Co-reporter:Alvin Kung, Marianne Schimpl, Arunika Ekanayake, Ying-Chu Chen, Ross Overman, and Chao Zhang
ACS Chemical Biology June 16, 2017 Volume 12(Issue 6) pp:1499-1499
Publication Date(Web):May 1, 2017
DOI:10.1021/acschembio.6b01083
Although a previously developed bump-hole approach has proven powerful in generating specific inhibitors for mapping functions of protein kinases, its application is limited by the intolerance of the large-to-small mutation by certain kinases and the inability to control two kinases separately in the same cells. Herein, we describe the development of an alternative chemical-genetic approach to overcome these limitations. Our approach features the use of an engineered cysteine residue at a particular position as a reactive feature to sensitize a kinase of interest to selective covalent blockade by electrophilic inhibitors and is thus termed the Ele-Cys approach. We successfully applied the Ele-Cys approach to identify selective covalent inhibitors of a receptor tyrosine kinase EphB1 and solved cocrystal structures to determine the mode of covalent binding. Importantly, the Ele-Cys and bump-hole approaches afforded orthogonal inhibition of two distinct kinases in the cell, opening the door to their combined use in the study of multikinase signaling pathways.
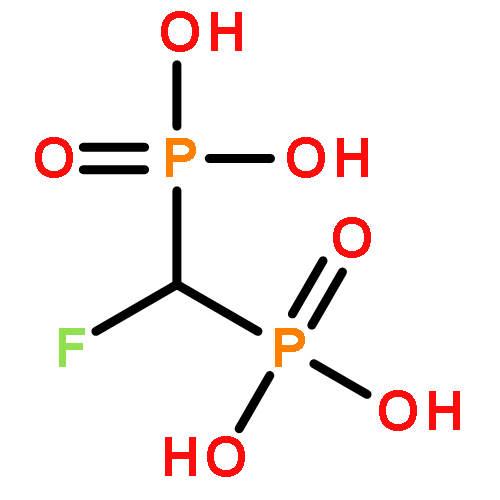
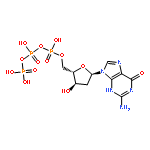
Co-reporter:Feng Ni, Alvin Kung, Yankun Duan, Vivek Shah, Carolina D. Amador, Ming Guo, Xuegong Fan, Lin Chen, Yongheng Chen, Charles E. McKenna, and Chao Zhang
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7701-7701
Publication Date(Web):May 23, 2017
DOI:10.1021/jacs.7b03266
ATP analogues containing a CXY group in place of the α,β-bridging oxygen atom are powerful chemical probes for studying ATP-dependent enzymes. A limitation of such probes has been that conventional synthetic methods generate a mixture of diastereomers when the bridging carbon substitution is nonequivalent (X ≠ Y). We report here a novel method based on derivatization of a bisphosphonate precursor with a d-phenylglycine chiral auxiliary that enables preparation of the individual diastereomers of α,β-CHF-ATP and α,β-CHCl-ATP, which differ only in the configuration at the CHX carbon. When tested on a dozen divergent protein kinases, these individual diastereomers exhibit remarkable diastereospecificity (up to over 1000-fold) in utilization by the enzymes. This high selectivity can be exploited in an enzymatic approach to obtain the otherwise inaccessible diastereomers of α,β-CHBr-ATP. The crystal structure of a tyrosine kinase Src bound to α,β-CHX-ADP establishes the absolute configuration of the CHX carbon and helps clarify the origin of the remarkable diastereospecificity observed. We further synthesized the individual diastereomers of α,β-CHF-γ-thiol-ATP and demonstrated their utility in labeling a wide spectrum of kinase substrates. The novel ATP substrate analogues afforded by these two complementary strategies should have broad application in the study of the structure and function of ATP-dependent enzymes.
Co-reporter:Ying-Chu Chen, Keriann M. Backus, Maria Merkulova, Christina Yang, Dennis Brown, Benjamin F. CravattChao Zhang
Journal of the American Chemical Society 2016 Volume 139(Issue 2) pp:639-642
Publication Date(Web):December 23, 2016
DOI:10.1021/jacs.6b12511
The vacuolar H+ ATPase (V-ATPase) is a complex multisubunit machine that regulates important cellular processes through controlling acidity of intracellular compartments in eukaryotes. Existing small-molecule modulators of V-ATPase either are restricted to targeting one membranous subunit of V-ATPase or have poorly understood mechanisms of action. Small molecules with novel and defined mechanisms of inhibition are thus needed to functionally characterize V-ATPase and to fully evaluate the therapeutic relevance of V-ATPase in human diseases. We have discovered electrophilic quinazolines that covalently modify a soluble catalytic subunit of V-ATPase with high potency and exquisite proteomic selectivity as revealed by fluorescence imaging and chemical proteomic activity-based profiling. The site of covalent modification was mapped to a cysteine residue located in a region of V-ATPase subunit A that is thought to regulate the dissociation of V-ATPase. We further demonstrate that a previously reported V-ATPase inhibitor, 3-bromopyruvate, also targets the same cysteine residue and that our electrophilic quinazolines modulate the function of V-ATPase in cells. With their well-defined mechanism of action and high proteomic specificity, the described quinazolines offer a powerful set of chemical probes to investigate the physiological and pathological roles of V-ATPase.
Co-reporter:Alvin Kung, Ying-Chu Chen, Marianne Schimpl, Feng Ni, Jianfa Zhu, Maurice Turner, Henrik Molina, Ross Overman, and Chao Zhang
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10554-10560
Publication Date(Web):August 1, 2016
DOI:10.1021/jacs.6b05483
Erythropoietin-producing human hepatocellular carcinoma (Eph) receptor tyrosine kinases (RTKs) regulate a variety of dynamic cellular events, including cell protrusion, migration, proliferation, and cell-fate determination. Small-molecule inhibitors of Eph kinases are valuable tools for dissecting the physiological and pathological roles of Eph. However, there is a lack of small-molecule inhibitors that are selective for individual Eph isoforms due to the high homology within the family. Herein, we report the development of the first potent and specific inhibitors of a single Eph isoform, EphB3. Through structural bioinformatic analysis, we identified a cysteine in the hinge region of the EphB3 kinase domain, a feature that is not shared with any other human kinases. We synthesized and characterized a series of electrophilic quinazolines to target this unique, reactive feature in EphB3. Some of the electrophilic quinazolines selectively and potently inhibited EphB3 both in vitro and in cells. Cocrystal structures of EphB3 in complex with two quinazolines confirmed the covalent linkage between the protein and the inhibitors. A “clickable” version of an optimized inhibitor was created and employed to verify specific target engagement in the whole proteome and to probe the extent and kinetics of target engagement of existing EphB3 inhibitors. Furthermore, we demonstrate that the autophosphorylation of EphB3 within the juxtamembrane region occurs in trans using a specific inhibitor. These exquisitely specific inhibitors will facilitate the dissection of EphB3’s role in various biological processes and disease contribution.
Co-reporter:Xu Liu; Alvin Kung; Brock Malinoski; G. K. Surya Prakash
Journal of Medicinal Chemistry 2015 Volume 58(Issue 23) pp:9228-9237
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jmedchem.5b01125
Despite the success of imatinib at inhibiting Bcr-Abl and treating chronic myelogenous leukemia (CML), resistance to the therapy occurs over time in patients. In particular, the resistance to imatinib caused by the gatekeeper mutation T315I in Bcr-Abl remains a challenge in the clinic. Inspired by the successful development of ponatinib to curb drug resistance, we hypothesize that the incorporation of an alkyne linker in other heterocyclic scaffolds can also achieve potent inhibition of Bcr-AblT315I by allowing for simultaneous occupancy of both the active site and the allosteric pocket in the Abl kinase domain. Herein, we describe the design, synthesis, and characterization of a series of alkyne-containing pyrazolopyrimidines as Bcr-Abl inhibitors. Our results demonstrate that some alkyne-containing pyrazolopyrimidines potently inhibit not only AblT315I in vitro but also Bcr-AblT315I in cells. These pyrazolopyrimidines can serve as lead compounds for future development of novel targeted therapy to overcome drug resistance of CML.
Co-reporter:Liang Xu, Ying-Chu Chen, Satoshi Nakajima, Jenny Chong, Lanfeng Wang, Li Lan, Chao Zhang and Dong Wang
Chemical Science 2014 vol. 5(Issue 2) pp:567-574
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3SC51849C
Dynamic regulation and faithful maintenance of proper DNA methylation patterns are essential for many cellular functions. 5-Formylcytosine (5fC), a newly discovered oxidized form of methylcytosine (mC), is involved in active DNA demethylation processes. The latest progress suggests exciting novel functional roles of this residue. Chemical tools are desired to further elucidate the functional roles of 5fC and to modulate dynamics of DNA demethylation and downstream biological processes. Here we designed and constructed a chemical probe, consisting of an aldehyde-targeting group and an intercalation group. This molecule can selectively react with 5fC and subsequently inhibit base excision by thymine DNA glycosylase (TDG) and cause significant pausing for both DNA and RNA polymerase elongation. Further investigation using a GFP reporter system in living cells revealed that the covalent modification in 5fC sites at 5′-UTR of the GFP gene greatly inhibited the GFP expression level. These results altogether confirmed our successful design and established a new approach for generating functional probes that target the formylcytosine sites and modulate 5fC-related biological processes.
Co-reporter:Liang Xu, Ying-Chu Chen, Satoshi Nakajima, Jenny Chong, Lanfeng Wang, Li Lan, Chao Zhang and Dong Wang
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN574-574
Publication Date(Web):2013/10/09
DOI:10.1039/C3SC51849C
Dynamic regulation and faithful maintenance of proper DNA methylation patterns are essential for many cellular functions. 5-Formylcytosine (5fC), a newly discovered oxidized form of methylcytosine (mC), is involved in active DNA demethylation processes. The latest progress suggests exciting novel functional roles of this residue. Chemical tools are desired to further elucidate the functional roles of 5fC and to modulate dynamics of DNA demethylation and downstream biological processes. Here we designed and constructed a chemical probe, consisting of an aldehyde-targeting group and an intercalation group. This molecule can selectively react with 5fC and subsequently inhibit base excision by thymine DNA glycosylase (TDG) and cause significant pausing for both DNA and RNA polymerase elongation. Further investigation using a GFP reporter system in living cells revealed that the covalent modification in 5fC sites at 5′-UTR of the GFP gene greatly inhibited the GFP expression level. These results altogether confirmed our successful design and established a new approach for generating functional probes that target the formylcytosine sites and modulate 5fC-related biological processes.
.jpg)





![3-((4-AMINO-1-ISOPROPYL-1H-PYRAZOLO[3,4-D]PYRIMIDIN-3-YL)ETHYNYL)PHENOL](http://img.cochemist.com/ccimg/1217500/1217488-30-6.png)
![3-((4-AMINO-1-ISOPROPYL-1H-PYRAZOLO[3,4-D]PYRIMIDIN-3-YL)ETHYNYL)PHENOL](http://img.cochemist.com/ccimg/1217500/1217488-30-6_b.png)








![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6,12-dihydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12R,13E,15R,15aR)-](http://img.cochemist.com/ccimg/22200/22144-77-0.png)
![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6,12-dihydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12R,13E,15R,15aR)-](http://img.cochemist.com/ccimg/22200/22144-77-0_b.png)