Co-reporter:James D. Osborne; Thomas P. Matthews; Tatiana McHardy; Nicolas Proisy; Kwai-Ming J. Cheung; Michael Lainchbury; Nathan Brown; Michael I. Walton; Paul D. Eve; Katherine J. Boxall; Angela Hayes; Alan T. Henley; Melanie R. Valenti; Alexis K. De Haven Brandon; Gary Box; Yann Jamin; Simon P. Robinson; Isaac M. Westwood; Rob L. M. van Montfort; Philip M. Leonard; Marieke B. A. C. Lamers; John C. Reader; G. Wynne Aherne; Florence I. Raynaud; Suzanne A. Eccles; Michelle D. Garrett
Journal of Medicinal Chemistry 2016 Volume 59(Issue 11) pp:5221-5237
Publication Date(Web):May 11, 2016
DOI:10.1021/acs.jmedchem.5b01938
Multiparameter optimization of a series of 5-((4-aminopyridin-2-yl)amino)pyrazine-2-carbonitriles resulted in the identification of a potent and selective oral CHK1 preclinical development candidate with in vivo efficacy as a potentiator of deoxyribonucleic acid (DNA) damaging chemotherapy and as a single agent. Cellular mechanism of action assays were used to give an integrated assessment of compound selectivity during optimization resulting in a highly CHK1 selective adenosine triphosphate (ATP) competitive inhibitor. A single substituent vector directed away from the CHK1 kinase active site was unexpectedly found to drive the selective cellular efficacy of the compounds. Both CHK1 potency and off-target human ether-a-go-go-related gene (hERG) ion channel inhibition were dependent on lipophilicity and basicity in this series. Optimization of CHK1 cellular potency and in vivo pharmacokinetic–pharmacodynamic (PK–PD) properties gave a compound with low predicted doses and exposures in humans which mitigated the residual weak in vitro hERG inhibition.
Co-reporter:Charlotte E. Allen, Amanda J. Welford, Thomas P. Matthews, John J. Caldwell and Ian Collins
MedChemComm 2014 vol. 5(Issue 2) pp:180-185
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3MD00308F
The activity patterns of kinase hinge-binding fragments can be retained or redirected in fragment growing strategies. Targeting conserved kinase features preserved the selectivity pattern of a PKB hinge-binding fragment over a 5000-fold increase in potency, while late-stage modification of a CHK1 hinge-binding fragment substantially changed the pattern.
Co-reporter:Charlotte E. Allen, Chiau L. Chow, John J. Caldwell, Isaac M. Westwood, Rob L. M. van Montfort, Ian Collins
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 18) pp:5707-5724
Publication Date(Web):15 September 2013
DOI:10.1016/j.bmc.2013.07.021
With the success of protein kinase inhibitors as drugs to target cancer, there is a continued need for new kinase inhibitor scaffolds. We have investigated the synthesis and kinase inhibition of new heteroaryl-substituted diazaspirocyclic compounds that mimic ATP. Versatile syntheses of substituted diazaspirocycles through ring-closing metathesis were demonstrated. Diazaspirocycles directly linked to heteroaromatic hinge binder groups provided ligand efficient inhibitors of multiple kinases, suitable as starting points for further optimization. The binding modes of representative diazaspirocyclic motifs were confirmed by protein crystallography. Selectivity profiles were influenced by the hinge binder group and the interactions of basic nitrogen atoms in the scaffold with acidic side-chains of residues in the ATP pocket. The introduction of more complex substitution to the diazaspirocycles increased potency and varied the selectivity profiles of these initial hits through engagement of the P-loop and changes to the spirocycle conformation, demonstrating the potential of these core scaffolds for future application to kinase inhibitor discovery.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Michael Lainchbury ; Thomas P. Matthews ; Tatiana McHardy ; Kathy J. Boxall ; Michael I. Walton ; Paul D. Eve ; Angela Hayes ; Melanie R. Valenti ; Alexis K. de Haven Brandon ; Gary Box ; G. Wynne Aherne ; John C. Reader ; Florence I. Raynaud ; Suzanne A. Eccles ; Michelle D. Garrett
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:10229-10240
Publication Date(Web):October 19, 2012
DOI:10.1021/jm3012933
Inhibitors of checkpoint kinase 1 (CHK1) are of current interest as potential antitumor agents, but the most advanced inhibitor series reported to date are not orally bioavailable. A novel series of potent and orally bioavailable 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitrile CHK1 inhibitors was generated by hybridization of two lead scaffolds derived from fragment-based drug design and optimized for CHK1 potency and high selectivity using a cell-based assay cascade. Efficient in vivo pharmacokinetic assessment was used to identify compounds with prolonged exposure following oral dosing. The optimized compound (CCT244747) was a potent and highly selective CHK1 inhibitor, which modulated the DNA damage response pathway in human tumor xenografts and showed antitumor activity in combination with genotoxic chemotherapies and as a single agent.
Co-reporter:John C. Reader ; Thomas P. Matthews ; Suki Klair ; Kwai-Ming J. Cheung ; Jane Scanlon ; Nicolas Proisy ; Glynn Addison ; John Ellard ; Nelly Piton ; Suzanne Taylor ; Michael Cherry ; Martin Fisher ; Kathy Boxall ; Samantha Burns ; Michael I. Walton ; Isaac M. Westwood ; Angela Hayes ; Paul Eve ; Melanie Valenti ; Alexis de Haven Brandon ; Gary Box ; Rob L. M. van Montfort ; David H. Williams ; G. Wynne Aherne ; Florence I. Raynaud ; Suzanne A. Eccles ; Michelle D. Garrett
Journal of Medicinal Chemistry 2011 Volume 54(Issue 24) pp:8328-8342
Publication Date(Web):November 23, 2011
DOI:10.1021/jm2007326
Pyrazolopyridine inhibitors with low micromolar potency for CHK1 and good selectivity against CHK2 were previously identified by fragment-based screening. The optimization of the pyrazolopyridines to a series of potent and CHK1-selective isoquinolines demonstrates how fragment-growing and scaffold morphing strategies arising from a structure-based understanding of CHK1 inhibitor binding can be combined to successfully progress fragment-derived hit matter to compounds with activity in vivo. The challenges of improving CHK1 potency and selectivity, addressing synthetic tractability, and achieving novelty in the crowded kinase inhibitor chemical space were tackled by multiple scaffold morphing steps, which progressed through tricyclic pyrimido[2,3-b]azaindoles to N-(pyrazin-2-yl)pyrimidin-4-amines and ultimately to imidazo[4,5-c]pyridines and isoquinolines. A potent and highly selective isoquinoline CHK1 inhibitor (SAR-020106) was identified, which potentiated the efficacies of irinotecan and gemcitabine in SW620 human colon carcinoma xenografts in nude mice.
Co-reporter:Amanda J. Welford and Ian Collins
Journal of Natural Products 2011 Volume 74(Issue 10) pp:2318-2328
Publication Date(Web):October 4, 2011
DOI:10.1021/np200125v
The 2,11-cyclized cembranoids are isolated from marine invertebrates of Octocorallia species. They are a very interesting class of natural products sharing a common oxatricyclo[6.6.1.02,7]pentadecane core and carrying a varied substituent pattern. This review presents their structural diversity along with the reported biological activities. The 2,11-cyclized cembranoids were comprehensively reviewed previously in 1998, and this contribution will serve as an update of that work. Since 1998 a number of structural assignments of the isolated products have been revised, some as a result of total synthesis efforts. The chemical reactivity of several of the natural compounds has been studied, and the relevance of these findings to the biosynthesis or the generation of isolation artifacts is discussed. The wide range of biological activities displayed by the 2,11-cyclized cembranoids justifies the interest shown within the synthetic chemistry community and suggests that this class of natural products remains a fruitful area for future synthetic and biological research.
Co-reporter:Lynette A. Smyth, Thomas P. Matthews, Ian Collins
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 11) pp:3569-3578
Publication Date(Web):1 June 2011
DOI:10.1016/j.bmc.2011.03.069
A lead-like kinase inhibitor screening library containing new 3-aminopyrazolopyridinones and closely related compounds was designed that contained hydrogen-bond donor-acceptor motifs and substitution vectors inspired by the natural product kinase inhibitor indirubin. The solubility of the 3-aminopyrazolopyridinone scaffold was more than 1000-fold greater than that of indirubin itself, and solubility was enhanced by reduction of the proportion of lipophilic aryl substituents or the introduction of basic groups. Several components of the library showed kinase inhibitory activity. A subset of diaryl-substituted analogues preferentially inhibited tyrosine kinases with low micromolar activity and good ligand efficiency, and showed cellular antiproliferative activity. The evaluation of the library shows that new, non-natural compounds with relevant biological activity and improved physicochemical properties can be generated from the natural product indirubin, providing compounds that may be useful for kinase inhibitor drug discovery.
Co-reporter:Tatiana McHardy ; John J. Caldwell ; Kwai-Ming Cheung ; Lisa J. Hunter ; Kevin Taylor ; Martin Rowlands ; Ruth Ruddle ; Alan Henley ; Alexis de Haven Brandon ; Melanie Valenti ; Thomas G. Davies ; Lynsey Fazal ; Lisa Seavers ; Florence I. Raynaud ; Suzanne A. Eccles ; G. Wynne Aherne ; Michelle D. Garrett
Journal of Medicinal Chemistry 2010 Volume 53(Issue 5) pp:2239-2249
Publication Date(Web):February 12, 2010
DOI:10.1021/jm901788j
Protein kinase B (PKB or Akt) is an important component of intracellular signaling pathways regulating growth and survival. Signaling through PKB is frequently deregulated in cancer, and inhibitors of PKB therefore have potential as antitumor agents. The optimization of lipophilic substitution within a series of 4-benzyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amines provided ATP-competitive, nanomolar inhibitors with up to 150-fold selectivity for inhibition of PKB over the closely related kinase PKA. Although active in cellular assays, compounds containing 4-amino-4-benzylpiperidines underwent metabolism in vivo, leading to rapid clearance and low oral bioavailability. Variation of the linker group between the piperidine and the lipophilic substituent identified 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as potent and orally bioavailable inhibitors of PKB. Representative compounds modulated biomarkers of signaling through PKB in vivo and strongly inhibited the growth of human tumor xenografts in nude mice at well-tolerated doses.
Co-reporter:Paul Workman, Ian Collins
Chemistry & Biology 2010 Volume 17(Issue 6) pp:561-577
Publication Date(Web):25 June 2010
DOI:10.1016/j.chembiol.2010.05.013
Chemical probes for interrogating biological processes are of considerable current interest. Cell permeable small molecule tools have a major role in facilitating the functional annotation of the human genome, understanding both physiological and pathological processes, and validating new molecular targets. To be valuable, chemical tools must satisfy necessary criteria and recent publications have suggested objective guidelines for what makes a useful chemical probe. Although recognizing that such guidelines may be valuable, we caution against overly restrictive rules that may stifle innovation in favor of a “fit-for-purpose” approach. Reviewing the literature and providing examples from the cancer field, we recommend a series of “fitness factors” to be considered when assessing chemical probes. We hope this will encourage innovative chemical biology research while minimizing the generation of poor quality and misleading biological data, thus increasing understanding of the particular biological area, to the benefit of basic research and drug discovery.
Co-reporter:Stephen Hilton, Sebastien Naud, John J. Caldwell, Kathy Boxall, Samantha Burns, Victoria E. Anderson, Laurent Antoni, Charlotte E. Allen, Laurence H. Pearl, Antony W. Oliver, G. Wynne Aherne, Michelle D. Garrett, Ian Collins
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 2) pp:707-718
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmc.2009.11.058
5-(Hetero)aryl-3-(4-carboxamidophenyl)-2-aminopyridine inhibitors of CHK2 were identified from high throughput screening of a kinase-focussed compound library. Rapid exploration of the hits through straightforward chemistry established structure–activity relationships and a proposed ATP-competitive binding mode which was verified by X-ray crystallography of several analogues bound to CHK2. Variation of the 5-(hetero)aryl substituent identified bicyclic dioxolane and dioxane groups which improved the affinity and the selectivity of the compounds for CHK2 versus CHK1. The 3-(4-carboxamidophenyl) substituent could be successfully replaced by acyclic ω-aminoalkylamides, which made additional polar interactions within the binding site and led to more potent inhibitors of CHK2. Compounds from this series showed activity in cell-based mechanistic assays for inhibition of CHK2.
Co-reporter:Stephen Hilton, Sebastien Naud, John J. Caldwell, Kathy Boxall, Samantha Burns, Victoria E. Anderson, Laurent Antoni, Charlotte E. Allen, Laurence H. Pearl, Antony W. Oliver, G. Wynne Aherne, Michelle D. Garrett, Ian Collins
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 12) pp:4591
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmc.2010.05.014
Co-reporter:Lynette A. Smyth, Thomas P. Matthews, Peter N. Horton, Michael B. Hursthouse, Ian Collins
Tetrahedron 2010 66(15) pp: 2843-2854
Publication Date(Web):
DOI:10.1016/j.tet.2010.02.047
Co-reporter:Thomas P. Matthews ; Suki Klair ; Samantha Burns ; Kathy Boxall ; Michael Cherry ; Martin Fisher ; Isaac M. Westwood ; Michael I. Walton ; Tatiana McHardy ; Kwai-Ming J. Cheung ; Rob Van Montfort ; David Williams ; G. Wynne Aherne ; Michelle D. Garrett ; John Reader
Journal of Medicinal Chemistry 2009 Volume 52(Issue 15) pp:4810-4819
Publication Date(Web):July 2, 2009
DOI:10.1021/jm900314j
Checkpoint kinase 1 (CHK1) is an oncology target of significant current interest. Inhibition of CHK1 abrogates DNA damage-induced cell cycle checkpoints and sensitizes p53 deficient cancer cells to genotoxic therapies. Using template screening, a fragment-based approach to small molecule hit generation, we have identified multiple CHK1 inhibitor scaffolds suitable for further optimization. The sequential combination of in silico low molecular weight template selection, a high concentration biochemical assay and hit validation through protein−ligand X-ray crystallography provided 13 template hits from an initial in silico screening library of ca. 15000 compounds. The use of appropriate counter-screening to rule out nonspecific aggregation by test compounds was essential for optimum performance of the high concentration bioassay. One low molecular weight, weakly active purine template hit was progressed by iterative structure-based design to give submicromolar pyrazolopyridines with good ligand efficiency and appropriate CHK1-mediated cellular activity in HT29 colon cancer cells.
Co-reporter:John J. Caldwell ; Thomas G. Davies ; Alastair Donald ; Tatiana McHardy ; Martin G. Rowlands ; G. Wynne Aherne ; Lisa K. Hunter ; Kevin Taylor ; Ruth Ruddle ; Florence I. Raynaud ; Marcel Verdonk ; Paul Workman ; Michelle D. Garrett
Journal of Medicinal Chemistry 2008 Volume 51(Issue 7) pp:2147-2157
Publication Date(Web):March 18, 2008
DOI:10.1021/jm701437d
Fragment-based screening identified 7-azaindole as a protein kinase B inhibitor scaffold. Fragment elaboration using iterative crystallography of inhibitor−PKA−PKB chimera complexes efficiently guided improvements in the potency and selectivity of the compounds, resulting in the identification of nanomolar 6-(piperidin-1-yl)purine, 4-(piperidin-1-yl)-7-azaindole, and 4-(piperidin-1-yl)pyrrolo[2,3-d]pyrimidine inhibitors of PKBβ with antiproliferative activity and showing pathway inhibition in cells. A divergence in the binding mode was seen between 4-aminomethylpiperidine and 4-aminopiperidine containing molecules. Selectivity for PKB vs PKA was observed with 4-aminopiperidine derivatives, and the most PKB-selective inhibitor (30-fold) showed significantly different bound conformations between PKA and PKA−PKB chimera.
Co-reporter:Michelle D. Garrett, Ian Collins
Trends in Pharmacological Sciences (May 2011) Volume 32(Issue 5) pp:308-316
Publication Date(Web):1 May 2011
DOI:10.1016/j.tips.2011.02.014
Research into inhibitors of the protein kinases controlling the cellular response to DNA damage has reached an exciting stage, particularly for the checkpoint kinases CHK1 and CHK2. Selective inhibitors are now being tested in clinical trials in cancer patients. In this review, we highlight recent data from cellular and in vivo preclinical models that provide insight into the clinical contexts for checkpoint kinase inhibition (e.g. the timing of treatment and what type of inhibitor would be most appropriate). Although it has been shown that CHK1 inhibition potentiates the efficacy of various DNA-damaging therapies, the context for selective CHK2 inhibition is not yet as well defined. Distinct effects of selective CHK1 or CHK2 inhibition are observed when combined with DNA-damaging agents. It has also been shown that both CHK1 and CHK2 inhibitors potentiate the effects of other molecular targeted therapeutics [e.g. poly(ADP-ribose) polymerase inhibitors]. We also consider the single-agent activity of checkpoint kinase inhibitors for tumours with defined genetic backgrounds.





![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)








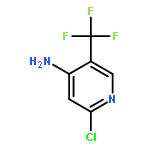





![1H-Purine, 6-[4-[4-(4-chlorophenyl)-4-piperidinyl]phenyl]-](http://img.cochemist.com/ccimg/885600/885594-13-8.png)
![1H-Purine, 6-[4-[4-(4-chlorophenyl)-4-piperidinyl]phenyl]-](http://img.cochemist.com/ccimg/885600/885594-13-8_b.png)
![4-PiperidineMethanaMine, 1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)-](http://img.cochemist.com/ccimg/885600/885594-09-2.png)
![4-PiperidineMethanaMine, 1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)-](http://img.cochemist.com/ccimg/885600/885594-09-2_b.png)




![4-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)piperidin-4-aMine](http://img.cochemist.com/ccimg/885500/885499-61-6.png)
![4-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)piperidin-4-aMine](http://img.cochemist.com/ccimg/885500/885499-61-6_b.png)
![4-PiperidinaMine, 1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)-](http://img.cochemist.com/ccimg/885500/885499-56-9.png)
![4-PiperidinaMine, 1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)-](http://img.cochemist.com/ccimg/885500/885499-56-9_b.png)


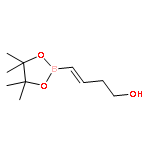
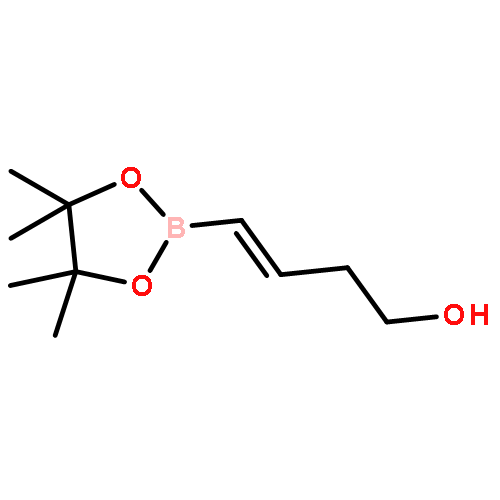


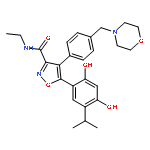









![2(5H)-Furanone, 5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-, (S)-](http://img.cochemist.com/ccimg/99400/99315-76-1.png)
![2(5H)-Furanone, 5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]-, (S)-](http://img.cochemist.com/ccimg/99400/99315-76-1_b.png)


![8-Azabicyclo[3.2.1]octan-3-amine, 8-methyl-, exo-](http://img.cochemist.com/ccimg/81500/81487-04-9.png)
![8-Azabicyclo[3.2.1]octan-3-amine, 8-methyl-, exo-](http://img.cochemist.com/ccimg/81500/81487-04-9_b.png)




![2-[6-amino-8-(benzylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/65500/65456-83-9.png)
![2-[6-amino-8-(benzylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/65500/65456-83-9_b.png)


![Silane, [(1-ethynylcyclopentyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/62800/62785-89-1.png)
![Silane, [(1-ethynylcyclopentyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/62800/62785-89-1_b.png)




![Silane, [(1,2-dimethyl-1-propenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/17600/17510-44-0.png)
![Silane, [(1,2-dimethyl-1-propenyl)oxy]trimethyl-](http://img.cochemist.com/ccimg/17600/17510-44-0_b.png)


![2-[6-amino-8-(methylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/13400/13389-13-4.png)
![2-[6-amino-8-(methylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol](http://img.cochemist.com/ccimg/13400/13389-13-4_b.png)
![1H-Imidazo[4,5-c]pyridin-4-amine,1-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/6800/6736-58-9.png)
![1H-Imidazo[4,5-c]pyridin-4-amine,1-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/6800/6736-58-9_b.png)










![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6_b.png)


![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0_b.png)



