaction monitoring mode. The ion transitions monitored were m/z 523.5 to 342.3 for HZ08 and 277.1 to 165.1 for the internal standard, respectively. The calibration curves obtained were linear in different matrices, and the lower limit of quantification (LLOQ) achieved was 1 ng/ml for rat plasma and 0.25 ng/ml for rat tissues, respectively. The RSDs for intra- and inter-day precision were less than 15%. Extraction recovery, matrix effect and stability were satisfactory in rat plasma and tissues. The developed method was successfully applied to a pharmacokinetic study of HZ08 liposome injection following intravenous administration of 1, 3, 10 mg/kg to Sprague-Dawley rats. The data profiles revealed that HZ08 had linear pharmacokinetic properties at the tested doses, and was rapidly distributed into the systemic circulation with wide distribution throughout the body followed by a rapid elimination phase. The major distribution tissues of HZ08 in rats were lung, spleen and liver. These results provided constructive contribution to support the clinical evaluation.
Co-reporter:Meng-xiang Su;Wen-di Zhou;Juan Lan;Bin Di
Journal of Separation Science 2015 Volume 38( Issue 5) pp:804-812
Publication Date(Web):
DOI:10.1002/jssc.201400946
A simultaneous determination method based on liquid chromatography coupled with time-of-flight mass spectrometry was developed for the analysis of 11 bioactive constituents in tripterygium glycosides tablets, an immune and inflammatory prescription used in China. The analysis was fully optimized on a 1.8 μm particle size C18 column with linear gradient elution, permitting good separation of the 11 analytes and two internal standards in 21 min. The quantitation of each target constituent was carried out using the narrow window extracted ion chromatograms with a ±l0 ppm extraction window, yielding good linearity (r2 > 0.996) with a linear range of 10–1000 ng/mL. The limits of quantitation were low ranging from 0.25 to 5.02 ng/mL for the 11 analytes, and the precisions and repeatability were better than 1.6 and 5.3%, respectively. The acceptable recoveries obtained were in the range of 93.4–107.4%. This proposed method was successfully applied to quantify the 11 bioactive constituents in commercial samples produced by nine pharmaceutical manufacturers to profile the quality of these preparations. The overall results demonstrate that the contents of the 11 bioactive constituents in different samples were in great diversity, therefore, the quality, clinical safety, and efficacy of this drug needs further research and evaluation.
Co-reporter:Chengyan Li;Gongjian Lan;Jinyuan Jiang;Mingjie Sun;Taijun Hang
Chromatographia 2015 Volume 78( Issue 11-12) pp:825-831
Publication Date(Web):2015 June
DOI:10.1007/s10337-015-2900-4
A novel stability-indicating high-performance liquid chromatographic (HPLC) method has been developed and validated for the analysis of the impurities A–G in cabazitaxel. Chromatographic separation was achieved on a Welch Xtimate™ C18 (250 × 4.6 mm; 5 μm) column, using the mixture of 0.02 mol L−1 sodium dihydrogen phosphate buffer pH 3.0 (pH value was adjusted with phosphoric acid) and acetonitrile as mobile phase by gradient elution with a flow rate of 1.0 mL min−1, and UV detection was performed at 230 nm. The column temperature was maintained at 40 °C by column oven. The method was validated according to the International Conference on Harmonization (ICH) guidelines. Linearity (r > 0.9990) was observed over the concentration ranges of 25.0–1500.0, 31.5–1518.0, 74.9–1796.8, 65.6–1573.2, 59.4–1425.6, 22.2–1332.0, 1.3–1570.8, 30.8–1476.0 ng mL−1 of cabazitaxel and its impurities A–G, respectively. The RSD value of the recovery for each impurity was <5.0 % (n = 9). The method was found simple and rapid with good specificity and robustness, which can be suitable for the determination of the impurities in cabazitaxel.
Co-reporter:Meng-xiang Su, Min Song, Da-song Yang, Jin-fang Shi, Bin Di, Tai-jun Hang
Journal of Chromatography B 2015 990() pp: 31-38
Publication Date(Web):
DOI:10.1016/j.jchromb.2015.03.011
Co-reporter:Min Song;Yue-Qin Chen;Ping-Bo Lu
Journal of Separation Science 2014 Volume 37( Issue 7) pp:758-763
Publication Date(Web):
DOI:10.1002/jssc.201301271
Following the underlying principles of quality by design mentioned in the ICH Q8 guidance, systematic approaches for the control of process-related impurities have been taken in the manufacturing process of fasudil hydrochloride, a potent Rho-kinase inhibitor and vasodilator. Three related impurities were found in fasudil hydrochloride lab samples by a newly developed RP-HPLC with volatile mobile phase gradient elution and UV detection method. The elemental compositions of the impurities were determined by positive ESI high-resolution TOF-MS analysis of their [M + H]+ ions and their structures were identified through the elucidation of the product mass spectra obtained by a triple quadrupole mass spectrometer. The key impurity was further verified through synthesis and organic spectroscopy including NMR and IR spectroscopy. The origins of these impurities were located and the effective approaches to eliminate them were proposed based on the redesign of the synthetic conditions. The results obtained are important for quality control in the manufacture of fasudil hydrochloride bulk drug substance and injection.
Co-reporter:Yan-Yan Jia;Cheng-Tao Lu;Juan Feng;Ying Song;Jin-Yi Zhao;Shan Wang;Yuan Sun;Ai-Dong Wen;Zhi-Fu Yang
Basic & Clinical Pharmacology & Toxicology 2013 Volume 113( Issue 6) pp:431-435
Publication Date(Web):
DOI:10.1111/bcpt.12112
Abstract
Pivalate-generating prodrugs have been suggested to cause clinically significant hypocarnitinaemia. Tenofovir dipivoxil, a novel ester prodrug of tenofovir, can be used for treatment for hepatitis B and HIV infection and it was necessary to evaluate the effect of its treatment on carnitine homeostasis. We sought to investigate the effect of Class 1 drug tenofovir dipivoxil on endogenous L-carnitine level during a 72-hr test in healthy Chinese volunteers and to establish a suitable dose of L-carnitine nutritional supplement for patients who were administered short-term tenofovir dipivoxil tablets for treatment for hepatitis B and herpes simplex virus infection. Tenofovir dipivoxil was administered in one of eight dosing regimens (single dose 150, 300 and 600 mg, multiple dose 300, 450, and 600 mg, multiple dose 450 (600) mg tenofovir dipivoxil and 0.5 g L-carnitine) to gender-balanced groups of 84 healthy Chinese volunteers. Plasma concentrations of L-carnitine were quantified before, during and after treatment. Plasma L-carnitine concentrations fell during tenofovir dipivoxil dosing. The nadir in L-carnitine concentration was dependent on the dose of tenofovir dipivoxil and it decreased from 6.1 ± 0.6 to 4.4 ± 0.8 μg/ml, 6.1 ± 1.8 to 3.3 ± 1.2 μg/ml, 6.2 ± 0.6 to 2.5 ± 0.5 μg/ml for single doses of 150, 300, 600 mg tenofovir dipivoxil tablets and from 6.0 ± 1.4 to 2.1 ± 1.5 μg/ml, 6.2 ± 0.4 to 0.9 ± 0.5 μg/ml for multiple doses of 450, 600 mg tenofovir dipivoxil tablets, respectively. Short-term administration of tenofovir dipivoxil results in hypocarnitinaemia and increased losses of carnitine in resulting of minor adverse events of decreased food appetite, nausea, abdominal distention and muscle weakness.
Co-reporter:Yunjing Zhang, Shuping Qiang, Jing Sun, Min Song, Taijun Hang
Journal of Chromatography B 2013 Volumes 917–918() pp:93-99
Publication Date(Web):15 February 2013
DOI:10.1016/j.jchromb.2012.12.029
A high performance liquid chromatography–hydride generation–atomic fluorescence spectrometry (HPLC–HG–AFS) method was developed for the simultaneous determination of four arsenic species (As(III), dimethylarsinic acid (DMA), monomethylarsonic acid (MMA) and arsenate As(V)) in dog plasma. Good separation of the four arsenic species was achieved within 15 min on an anion-exchange column with isocratic elution using 15 mmol/L KH2PO4 (pH 5.9) as eluent at a flow rate of 1.0 mL/min. The assay was linear over the range of 1.25–200, 1.56–200, 1.34–172, and 2.50–200 ng/mL with the detection limits of 0.80, 1.00, 0.86 and 2.00 ng/mL for As(III), DMA, MMA and As(V), respectively. The method was validated for selectivity, precision, accuracy and recovery and then applied to a comparative pharmacokinetic study of the arsenic species in beagle dogs after a single oral administration of Realgar (24.32 mg/kg, equivalent to 11.31 mg As/kg) alone or Niu Huang Jie Du Pian (a patent traditional Chinese medicine (TCM), 380 mg/kg, equivalent to 28.45 mg As/kg), respectively. DMA was found to be the predominant species in the dog plasma after dosing, with As(V) appeared as the quickly eliminating one. No traces of MMA and As(III) were detected at any sampling time points. The main pharmacokinetic parameters found for DMA p.o. administration of Realgar and Niu Huang Jie Du Pian were as follows: Cmax (14.7 ± 4.2) and (57.0 ± 32.0) ng/mL, Tmax (2.4 ± 0.5) and (2.5 ± 0.5) h, AUC0–36 (151.1 ± 12.9) and (635.9 ± 418.2) ng h/mL, AUC0–∞ (206.0 ± 44.5) and (687.2 ± 425.1) ng h/mL, t1/2 (16.2 ± 7.9) and (9.4 ± 2.2) h, respectively. The influence of compounding in Niu Huang Jie Du Pian on the pharmacokinetics of arsenics was shown with increased transformation of DMA and its faster elimination rate.Highlights► An HPLC–HG–AFS method for determination of 4 arsenic species in dog plasma was developed. ► The comparative pharmacokinetic study of arsenic species was carried out after oral administration of Realgar and NHJDP. ► DMA and As(V) were found to be the main arsenic species in dog plasma after oral administration of both Realgar and NHJDP. ► The concentration–time profiles and pharmacokinetic parameters of DMA in beagle dogs were presented.
Co-reporter:Ji Li, Li Wang, Zhao Chen, Rui Xie, You Li, Taijun Hang, Guorong Fan
Journal of Chromatography B 2012 Volumes 895–896() pp:83-88
Publication Date(Web):1 May 2012
DOI:10.1016/j.jchromb.2012.03.018
The high-performance liquid chromatography (HPLC) coupled with on-line solid phase extraction (SPE) and ultraviolet (UV) detection was developed for determining cefdinir in beagle dog plasma. After simple pretreatment for plasma with 6% perchloric acid, a volume of 100 μL upper layer of the plasma sample was injected into the self-made on-line SPE column. The analytes were retained on the trap column (Lichrospher C18, 4.6 mm × 37 mm, 25 μm), and the biological matrix was washed out with the solvent (20 mM KH2PO4 adjusted pH 3.0) at flow rate of 2 mL/min. By rotation of the switching valve, the target analytes could be eluted from trap column to analytical column in the back-flush mode by the mobile phase (methanol–acetonitrile–20 mM KH2PO4 adjusted pH 3.0, 11.25:6.75:82, v/v/v) at flow rate of 1.5 mL/min, and then separated on the analytical column (Ultimate™ XB-C18, 4.6 mm × 50 mm, 5 μm). The complete cycle of the on-line SPE preconcentration, purification and HPLC separation of the analytes was 4 min. The UV detection was performed at 286 nm. The calibration curves showed excellent linear relationship (R2 = 0.9995) over the concentration range of 0.05–50 μg/mL. The optimized method showed good performance in terms of specificity, linearity, detection and quantification limits, precision and accuracy. This method was successfully applied to quantify cefdinir in beagle dog plasma to support the pre-clinical pharmacokinetic trial.Highlights► A rapid on line SPE-HPLC method was developed to determine cefdinir in dog plasma. ► On line SPE following 96-Well protein precipitation could reduce more interference. ► Application of 96-well format improved the extraction efficiency. ► The method was applied to the pharmacokinetic study of cefdinir in beagle dogs. ► This method is economic.
Co-reporter:Meng-Xiang Su, Min Song, De-Zhu Sun, Hua Zhao, Xiao Gu, Ling Zhu, Xiao-Le Zhan, Zhong-Nan Xu, Ai-Dong Wen, Tai-Jun Hang
Journal of Chromatography B 2012 s 889–890() pp: 39-43
Publication Date(Web):15 March 2012
DOI:10.1016/j.jchromb.2012.01.027
A sensitive and selective liquid chromatographic tandem mass spectrometric method was developed and validated for the determination of sinomenine in human plasma. Plasma samples were precipitated using methanol with metronidazole as internal standard. Separation was carried out on an Inertsil ODS-3 column using a mixture of 0.2% ammonium acetate solution (A) and methanol (B) as the mobile phase with linear gradient elution as follows: 0 min (50%B) → 1.5 min (80%B) → 4.5 min (80%B) → 4.6 min (50%B) → 6.0 min (50%B). All mass data were obtained in the positive ion mode, and the fragmentation transitions for the selective multiple reaction monitoring were m/z 330 → 181 and 172 → 128 for sinomenine and metronidazole, respectively. The method was fully validated to be accurate and precise with a linear range of 0.5–500 ng/mL and applied to a single- and multiple-dose pharmacokinetics study of sustained-release capsules of sinomenine hydrochloride in 20 healthy Chinese volunteers. After oral administration of a single 60-mg dose, the Tmax, Cmax, AUC0–96 and t1/2 were 7.9 ± 2.0 h, 123 ± 22 ng/mL, 3032 ± 682 ng h/mL and 13.4 ± 1.6 h, respectively. After oral administration of the 60 mg capsules twice-daily for 7 consecutive days, these parameters were 4.4 ± 3.6 h, 279 ± 69 ng/mL, 7333 ± 2096 ng h/mL and 15.1 ± 1.3 h, respectively. The AUC and Cmax values after multiple-dose treatment were significantly higher than those after a single-dose treatment (P < 0.01), with an accumulation factor of 2.49 ± 0.77.Highlights► LC–MS/MS quantification of sinomenon in human plasma was developed. ► This method achieved a LLOQ as low as 0.5 ng/mL with a simple pretreatment procedure. ► Clinical pharmacokinetic of sinomenine sustained-release capsules was characterized. ► The accumulation of sinomenine in body was observed after repeated dosing.
Co-reporter:Meng-xiang Su, Min Song, Ying Zhuang, Yong-qing Wang, Tai-jun Hang
Journal of Pharmaceutical and Biomedical Analysis 2011 55(1) pp: 230-235
Publication Date(Web):
DOI:10.1016/j.jpba.2011.01.009
Co-reporter:Meng-xiang Su;Lin-jun You;Bin Di;Lan-jin Qu
Chromatographia 2011 Volume 74( Issue 9-10) pp:
Publication Date(Web):2011 November
DOI:10.1007/s10337-011-2131-2
A simple and sensitive liquid chromatography with ultraviolet detection (LC–UV) method was developed for the determination of three impurities with a content over 0.1% (w/w) in technical triadimefon. A Gemini C18 column (5 μm, 250 mm × 4.6 mm i.d.) was used for the chromatographic separations. The samples were separated by gradient elution with water (solvent A) and methanol (solvent B) using the following conditions: 70% A isocratic for 12 min, linear to 0% A within 8 min, and isocratic for 10 min at 0% A with a flow rate of 1.0 mL min−1. Chromatograms were recorded at an absorption wavelength of 280 nm. The chromatographic resolutions between triadimefon and its potential impurities A, B, and C were greater than 3. The developed LC method was validated with respect to linearity, accuracy, precision, and robustness. This method was successfully applied to analyze the impurities in commercial technical triadimefon. In addition, the structures of the three impurities were identified to be (A) 4-chlorophenol, (B) 1-(2,4-dichlorophenoxy)-3,3-dimethyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone, and (C) 1,1-bis(4-chlorophenoxy)-3,3-dimethyl-2-butanone.
Co-reporter:Yu Wang;Min Song;Taijun Hang;Aidong Wen;Lin Yang
Chromatographia 2010 Volume 72( Issue 3-4) pp:245-253
Publication Date(Web):2010 August
DOI:10.1365/s10337-010-1645-3
A highly selective and sensitive liquid chromatographic tandem mass spectrometric (LC-MS–MS) method was developed and validated for the quantitation and pharmacokinetic study of niacin (NA) and its two metabolites niacinamide (NAM) and nicotinuric acid (NUR) in human plasma. Protein precipitation with 14% perchloric acid solution was selected for sample preparation, and ganciclovir was used as an internal standard. Separation was on a Phenomenex Curosil-PFP (250 mm × 4.6 mm, 5 μm) column by a multiple steep steps linear gradient elution with mobile phase consisting of water and methanol, both containing 0.1% formic acid, pumped at a flow rate of 1 mL min−1. The determination was optimized and carried out with positive electrospray ionization by selective multiple reaction monitoring. The method was linear in the concentration range of 15–2,000 ng mL−1 for NA, 70–2,000 ng mL−1 for NAM and 10–2,000 ng mL−1 for NUR, by standard addition calibration. The application of LC-MS–MS was demonstrated for the specific and quantitative analysis of NA, NAM and NUR in human plasma from a pharmacokinetic study in 12 healthy Chinese volunteers treated with three incremental doses of niacin extended-release/lovastatin tablets and an additional steady-state regime.
Co-reporter:Min Song, Siyun Zhang, Xiaoyan Xu, Taijun Hang, Lee Jia
Journal of Chromatography B 2010 Volume 878(Issue 32) pp:3331-3337
Publication Date(Web):15 December 2010
DOI:10.1016/j.jchromb.2010.10.007
Compound Danshen tablets are composed of Panax notoginseng, Salvia miltiorrhiza and Borneol. The tablets are prescribed for treatment of cardiovascular diseases in China. The present study aimed at developing a specific and sensitive LC–MS/MS method to simultaneously determine three bioactive P. notoginseng saponins, i.e., notoginsenoside R1, ginsenoside Rg1 and Rb1, in dogs after a single oral administration of the compound tablets in order to obtain the clinically relevant saponin-related pharmacodynamics of the tablets in patients. The R1, Rg1 and Rb1 were extracted from dog plasma with acetone–methanol (80:20, v/v), separated by reversed phase liquid chromatography and determined by tandem mass spectrometry (LC–MS/MS) with positive electrospray ionization (ESI). The developed method reached lower limit of quantitation (LLOQ) at 0.10 ng/ml for the three saponins. The method was validated in terms of selectivity, matrix effects, linearity, precision and accuracy, and then was applied to a pharmacokinetic study of the three bioactive saponins simultaneously in dogs after a single oral administration of compound Danshen tablets at a clinical equivalent dose. The Cmax and AUC(0−∞) for R1, Rg1 and Rb1 were 1.91, 3.34 and 28.6 ng/ml, and 7.5, 11.0, and 1712 (h ng/ml), respectively.
Co-reporter:Min Song, Xinxin Xu, Taijun Hang, Aidong Wen, Lin Yang
Analytical Biochemistry 2009 Volume 385(Issue 2) pp:270-277
Publication Date(Web):15 February 2009
DOI:10.1016/j.ab.2008.11.027
A selective, sensitive, and accurate liquid chromatography–tandem mass spectrometry (LC–MS/MS) method for the simultaneous determination of aripiprazole and its active metabolite dehydroaripiprazole in human plasma has been developed using papaverine as internal standard (IS). LC–MS/MS analysis was carried out on a Finnigan LC–TSQ Quantum mass spectrometer using positive ion electrospray ionization (ESI+) and selected reaction monitoring (SRM). The assays for aripiprazole and dehydroaripiprazole were linear over the ranges of 0.1 to 600 ng/ml and 0.01 to 60 ng/ml, respectively. The average recoveries in plasma samples both were better than 85%. The intra- and interrun precision and accuracy values were found to be within the assay variability criteria limits according to the US Food and Drug Administration guidelines. The developed method was proved to be suitable for use in a clinical pharmacokinetic study after a single oral administration of a 5-mg aripiprazole tablet in healthy Chinese volunteers.
Co-reporter:Min Song;Xiaorong Yu;Hua Zhao;Taijun Hang;Lin Yang;Wei Xu
Chromatographia 2009 Volume 69( Issue 9-10) pp:
Publication Date(Web):2009 May
DOI:10.1365/s10337-009-0975-5
A sensitive and selective liquid chromatographic method coupled with tandem mass spectrometry was established and validated for the determination and pharmacokinetic study of clozapine in human plasma. Ethyl acetate extraction was used for plasma sample preparation with mirtazapine as internal standard. Chromatographic separation was achieved on a Hanbon Kromasil C18 (250 mm × 4.6 mm, 5 μm) column by isocratic elution with a mixture of 70 volumes of methanol and 30 volumes of water containing 0.2% ammonium acetate and 0.1% formic acid as mobile phase delivered at 1.0 mL min−1. The MS-MS detection was carried out on a tandem mass spectrometer using positive electrospray ionization and multiple reaction monitoring with argon for collision-induced dissociation. The ion transitions were monitored as follows: m/z 327 to m/z 270 for clozapine and m/z 266 to m/z 195 for the internal standard (mirtazapine), respectively. Calibration curves were generated over the concentration range from 0.10 to 200 ng mL−1 with the lower limit of quantification of 0.10 ng mL−1, and two segments of linear calibration curves were established by regressing in the way of least-square in the range from 0.10 to 5.0 and 5.0 to 200 ng mL−1, respectively. The intra- and inter-day precision and accuracy were determined at three different concentration levels, 0.20, 10.0 and 100 ng mL−1, and were all better than 15% (n = 5). This specific and sensitive liquid chromatography coupled with tandem mass spectrometry has been successfully applied to a pharmacokinetic study of clozapine after a single oral dose of 25 mg in healthy Chinese volunteers.
Co-reporter:Min Song, Li Wang, Hua Zhao, Taijun Hang, Aidong Wen, Lin Yang, Lee Jia
Journal of Chromatography B 2008 Volume 875(Issue 2) pp:515-521
Publication Date(Web):15 November 2008
DOI:10.1016/j.jchromb.2008.10.005
Rasagiline is a highly potent, selective and irreversible second-generation monoamine oxidase inhibitor with selectivity for type B of the enzyme (MAO-B). The present studies aimed at developing and validating a rapid and sensitive liquid chromatography–tandem mass spectrometric (LC–MS/MS) method for determination of rasagiline in human plasma and urine. LC–MS/MS analysis was carried out on a Finnigan LC-TSQ Quantum mass spectrometer using positive ion electrospray ionization (ESI+) and selected reaction monitoring (SRM). The assay for rasagiline was linear over the range of 0.01–40 ng/mL in plasma and 0.025–40 ng/mL in urine. It took 5.5 min to analyze a sample. The average recoveries in plasma and urine samples were both >85%. The RSD of precision and bias of accuracy were less than 15% and 10%, respectively, of their nominal values based on the intra- and inter-day analysis. The developed method was proved to be suitable for use in clinical pharmacokinetic study after single oral administration of 0.5, 1 and 2 mg rasagiline mesylate tablets in healthy Chinese volunteers.
Co-reporter:Jing-yan Weng, Min Song, Tai-jun Hang, Wen-long Huang, Yun Du
Journal of Chromatography B 2007 Volume 856(1–2) pp:29-34
Publication Date(Web):1 September 2007
DOI:10.1016/j.jchromb.2007.05.013
A selective and sensitive liquid chromatographic method coupled with ion spray tandem mass spectrometry detection (LC–MS/MS) was developed for the determination and pharmacokinetic study of N-cyano-1-[(3,4-dimethoxyphenyl)methyl]-3,4-dihydro-6,7-dimethoxy-N′-octyl-2(1H)-isoquinoline-carboximidamide (HZ08, a candidate reversing agent for multidrug resistance of cancer) liposome injection in rat plasma. The analyte was extracted from plasma using liquid–liquid extraction by methyl tert-butyl ether with drotaverine as internal standard. The chromatographic separation was performed on a Kromasil-C18 column (150 mm × 4.6 mm, i.d., 5 μm) with gradient elution. The tandem mass detection was made with electrospray ionization in positive ion selected reaction monitoring mode with argon collision-induced dissociation. The ion transitions were m/z 523.1 to 342.1 for HZ08 at 27 eV and m/z 398.1 to 326.1 at 35 eV for the internal standard, respectively. The determination was validated to be accurate and precise for the analysis in the concentration range of 5–10,000 ng/ml for HZ08 with the lower limit of detection (LOD) being 1 ng/ml, when 0.1 ml of rat plasma sample was processed. The main pharmacokinetic parameters found for HZ08 after intravenous (i.v.) administration of its liposome injection at doses of 2, 4 and 8 mg/kg were as follows: Cmax (4511 ± 681), (5553 ± 1600) and (6444 ± 950) ng/ml, Tmax (0.033 ± 0), (0.056 ± 0.048) and (0.033 ± 0) h, t1/2 (1.75 ± 0.19), (1.63 ± 0.12) and (1.56 ± 0.18) h, AUC0–6 (899 ± 112), (1238 ± 190) and (1707 ± 307) h ng/ml, AUC0-∞ (917 ± 110), (1256 ± 189) and (1723 ± 306) h ng/ml, MRT (1.14 ± 0.21), (1.01 ± 0.13) and (1.16 ± 0.17) h, CL (2.90 ± 0.15), (3.01 ± 0.74) and (4.11 ± 0.59) l/h/kg, respectively. The plasma concentration–time profiles of HZ08 were best fitted with two-compartment models. Linear pharmacokinetics was found for HZ08 in rats after intravenous administration of the liposome injection.
Co-reporter:Tai-Jun Hang, Wei Zhao, Jie Liu, Ming Song, Ying Xie, Zhengxing Zhang, Jianping Shen, Yingdi Zhang
Journal of Pharmaceutical and Biomedical Analysis 2006 Volume 40(Issue 1) pp:202-205
Publication Date(Web):23 January 2006
DOI:10.1016/j.jpba.2005.06.035
Indapamide was extracted from human whole blood with diethyl ether and was determined by a HPLC-UV method using an Inertsil ODS-3 column and an isocratic mobile phase consisting of 55% buffer solution (2 g KH2PO4, 3 ml H3PO4 and 3.5 ml triethylamine in 1 l of H2O), 40% acetonitrile and 5% methanol for 12.5 min, and then a gradient flush from 100% isocratic to a mixture of 20% isocratic mobile phase and 80% methanol for 3 min. Indapamide and glipizide (internal standard) were eluted from the column at about 10.5 and 12.8 min, respectively. The method had within day precision values in the range ±1.2 to ±9.7% (n = 5) and between day precision in the range ±3.3 to ±9.7%. The method was linear over the range of 10–400 ng/ml of indapamide in blood (R = 0.999). The LOQ (s/n = 10) of the method was 10 ng/ml. The method was applied in a study of the pharmacokinetics and bioequivalence of generic indapamide capsules (2.5 mg) in comparison with reference indapamide tablets (2.5 mg), in 20 healthy male Chinese volunteers. The mean values of major pharmacokinetic parameters of Cmax, AUC0–48, AUC0–∞, Tmax, and t1/2 of indapamide in healthy male Chinese volunteers after po a single 5 mg dose for the test product were 331.0 ± 39.2 ng/ml, 6193.7 ± 873.5 ng h/ml, 7311.8 ± 1232.3 ng h/ml, 3.2 ± 0.9 h, and 17.3 ± 2.8 h, respectively. There was no significant differences between the two formulations.
Co-reporter:Min Song, Tai-jun Hang, Ying Wang, Li Jiang, Xiao-luan Wu, Zhengxing Zhang, Jianping Shen, Yindi Zhang
Journal of Pharmaceutical and Biomedical Analysis 2006 Volume 40(Issue 1) pp:190-196
Publication Date(Web):23 January 2006
DOI:10.1016/j.jpba.2005.06.034
A highly selective and sensitive HPLC–ESI–MS–MS method was developed for the determination of oleanolic acid in human plasma. The oleanolic acid and glycyrrhetinic acid (internal standard) were recovered from plasma with ethyl acetate liquid–liquid extraction. The organic extracts were dried under a stream of warm nitrogen, reconstituted in mobile phase and injected into a Zorbax-Extend ODS analytical column (150 mm × 4.6 mm i.d., 5 μm), with the mobile phase consisting of methanol–ammonium acetate (32.5 mM) (85:15, v/v) pumped at a flow rate of 1.0 ml/min, and 30% of the eluent was split into a MS system with electrospray ionization tandem mass (ESI–MS–MS) detection in negative ion mode. The tandem mass detection was performed on a Finnigan Surveyor LC-TSQ Quantum Ultra AM tandem mass spectrometer operated in selected reaction monitoring mode. The parent to product ion combinations of m/z 455.4 → 455.4 and 469.3 → 425.2 at 38 V 1.5 mTorr Ar CID were used to quantify oleanolic acid and glycyrrhetinic acid, respectively. The assay was validated in the concentration range of 0.02–30.0 ng/ml for oleacolic acid when 0.5 ml of plasma was processed. The precision of the assay (expressed as relative standard deviation, R.S.D.%) was less than 15% at all concentrations levels within the tested range and adequate accuracy, and the limit of quantification was 0.02 ng/ml. The established method was applied for the pharmacokinetics study of oleanolic acid capsules in 18 healthy male Chinese volunteers with the mean values of Cmax, Tmax, AUC0–48, AUC0–∞, t1/2, CL/F, and V/F of oleanolic acid after p.o. a single 40 mg dose obtained were 12.12 ± 6.84 ng/ml, 5.2 ± 2.9 h, 114.34 ± 74.87 ng h/ml, 124.29 ± 106.77 ng h/ml, 8.73 ± 6.11 h, 555.3 ± 347.7 L/h, and 3371.1 ± 1990.1 L, respectively.
Co-reporter:Min Song, Tai-Jun Hang, Cheng Wang, Lin Yang, Ai-Dong Wen
Journal of Pharmaceutical Analysis (February 2012) Volume 2(Issue 1) pp:19-28
Publication Date(Web):1 February 2012
DOI:10.1016/j.jpha.2011.08.003
A selective precolumn derivatization liquid chromatography–tandem mass spectrometric (LC–MS/MS) method for the determination of glucosamine in human plasma and urine has been developed and validated. Glucosamine was derivatized by o-phthalaldehyde/3-mercaptopropionic acid. Chromatographic separation was performed on a Phenomenex ODS column (150 mm×4.6 mm, 5 μm) using linear gradient elution by a mobile phase consisting of methanol (A), and an aqueous solution containing 0.2% ammonium acetate and 0.1% formic acid (B) at a flow rate of 1 mL/min. Tolterodine tartrate was used as the internal standard (IS). With protein precipitation by acetonitrile and then the simple one-step derivatization, a sensitive bio-assay was achieved with the lower limit of quantitation (LLOQ) as low as 12 ng/mL for plasma. The standard addition calibration curves suitable for clinical sample analysis showed good linearity over the range of 0.012–8.27 μg/mL in plasma and 1.80–84.1 μg/mL in urine. The fully validated method has been successfully applied to a pharmacokinetic study of compound glucosamine sulfate dispersible tablets in health Chinese volunteers receiving single oral doses at 500, 1000 and 1500 mg of glucosamine sulfate, as well as multiple oral doses of 500 mg t.i.d. for 7 consecutive days.
Co-reporter:Mo Chen, Yuan Zhang, Xiao-Ting Que, Ya Ding, ... Tai-Jun Hang
Journal of Pharmaceutical Analysis (December 2013) Volume 3(Issue 6) pp:387-393
Publication Date(Web):1 December 2013
DOI:10.1016/j.jpha.2013.03.001
Inosiplex is a compound formulation composed of inosine and p-acetaminobenzoic acid (PABA) salt of N,N-dimethylamino-2-propanol (DIP). This study was to investigate the clinical plasma pharmacokinetic properties of DIP and PABA after single and multiple oral doses of inosiplex tablets in healthy Chinese volunteers. The established LC/MS/MS method for plasma DIP determination had a linear range of 0.02–10 µg/mL, and the HPLC method for plasma PABA determination had a linear range of 0.05–40 µg/mL. Linear pharmacokinetic characteristics were found with single oral doses of 0.5, 1.0 and 2.0 g. No obvious accumulation effects were observed for DIP and PABA.

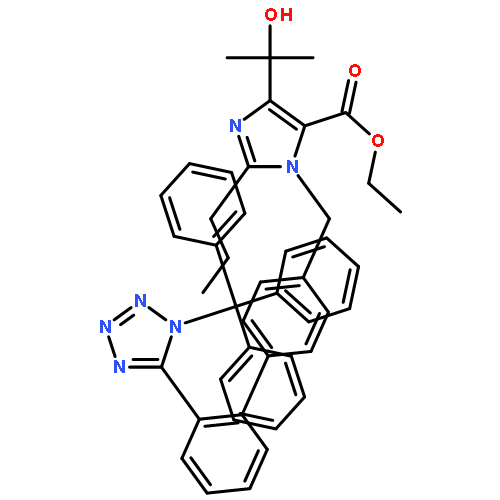
![(5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl 1-((2'-(2H-tetrazol-5-yl)-[1,1'-biphenyl]-4-yl)methyl)-4-(prop-1-en-2-yl)-2-propyl-1H-imidazole-5-carboxylate](http://img.cochemist.com/ccimg/879600/879562-26-2.png)
![(5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl 1-((2'-(2H-tetrazol-5-yl)-[1,1'-biphenyl]-4-yl)methyl)-4-(prop-1-en-2-yl)-2-propyl-1H-imidazole-5-carboxylate](http://img.cochemist.com/ccimg/879600/879562-26-2_b.png)
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6.png)
![Benzamide,N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-4-(1-piperazinylmethyl)-](http://img.cochemist.com/ccimg/404900/404844-02-6_b.png)


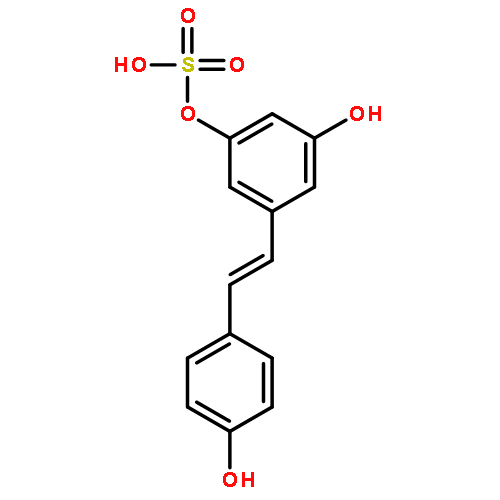
![b-D-Glucopyranosiduronic acid,3-hydroxy-5-[(1E)-2-(4-hydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-17-0.png)
![b-D-Glucopyranosiduronic acid,3-hydroxy-5-[(1E)-2-(4-hydroxyphenyl)ethenyl]phenyl](http://img.cochemist.com/ccimg/387400/387372-17-0_b.png)

![L-Alanine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-3-(methylsulfinyl)-](http://img.cochemist.com/ccimg/194100/194019-20-0.png)
![L-Alanine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-3-(methylsulfinyl)-](http://img.cochemist.com/ccimg/194100/194019-20-0_b.png)



![Benzenepropanoic acid, a-[[(2E)-3-[2-(carboxymethyl)-3,4-dihydroxyphenyl]-1-oxo-2-propen-1-yl]oxy]-3,4-dihydroxy-,(aR)-](http://img.cochemist.com/ccimg/143000/142998-47-8.png)
![Benzenepropanoic acid, a-[[(2E)-3-[2-(carboxymethyl)-3,4-dihydroxyphenyl]-1-oxo-2-propen-1-yl]oxy]-3,4-dihydroxy-,(aR)-](http://img.cochemist.com/ccimg/143000/142998-47-8_b.png)


![Glycine,N-[2-[(acetyloxy)methoxy]-2-oxoethyl]-N-[2-[2-[2-[bis(carboxymethyl)amino]phenoxy]ethoxy]phenyl]-](http://img.cochemist.com/ccimg/139900/139890-68-9.png)
![Glycine,N-[2-[(acetyloxy)methoxy]-2-oxoethyl]-N-[2-[2-[2-[bis(carboxymethyl)amino]phenoxy]ethoxy]phenyl]-](http://img.cochemist.com/ccimg/139900/139890-68-9_b.png)
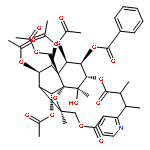
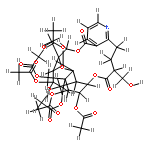
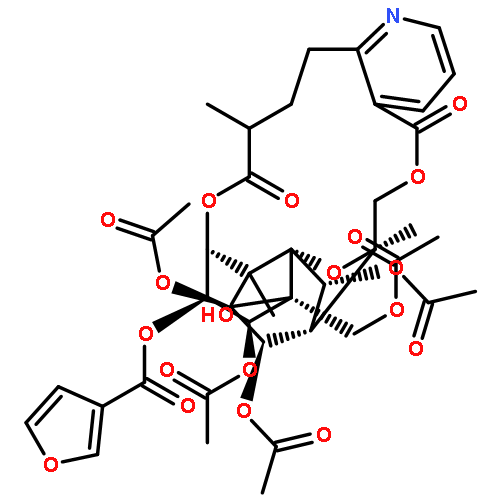
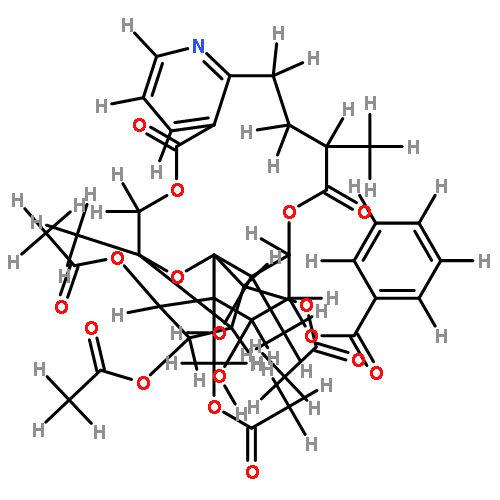
![8,11-Epoxy-9,12-ethano-11,15-methano-11H-[1,8]dioxacycloheptadecino[4,3-b]pyridine-5,17-dione,14,21,22-tris(acetyloxy)-12-[(acetyloxy)methyl]-10,13-bis(benzoyloxy)-7,8,9,10,12,13,14,15,18,19-decahydro-20-hydroxy-8,18,19,20-tetramethyl-,(8R,9R,10R,11R,12S,13R,14R,15S,18S,19S,20S,21S,22R)- (9CI)](http://img.cochemist.com/ccimg/133800/133740-16-6.png)
![8,11-Epoxy-9,12-ethano-11,15-methano-11H-[1,8]dioxacycloheptadecino[4,3-b]pyridine-5,17-dione,14,21,22-tris(acetyloxy)-12-[(acetyloxy)methyl]-10,13-bis(benzoyloxy)-7,8,9,10,12,13,14,15,18,19-decahydro-20-hydroxy-8,18,19,20-tetramethyl-,(8R,9R,10R,11R,12S,13R,14R,15S,18S,19S,20S,21S,22R)- (9CI)](http://img.cochemist.com/ccimg/133800/133740-16-6_b.png)
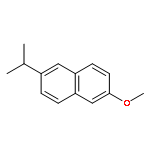
![Isoquinoline,1,2,3,4-tetrahydro-6,7-dimethoxy-1-[1-(6-methoxy-2-naphthalenyl)ethyl]-](http://img.cochemist.com/ccimg/132900/132836-13-6.png)
![Isoquinoline,1,2,3,4-tetrahydro-6,7-dimethoxy-1-[1-(6-methoxy-2-naphthalenyl)ethyl]-](http://img.cochemist.com/ccimg/132900/132836-13-6_b.png)
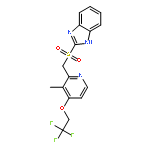
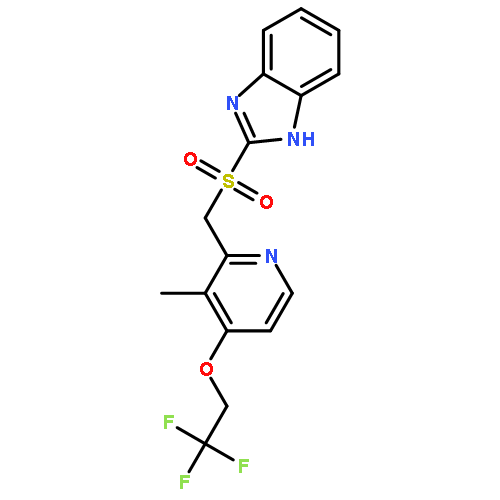
![1H-Benzimidazol-6-ol,2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridinyl]methyl]sulfinyl]-](http://img.cochemist.com/ccimg/132000/131926-98-2.png)
![1H-Benzimidazol-6-ol,2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridinyl]methyl]sulfinyl]-](http://img.cochemist.com/ccimg/132000/131926-98-2_b.png)





![Benzoic acid,4-[(aminoiminomethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/122300/122228-09-5.png)
![Benzoic acid,4-[(aminoiminomethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/122300/122228-09-5_b.png)


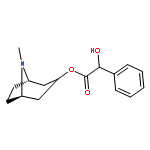
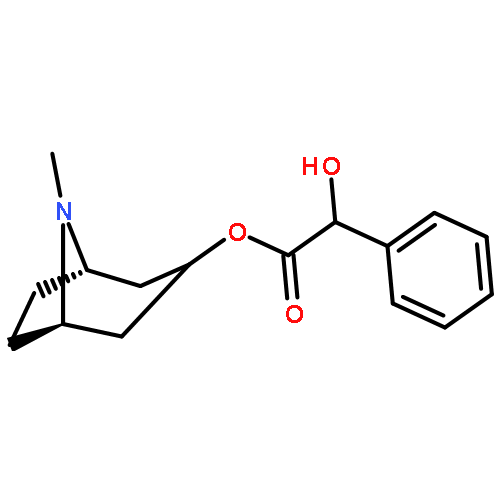
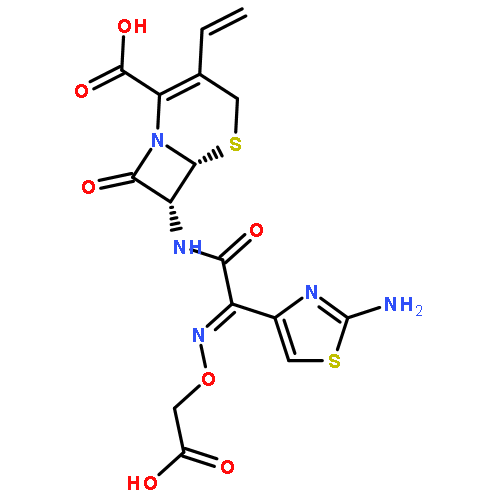
![(6R,7S)-7-[[(2Z)-2-(2-AMINO-1,3-THIAZOL-4-YL)-2-(CARBOXYMETHOXYIMINO)ACETYL]AMINO]-3-ETHENYL-8-OXO-5-THIA-1-AZABICYCLO[4.2.0]OCT-2-ENE-2-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/108700/108691-83-4.png)
![(6R,7S)-7-[[(2Z)-2-(2-AMINO-1,3-THIAZOL-4-YL)-2-(CARBOXYMETHOXYIMINO)ACETYL]AMINO]-3-ETHENYL-8-OXO-5-THIA-1-AZABICYCLO[4.2.0]OCT-2-ENE-2-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/108700/108691-83-4_b.png)
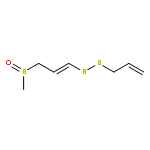
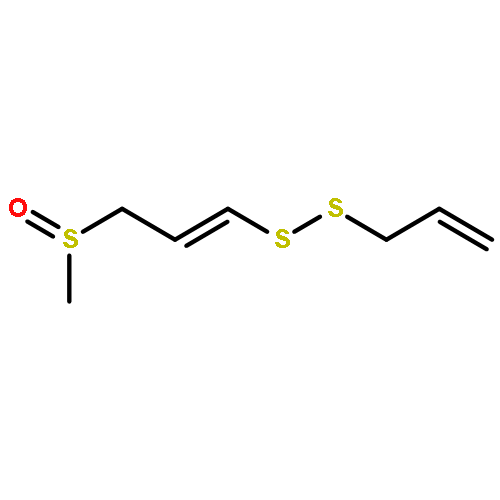

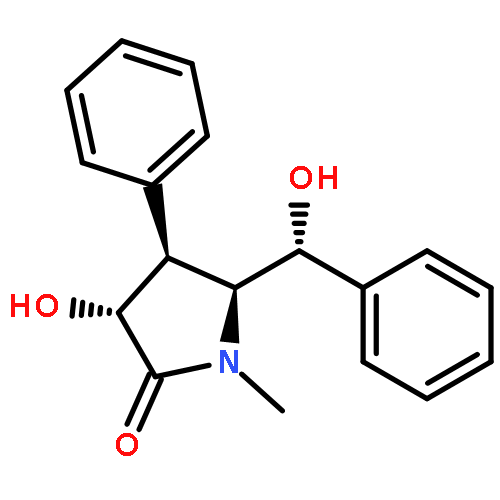
![Benzoic acid,4-[(aminoiminomethyl)amino]-, 4-(methoxycarbonyl)phenyl ester](http://img.cochemist.com/ccimg/99500/99450-77-8.png)
![Benzoic acid,4-[(aminoiminomethyl)amino]-, 4-(methoxycarbonyl)phenyl ester](http://img.cochemist.com/ccimg/99500/99450-77-8_b.png)




![7-{2-[(2-aminothiazol)-4-yl]-2-[(Z)(methoxycarbonyl)methoxyimino]acetamido}-3-vinyl-ceph-3-em-4-carboxylic acid](http://img.cochemist.com/ccimg/88700/88621-01-6.png)
![7-{2-[(2-aminothiazol)-4-yl]-2-[(Z)(methoxycarbonyl)methoxyimino]acetamido}-3-vinyl-ceph-3-em-4-carboxylic acid](http://img.cochemist.com/ccimg/88700/88621-01-6_b.png)


![[(2R)-2-[(Z)-octadec-9-enoyl]oxy-3-phosphonooxy-propyl] (Z)-octadec-9-enoate](http://img.cochemist.com/ccimg/61700/61617-08-1.png)
![[(2R)-2-[(Z)-octadec-9-enoyl]oxy-3-phosphonooxy-propyl] (Z)-octadec-9-enoate](http://img.cochemist.com/ccimg/61700/61617-08-1_b.png)
![1,3-Benzenediol, 5-[2-(4-hydroxyphenyl)ethyl]-](/data/chemimg/1590700/58436-28-5.png)
![1,3-Benzenediol, 5-[2-(4-hydroxyphenyl)ethyl]-](/data/chemimg/1590700/58436-28-5_b.png)
![Isoquinolinium,3,4-dihydro-6,7-dimethoxy-2-[(4-nitrophenyl)methyl]-, bromide (1:1)](http://img.cochemist.com/ccimg/57700/57687-28-2.png)
![Isoquinolinium,3,4-dihydro-6,7-dimethoxy-2-[(4-nitrophenyl)methyl]-, bromide (1:1)](http://img.cochemist.com/ccimg/57700/57687-28-2_b.png)



![ETHYL {2-AMINO-6-[(4-FLUOROBENZYL)AMINO]-3-PYRIDINYL}CARBAMATE HY<WBR />DROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/37300/37270-69-2.png)
![ETHYL {2-AMINO-6-[(4-FLUOROBENZYL)AMINO]-3-PYRIDINYL}CARBAMATE HY<WBR />DROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/37300/37270-69-2_b.png)
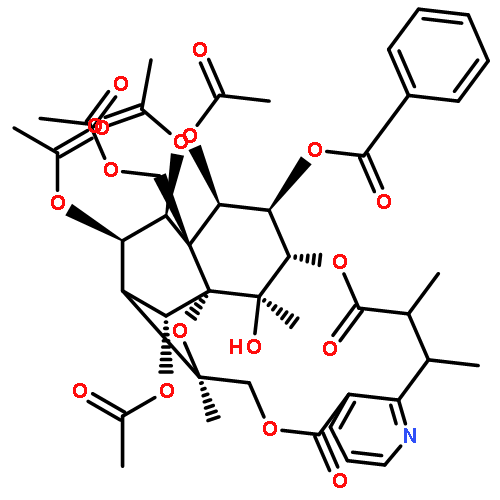
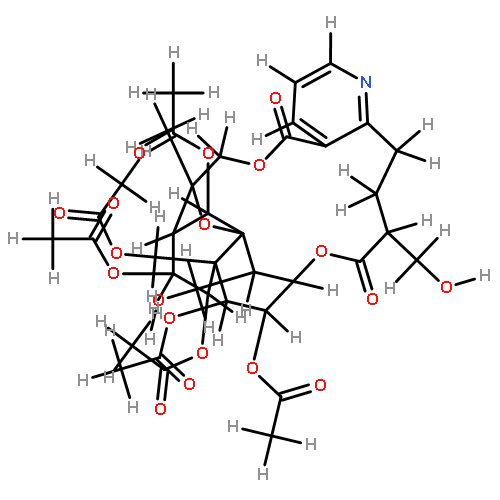
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-18,21-dihydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,21S,22S,23R)-](http://img.cochemist.com/ccimg/37300/37239-51-3.png)
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-18,21-dihydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,21S,22S,23R)-](http://img.cochemist.com/ccimg/37300/37239-51-3_b.png)
![3-Furancarboxylic acid,(8R,9R,10R,11S,12R,13R,14R,15S,21S,22S,23R)-10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-7,8,9,10,12,13,14,15,17,18,19,20-dodecahydro-18,21-dihydroxy-8,18,21-trimethyl-5,17-dioxo-8,11-epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridin-14-ylester](http://img.cochemist.com/ccimg/37300/37239-48-8.png)
![3-Furancarboxylic acid,(8R,9R,10R,11S,12R,13R,14R,15S,21S,22S,23R)-10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-7,8,9,10,12,13,14,15,17,18,19,20-dodecahydro-18,21-dihydroxy-8,18,21-trimethyl-5,17-dioxo-8,11-epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridin-14-ylester](http://img.cochemist.com/ccimg/37300/37239-48-8_b.png)



![L-Cysteine, S-methyl-,S-oxide, [S(S)]-](http://img.cochemist.com/ccimg/32800/32726-14-0.png)
![L-Cysteine, S-methyl-,S-oxide, [S(S)]-](http://img.cochemist.com/ccimg/32800/32726-14-0_b.png)
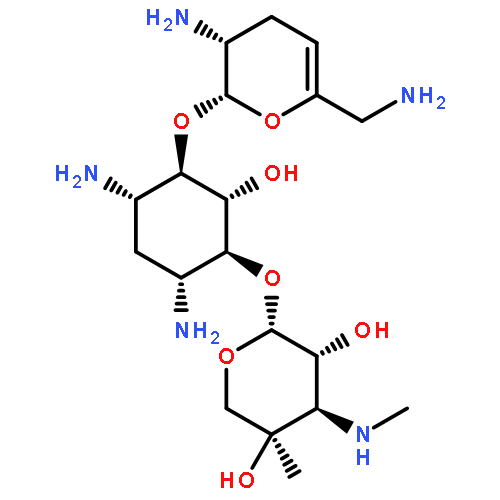

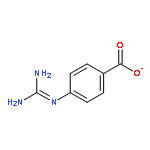
![L-Cysteine,S-(1E)-1-propen-1-yl-, S-oxide, [S(R)]-](http://img.cochemist.com/ccimg/16800/16718-23-3.png)
![L-Cysteine,S-(1E)-1-propen-1-yl-, S-oxide, [S(R)]-](http://img.cochemist.com/ccimg/16800/16718-23-3_b.png)
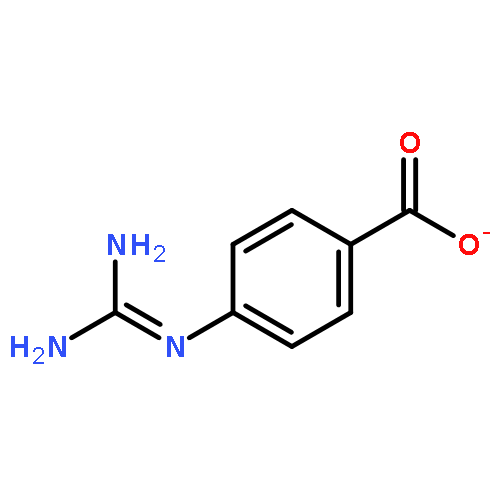






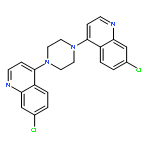
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-21-hydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,18S,21S,22S,23R)-](http://img.cochemist.com/ccimg/11100/11088-09-8.png)
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-21-hydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,18S,21S,22S,23R)-](http://img.cochemist.com/ccimg/11100/11088-09-8_b.png)

















![1,7,7-TRIMETHYLBICYCLO[2.2.1]HEPTAN-2-OL ACETATE](http://img.cochemist.com/ccimg/100/76-49-3.png)
![1,7,7-TRIMETHYLBICYCLO[2.2.1]HEPTAN-2-OL ACETATE](http://img.cochemist.com/ccimg/100/76-49-3_b.png)










![1,6-DIMETHYLPHENANTHRO[1,2-B]FURAN-10,11-DIONE](http://img.cochemist.com/ccimg/54700/54693-68-4.png)
![1,6-DIMETHYLPHENANTHRO[1,2-B]FURAN-10,11-DIONE](http://img.cochemist.com/ccimg/54700/54693-68-4_b.png)




![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4.png)
![(2s,3s)-4-[(e)-3-[(1r)-1-carboxy-2-(3,4-dihydroxyphenyl)ethoxy]-3-oxoprop-1-enyl]-2-(3,4-dihydroxyphenyl)-7-hydroxy-2,3-dihydro-1-benzofuran-3-carboxylic Acid](http://img.cochemist.com/ccimg/28900/28831-65-4_b.png)