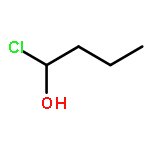
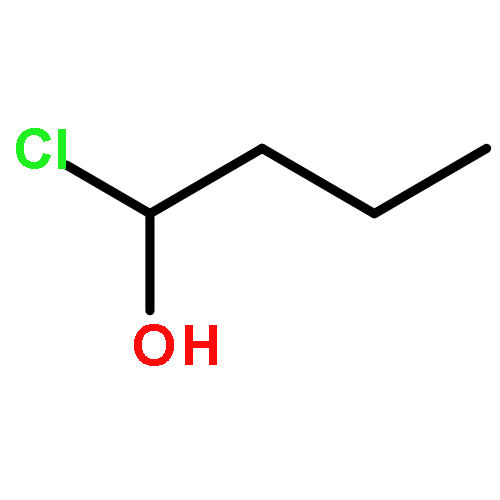
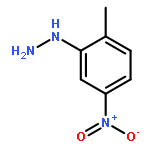
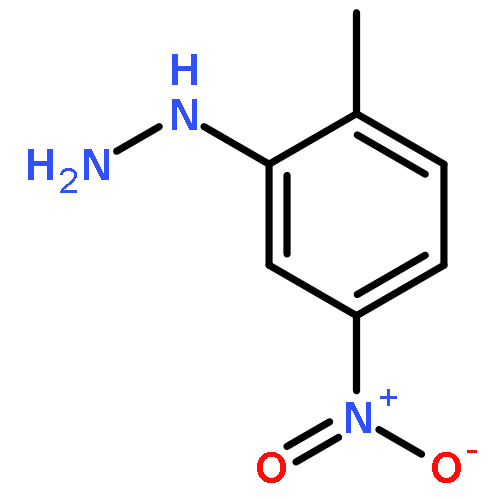
Co-reporter:Zhipeng Li, Yi-Feng Wang, Xu Zhang, Chengchu Zeng, Liming Hu, Xing-Jie Liang
Sensors and Actuators B: Chemical 2017 Volume 242() pp:189-194
Publication Date(Web):April 2017
DOI:10.1016/j.snb.2016.11.011
•A series of “turn-on” response mechanism compounds (F1-F4) to TYR have been designed and synthesized.•Based on tyrosinase-triggered oxidative reaction and urea hydrolysis reaction, F2 have excellent sensitivity and selectivity toward tyrosinase in vitro.•F2 was successfully used to imaging living melanoma cells as a result of the conversion of F2 to F0 by TYR catalysis.Melanoma, with poor prognosis and highly metastatic spread, is the most deadly skin cancer, but the monitoring and diagnosis of melanoma is still a challenging. The rate-limiting enzyme tyrosinase is crucial for controlling melanin production, and is a well-known biomarker closely associated with the level of malignancy. However, effective probes for detecting tyrosinase in living cells are currently lacking. This paper describes the design, synthesis and characterization of F2, a high sensitive type of “turn-on” fluorescent probe for imaging living melanoma cells. F2 could be activated by tyrosinase-catalyzed oxidation followed by hydrolysis of a urea linkage. The results demonstrate that F2 displays cyan fluorescence when activated by tyrosinase, and has sufficient sensitivity and selectivity to detect tyrosinase in aqueous solution and in living cells. F2 has potential for the noninvasive real-time diagnosis and tracking of melanoma.Download high-res image (216KB)Download full-size image
Co-reporter:Ming Bu, Tingting Cao, Hongxia Li, Mingzhou Guo, Burton B. Yang, Chengchu Zeng, Yue Zhou, Na Zhang, Liming Hu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.06.048
Inspired by the significant anti-cancer activity of our previously screened natural ergosterol peroxide (EP, 1), we synthesized and characterized a series of novel 5α,8α-epidioxyandrost-3β-ol-17-(O-phenylacetamide)oxime derivatives (9a–o). The anti-proliferative activity of the synthesized compounds against human hepatocellular carcinoma cells (HepG2, Sk-Hep1) and human breast cancer cells (MCF-7, MDA-MB231) were investigated. Compounds 9d, 9f, 9h, 9j and 9m displayed good anti-proliferative activity (most IC50 < 20 μM) in vitro. Furthermore, fluorescence imaging showed that the designed coumarin-9d conjugate (12) localized mainly in mitochondria, leading to enhanced anticancer activities over the parent structure.Download high-res image (105KB)Download full-size image
Co-reporter:Ming Bu, Tingting Cao, Hongxia Li, Mingzhou Guo, Burton B. Yang, Yue Zhou, Na Zhang, Chengchu Zeng, Liming Hu
Steroids 2017 Volume 124(Volume 124) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.steroids.2017.05.013
•Ergosterol peroxide was synthesized by photooxidation reaction.•A series of steroidal 5α,8α-endoperoxide derivatives with aliphatic side-chain were synthesized.•X-ray crystallography to support the stereochemistry of the endoperoxide.•Compounds 5b and 14d exhibited good anti-proliferative activity against HepG2 cells.By inspiration of significant anti-cancer activity of our previously screened natural ergosterol peroxide (EP), a series of novel steroidal 5α,8α-endoperoxide derivatives 5a–d and 14a–f were designed, synthesized, and biologically evaluated for their in vitro anti-proliferative inhibitory and cytotoxic activity. The results revealed that most of these compounds showed moderate-to-excellent anti-proliferative effects against the tested cancer cell lines (i.e. HepG2, SK-Hep1, MDA-MB-231 and MCF-7). Among them, compound 5b and 14d exhibited preferable inhibitory activities (IC50 of 5b and 14d are 8.07 and 9.50 μM against HepG2, respectively). The structure-activity relationships indicated that incorporation the peroxidic bridge to the steroid scaffolds at C-5 and C-8 positions together with the aliphatic side-chain at the C-17 position would provide synergistic effect for the bioactivity.Download high-res image (124KB)Download full-size image
Co-reporter:Xuemei Qin, Zhipeng Li, Leifu Yang, Peng Liu, Liming Hu, Chengchu Zeng, Zhiyong Pan
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 13) pp:2871-2881
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmc.2016.01.003
A novel series of 2,3-dihydro-[1,4]dioxino[2,3-f]quinazoline derivatives were designed, synthesized and evaluated as reversible and noncovalent epidermal growth factor receptor (EGFR) inhibitors. Most of the compounds exhibited good potency against EGFRwt and some showed moderate to excellent potency against EGFRT790M/L858R mutant. The half-maximal inhibitory concentration (IC50) values of twenty-one compounds against EGFRwt were less than 50 nM, and those of six compounds were less than 10 nM. The IC50 values of eleven compounds against EGFRT790M/L858R were less than 100 nM. Among these, compound b1 displayed the most potent inhibitory activity against EGFRwt (IC50 = 2.0 nM) and EGFRT790M/L858R (IC50 = 6.9 nM). Compounds with excellent inhibitory activities against EGFRwt and EGFRT790M/L858R kinase inhibitory activities showed good antiproliferative activities against H358 and A549 cells. Docking study was performed to position compound b1 into the EGFR active pocket to determine the probable binding conformation.
Co-reporter:Liming Hu, Tingting Cao, Yongjuan Lv, Yiming Ding, Leifu Yang, Qiang Zhang, Mingzhou Guo
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 23) pp:5830-5835
Publication Date(Web):1 December 2016
DOI:10.1016/j.bmcl.2016.10.007
A series of 4-((pyrazolo[1,5-a]pyrimidin-6-yl)-1H-pyrazol-1-yl)phenyl-3-benzamide derivatives and 4-((imidazo[1,2-b]pyridazin-3-yl)-1H-pyrazol-1-yl-)phenyl-3-benzamide derivatives were designed, synthesized as new BCR–ABL tyrosine kinase inhibitors by using combinational strategies of scaffold hopping and conformational constraint. These new compounds were screened for BCR–ABL1 kinase inhibitory activity, and most of them appeared good inhibitory activity against BCR–ABL1 kinase. One of the most potent compounds 16a strongly suppressed BCR–ABL1 kinase with IC50 value of 8.5 nM. The tested compounds 16a and 16i showed strong inhibitory activities against K562 with IC50 value of less than 2 nM. Molecular docking studies indicated that these compounds fitted well with the active site of BCR–ABL1 protein. The results showed these inhibitors may serve as lead compounds for further developing new drugs targeted BCR–ABL kinase.
Co-reporter:Xuemei Qin, Yongjuan Lv, Peng Liu, Zhipeng Li, Liming Hu, Chengchu Zeng, Leifu Yang
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 6) pp:1571-1575
Publication Date(Web):15 March 2016
DOI:10.1016/j.bmcl.2016.02.009
A series of novel morpholin-3-one-fused quinazoline derivatives were designed, synthesized and evaluated as EGFR tyrosine kinase inhibitors. Nineteen compounds showed significant inhibitory activities against EGFRwt kinase (IC50 < 1 μM). Compound a8 demonstrated the most potent inhibitory activity toward EGFRwt (IC50 = 53.1 nM). Compound a7 and a8 showed excellent inhibitory activities against mutant EGFRT790M/L858R and strong antiproliferative activity against H358 and A549 cell lines. Finally, molecular docking studies were performed to predict the possible binding mode of the target compounds. It is believed that this work would be very useful for designing a new series of tyrosine kinase inhibitors targeting EGFR.
Co-reporter:Gang Wang, Tao Peng, Shouguo Zhang, Junyi Wang, Xiaoxue Wen, Haiyan Yan, Liming Hu and Lin Wang
RSC Advances 2015 vol. 5(Issue 36) pp:28344-28348
Publication Date(Web):13 Mar 2015
DOI:10.1039/C5RA01210D
We developed a new efficient photolabile protecting group, 2-(2-nitrophenyl) propyl (Npp) that blocks the carboxyl group in peptide synthesis. The Npp group is quite compatible with the Fmoc–t-Bu strategy and can be removed rapidly and quantitatively by irradiation with UV light. Using this method, we prepared a head-to-tail cycloheptapeptide.
Co-reporter:Yujie Wang, Jie Rong, Bin Zhang, Liming Hu, Xiaoli Wang, Chengchu Zeng
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:735-741
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.12.059
A series of novel β-diketo derivatives which combined the virtues of dihydroxypyrimidine carboxamide derived from the evolution of DKA and polyhydroxylated aromatics moieties, were designed and synthesized as potential HIV-1 integrase (IN) inhibitors and evaluated their inhibition to the strand transfer process of HIV-1 integrase and anti-HIV-1 activity. The result indicates that 3,4,5-trihydroxylated aromatic derivatives exhibit good inhibition to HIV-1 integrase, but dihydroxylated aromatic derivatives appear little inhibition to HIV-1 integrase. In addition, the preliminary structure–activity relationship (SAR) of these new derivatives was rationalized by docking studies.3,4,5-Trihydroxylated aromatic derivatives exhibit good inhibition to HIV-1 integrase, but dihydroxylated aromatic derivatives appear little inhibition to HIV-1 integrase. The preliminary structure–activity relationship (SAR) of these new derivatives was rationalized.
Co-reporter:Liming Hu, Yuyan Zheng, Zhipeng Li, Yujie Wang, Yongjuan Lv, Xuemei Qin, Chengchu Zeng
Bioorganic & Medicinal Chemistry 2015 23(13) pp: 3147-3152
Publication Date(Web):
DOI:10.1016/j.bmc.2015.04.083
Co-reporter:Liming Hu, Yun Sun, Shengliang Li, Xiaoli Wang, Kelei Hu, Lirong Wang, Xing-jie Liang, Yan Wu
Carbon 2014 Volume 67() pp:508-513
Publication Date(Web):February 2014
DOI:10.1016/j.carbon.2013.10.023
Carbon dots (CD) are luminescent nanomaterial with unique properties that show great potential in many applications. Herein, branched polyethyleneimine-based carbon dots (PCD) are prepared from branched polyethyleneimine by oxidation and a modified hydrothermal reaction. Structure and composition analysis indicate that obtained PCD possesses a 3–4 nm in diameter and a graphitic structure with lattice spacing of 0.30 nm. The PCD has a quantum yield of 54.3%. The bright photoluminescence shows that it can be used for cell imaging. The PCD exhibits extremely good biocompatibility and can be applied for gene delivery. Because of its specific nanostructure and photoluminescence property, the multifunctional PCD prepared shows potential for applications in bioimaging and gene delivery.
Co-reporter:Liming Hu, Yun Sun and Yan Wu
Nanoscale 2013 vol. 5(Issue 8) pp:3103-3111
Publication Date(Web):28 Feb 2013
DOI:10.1039/C3NR00338H
Within the past few years, chitosan-based drug delivery vehicles have become some of the most attractive to be studied. In contrast to all other polysaccharides, chitosan has demonstrated its unique characteristics for drug delivery platforms, including its active primary amino groups for chemical modification, simple and mild preparation methods for the encapsulation of biomolecules or drugs, mucoadhesion to facilitate transport across mucosal barriers and so on. In this review, an overview of the various types of chitosan-based drug delivery systems is provided, with special focus on polymeric drug conjugates and drug nanocarriers. The first part of the review is concerned with the development and applications of polymeric chitosan–drug conjugates. Then the chitosan-based nanocarrier systems as well as their preparation methods and applications are further discussed.
Co-reporter:Meng Wang, Yunhe Jin, Haijun Yang, Hua Fu and Liming Hu
RSC Advances 2013 vol. 3(Issue 22) pp:8211-8214
Publication Date(Web):10 Apr 2013
DOI:10.1039/C3RA40999F
A novel and efficient copper-catalyzed one-pot synthesis of indoloimidazoquinoline derivatives has been developed. The protocol uses the readily available substituted 2-(2-bromophenyl)-1H-indoles, imidazole and benzoimidazoles as the starting materials, inexpensive CuBr as the catalyst, air as the terminal oxidant, and the procedure underwent a sequential copper-catalyzed intermolecular N-arylation and an aerobic oxidative intramolecular C–H/C–H coupling.
Co-reporter:Liming Hu, Sulei Zhang, Xianzhuo He, Zaigang Luo, Xiaoli Wang, Wei Liu, Xuemei Qin
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:177-182
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.11.014
A series of novel β-diketo derivatives which combined the virtues of 1,3-diketo, 1,2,3-triazole and polyhydroxylated aromatics moieties, were designed and synthesized as potential HIV-1 integrase (IN) inhibitors and evaluated their inhibition to the strand transfer process of HIV-1 integrase. The result indicates that 3,4,5-trihydroxylated aromatic derivatives exhibit good inhibition to HIV-1 integrase, but dihydroxylated aromatic derivatives and corresponding methoxy aromatic derivatives appear little inhibition to HIV-1 integrase.A series of novel β-diketo derivatives which combined the virtues of 1,3-diketo, 1,2,3-triazole and polyhydroxylated aromatics moieties were prepared and their HIV-1 IN inhibitory activities were evaluated. The polyhydroxylated aromatic moiety plays an important role in the inhibitory of HIV integrase.
Co-reporter:Leifu Yang, Xuemei Xu, Yali Huang, Bin Zhang, Chengchu Zeng, Hongqiu He, Cunxin Wang, Liming Hu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 18) pp:5469-5471
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmcl.2010.07.087
(E)-N-[3-(4-Cinnamoylpiperazin-1-yl)propyl]-3,4-dihydroxybenzamide and (E)-N-[3-(4-cinnamoylpiperazin-1-yl)propyl]-3,4,5-trihydroxybenzamide were designed and synthesized as potential HIV-1 integrase inhibitors and evaluated their inhibition to the strand transfer process of HIV-1 integrase. The result indicates that 3,4,5-trihydroxylated aromatic derivatives exhibit good inhibition to HIV-1 integrase, however, corresponding 3,4-dihydroxylated aromatic derivatives appear little inhibition of HIV-1 integrase.A series of polyhydroxylated aromatics having amidation of piperazine nitrogen were prepared and their HIV-1 IN inhibitory activities were evaluated. The galloyl moiety was a core pharmacophore in the inhibitory of HIV integrase.
Co-reporter:Zai Gang Luo, Cheng Chu Zeng, Lei Fu Yang, Hong Qiu He, Cun Xin Wang, Li Ming Hu
Chinese Chemical Letters 2009 Volume 20(Issue 7) pp:789-792
Publication Date(Web):July 2009
DOI:10.1016/j.cclet.2009.03.014
A series of novel 6-sulfamoyl-4-oxoquinoline-3-carboxylic acids derivatives have been synthesized and screened for HIV integrase inhibition activity. Their structures were confirmed by ESI-MS, 1H NMR and 13C NMR.
Co-reporter:Zaigang LUO;Chengchu ZENG;Daoshan YANG;Yali HUANG;Fang WANG;Hongguang DU
Chinese Journal of Chemistry 2009 Volume 27( Issue 7) pp:1333-1338
Publication Date(Web):
DOI:10.1002/cjoc.200990223
Abstract
Based on the structure of the HIV integrase core domain, dipeptide derivatives, as a type of HIV integrase inhibitor, were synthesized, and their fragmentation pathways were investigated by electrospray ionization mass spectrometry (ESI-MSn) in conjunction with tandem mass spectrometry (MS/MS). In order to better understand the fragmentation pathways, the MS2 and MS3 spectra of the title compound were obtained. The main fragmentation pathways occur by the cleavage of the C–CO bonds between N-(benzothiazol-2-yl)aminocarbonyl and methylene, NH–CO bonds between the NH groups and carbonyl groups. Electrospray ionization was proven to be a good method for the structural characterization and identification of this kind of compound.
Co-reporter:Ren Kong, Xuemei Xu, Weizu Chen, Cunxin Wang, Liming Hu
Acta Physico-Chimica Sinica 2007 Volume 23(Issue 9) pp:1325-1331
Publication Date(Web):September 2007
DOI:10.1016/S1872-1508(07)60067-9
A three-dimensional pharmacophore model was developed for a considerable number of pyrrolidine-based and butane-based chemokine (C-C motif) receptor 5 (CCR5) antagonists, which can block the entry of human immunodeficiency virus type 1 (HIV-1) by inhibiting the interaction of HIV-1 envelope protein and CCR5. The pharmacophore model was generated using a training set consisting of 25 carefully selected antagonists with the diverse molecular architecture and bioactivity, as required by the Catalyst/HypoGen program. The activity of the training set molecules expressed in IC50 (half-inhibitory concentration) covered from 0.06 to 10000 nmol·L–1. The most predictive pharmacophore model (Hypo 1), consisting of two positive ionizable points and three hydrophobic groups, had a correlation of 0.924 and a root mean square of 1.068, and a cost difference of 63.67 bits between the null cost and the total cost. The model was applied in predicting the activity of 74 compounds as a test set. The results indicated that the model was able to provide clear guidelines and accurate activity prediction for novel antagonist design.



![4-[(diethylamino)methyl]benzoyl chloride](http://img.cochemist.com/ccimg/80400/80335-46-2.png)
![4-[(diethylamino)methyl]benzoyl chloride](http://img.cochemist.com/ccimg/80400/80335-46-2_b.png)
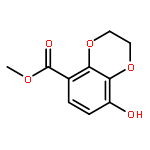
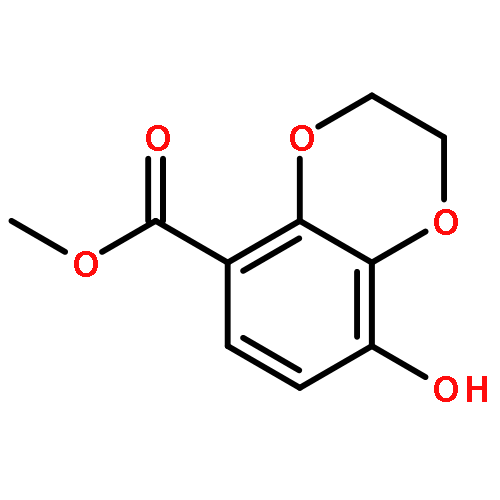
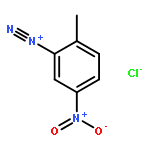
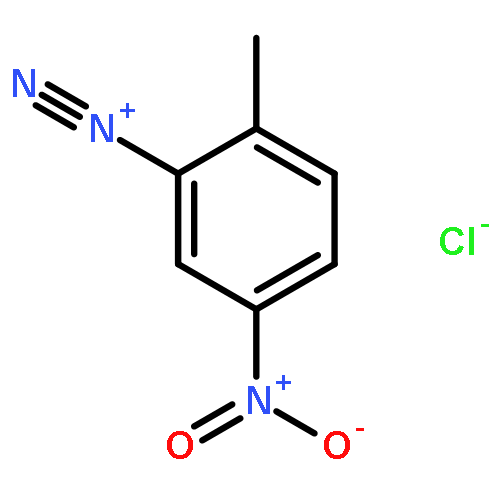


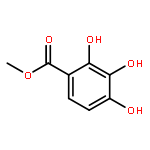
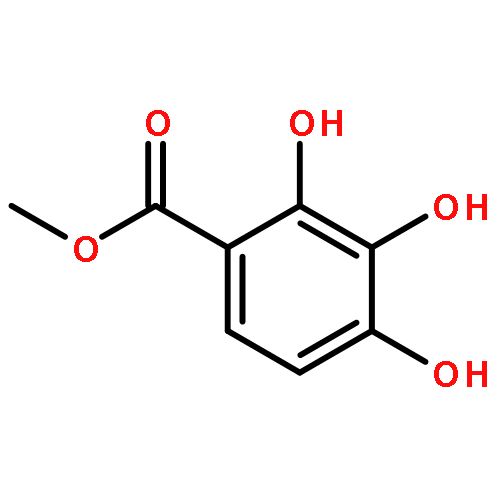
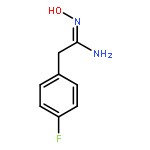
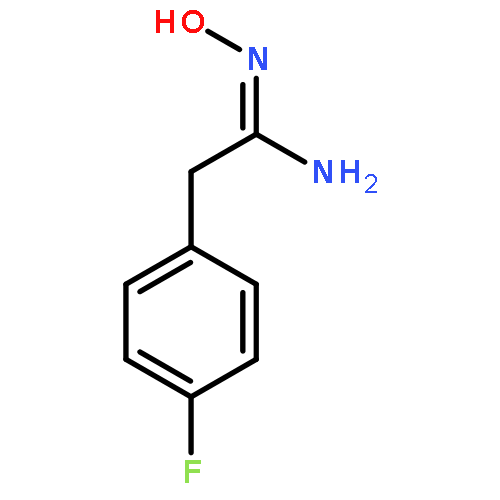
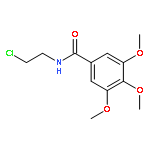
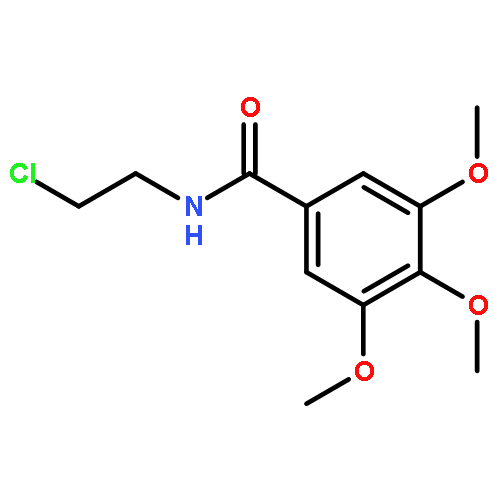
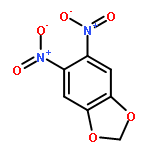
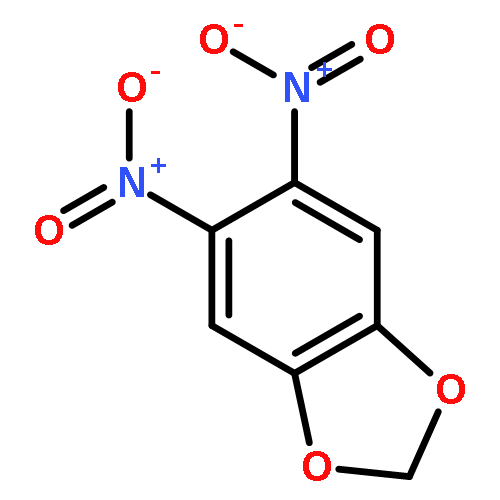
![4-CARBAMOYL-2,6-DIMETHYL-N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L<WBR />-PHENYLALANINE](http://img.cochemist.com/ccimg/7100/7081-78-9.png)
![4-CARBAMOYL-2,6-DIMETHYL-N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L<WBR />-PHENYLALANINE](http://img.cochemist.com/ccimg/7100/7081-78-9_b.png)