Co-reporter:U Siemoneit;A Koeberle;A Rossi;F Dehm;M Verhoff;S Reckel;TJ Maier;J Jauch;H Northoff;F Bernhard;V Doetsch;L Sautebin;O Werz
British Journal of Pharmacology 2011 Volume 162( Issue 1) pp:147-162
Publication Date(Web):
DOI:10.1111/j.1476-5381.2010.01020.x
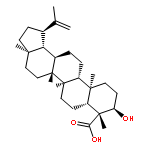
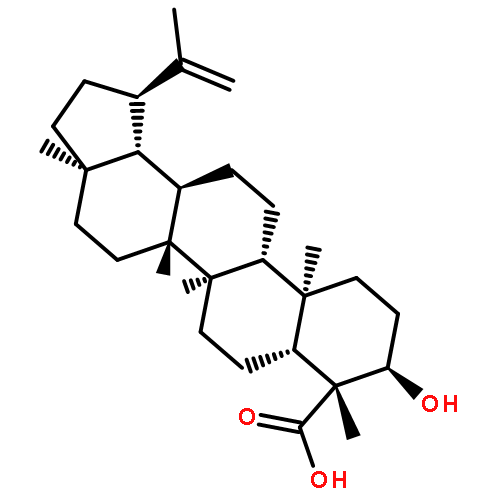
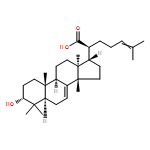
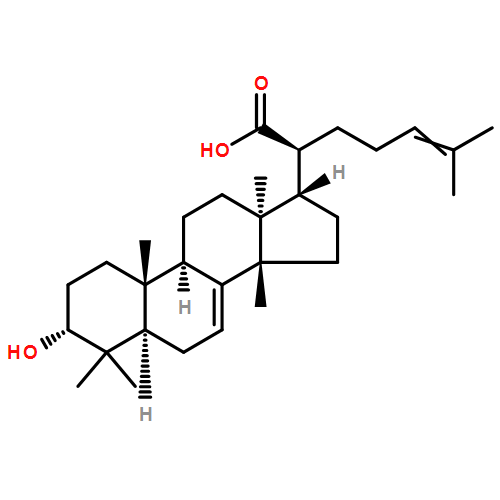
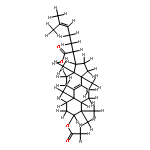
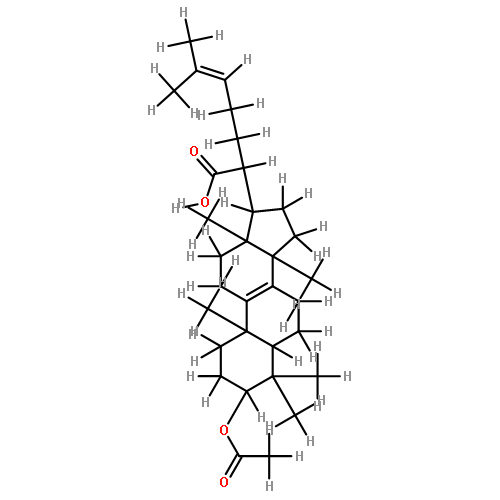
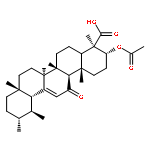
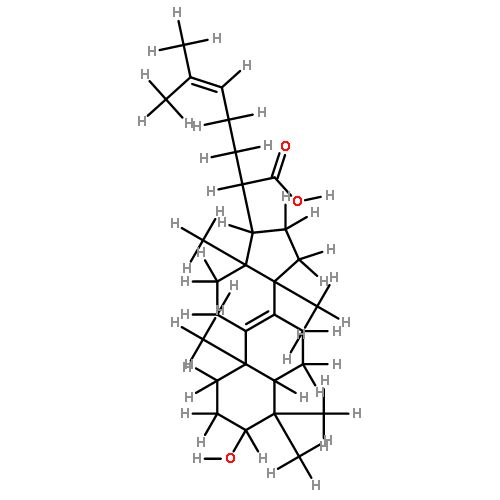
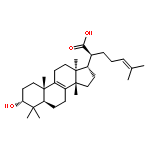
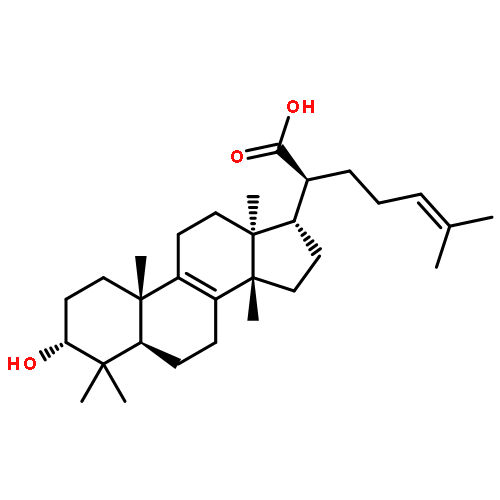
BACKGROUND AND PURPOSE Frankincense, the gum resin derived from Boswellia species, showed anti-inflammatory efficacy in animal models and in pilot clinical studies. Boswellic acids (BAs) are assumed to be responsible for these effects but their anti-inflammatory efficacy in vivo and their molecular modes of action are incompletely understood.
EXPERIMENTAL APPROACH A protein fishing approach using immobilized BA and surface plasmon resonance (SPR) spectroscopy were used to reveal microsomal prostaglandin E2 synthase-1 (mPGES1) as a BA-interacting protein. Cell-free and cell-based assays were applied to confirm the functional interference of BAs with mPGES1. Carrageenan-induced mouse paw oedema and rat pleurisy models were utilized to demonstrate the efficacy of defined BAs in vivo.
KEY RESULTS Human mPGES1 from A549 cells or in vitro-translated human enzyme selectively bound to BA affinity matrices and SPR spectroscopy confirmed these interactions. BAs reversibly suppressed the transformation of prostaglandin (PG)H2 to PGE2 mediated by mPGES1 (IC50 = 3–10 µM). Also, in intact A549 cells, BAs selectively inhibited PGE2 generation and, in human whole blood, β-BA reduced lipopolysaccharide-induced PGE2 biosynthesis without affecting formation of the COX-derived metabolites 6-keto PGF1α and thromboxane B2. Intraperitoneal or oral administration of β-BA (1 mg·kg−1) suppressed rat pleurisy, accompanied by impaired levels of PGE2 and β-BA (1 mg·kg−1, given i.p.) also reduced mouse paw oedema, both induced by carrageenan.
CONCLUSIONS AND IMPLICATIONS Suppression of PGE2 formation by BAs via interference with mPGES1 contribute to the anti-inflammatory effectiveness of BAs and of frankincense, and may constitute a biochemical basis for their anti-inflammatory properties.
Co-reporter:Eva-Maria Karg ; Susann Luderer ; Carlo Pergola ; Ulrike Bühring ; Antonietta Rossi ; Hinnak Northoff ; Lidia Sautebin ; Reinhard Troschütz
Journal of Medicinal Chemistry 2009 Volume 52(Issue 11) pp:3474-3483
Publication Date(Web):May 7, 2009
DOI:10.1021/jm900212y
Pharmacological suppression of leukotriene biosynthesis by inhibitors of 5-lipoxygenase (5-LO) is a strategy to intervene with inflammatory and allergic disorders. We recently presented 2-amino-5-hydroxy-1H-indoles as efficient 5-LO inhibitors in cell-based and cell-free assays. Structural optimization led to novel benzo[g]indole-3-carboxylates exemplified by ethyl 2-(3-chlorobenzyl)-5-hydroxy-1H-benzo[g]indole-3-carboxylate (compound 11a), which inhibits 5-LO activity in human neutrophils and recombinant human 5-LO with IC50 values of 0.23 and 0.086 μM, respectively. Notably, 11a efficiently blocks 5-LO product formation in human whole blood assays (IC50 = 0.83−1.6 μM) and significantly prevented leukotriene B4 production in pleural exudates of carrageenan-treated rats, associated with reduced severity of pleurisy. Together, on the basis of their high potency against 5-LO and the marked efficacy in biological systems, these novel and straightforward benzo[g]indole-3-carboxylates may have potential as anti-inflammatory therapeutics.
Co-reporter:A Koeberle;F Pollastro;H Northoff;O Werz
British Journal of Pharmacology 2009 Volume 156( Issue 6) pp:952-961
Publication Date(Web):
DOI:10.1111/j.1476-5381.2009.00070.x
Background and purpose: The selective inhibition of prostaglandin (PG)E2 formation via interference with microsomal PGE2 synthase (mPGES)-1 could have advantages in the treatment of PGE2-associated diseases, such as inflammation, fever and pain, compared with a general suppression of all PG biosynthesis, provided by inhibition of cyclooxygenase (COX)-1 and 2. Here, we addressed whether the naturally occurring acylphloroglucinol myrtucommulone (MC) from Myrtus communis L. (myrtle) affected mPGES-1.
Experimental approach: The effect of MC on PGE2 formation was investigated in a cell-free assay by using microsomal preparations of interleukin-1β-stimulated A549 cells as the source of mPGES-1, in intact A549 cells, and in lipopolysaccharide-stimulated human whole blood. Inhibition of COX-1 and COX-2 activity in cellular and cell-free assays was assessed by measuring 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid and 6-oxo PGF1α formation.
Key results: MC concentration-dependently inhibited cell-free mPGES-1-mediated conversion of PGH2 to PGE2 (IC50 = 1 µmol·L−1). PGE2 formation was also diminished in intact A549 cells as well as in human whole blood at low micromolar concentrations. Neither COX-2 activity in A549 cells nor isolated human recombinant COX-2 was significantly affected by MC up to 30 µmol·L−1, and only moderate inhibition of cellular or cell-free COX-1 was evident (IC50 > 15 µmol·L−1).
Conclusions and implications: MC is the first natural product to inhibit mPGES-1 that efficiently suppresses PGE2 formation without significant inhibition of the COX enzymes. This provides an interesting pharmacological profile suitable for interventions in inflammatory disorders, without the typical side effects of coxibs and non-steroidal anti-inflammatory drugs.
Co-reporter:Andreas Koeberle, Eva-Maria Haberl, Antonietta Rossi, Carlo Pergola, Friederike Dehm, Hinnak Northoff, Reinhard Troschuetz, Lidia Sautebin, Oliver Werz
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 23) pp:7924-7932
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmc.2009.10.025
Selective inhibition of pro-inflammatory prostaglandin (PG)E2 formation via microsomal PGE2 synthase-1 (mPGES-1) might be superior over inhibition of all cyclooxygenase (COX)-derived products by non-steroidal anti-inflammatory drugs (NSAIDs) and coxibs. We recently showed that benzo[g]indol-3-carboxylates potently suppress leukotriene biosynthesis by inhibiting 5-lipoxygenase. Here, we describe the discovery of benzo[g]indol-3-carboxylates as a novel class of potent mPGES-1 inhibitors (IC50 ⩾ 0.1 μM). Ethyl 2-(3-chlorobenzyl)-5-hydroxy-1H-benzo[g]indole-3-carboxylate (compound 7a) inhibits human mPGES-1 in a cell-free assay (IC50 = 0.6 μM) as well as in intact A549 cells (IC50 = 2 μM), and suppressed PGE2 pleural levels in rat carrageenan-induced pleurisy. Inhibition of cellular COX-1/2 activity was significantly less pronounced. Compound 7a significantly reduced inflammatory reactions in the carrageenan-induced mouse paw edema and rat pleurisy. Together, based on the select and potent inhibition of mPGES-1 and 5-lipoxygenase, benzo[g]indol-3-carboxylates possess potential as novel anti-inflammatory drugs with a valuable pharmacological profile.
Co-reporter:Christian Feißt;Carlo Pergola;Marija Rakonjac
Cellular and Molecular Life Sciences 2009 Volume 66( Issue 16) pp:2759-2771
Publication Date(Web):2009 August
DOI:10.1007/s00018-009-0078-3
We previously showed that, in vitro, hyperforin from St. John’s wort (Hypericum perforatum) inhibits 5-lipoxygenase (5-LO), the key enzyme in leukotriene biosynthesis. Here, we demonstrate that hyperforin possesses a novel and unique molecular pharmacological profile as a 5-LO inhibitor with remarkable efficacy in vivo. Hyperforin (4 mg/kg, i.p.) significantly suppressed leukotriene B4 formation in pleural exudates of carrageenan-treated rats associated with potent anti-inflammatory effectiveness. Inhibition of 5-LO by hyperforin, but not by the iron-ligand type 5-LO inhibitor BWA4C or the nonredox-type inhibitor ZM230487, was abolished in the presence of phosphatidylcholine and strongly reduced by mutation (W13A-W75A-W102A) of the 5-LO C2-like domain. Moreover, hyperforin impaired the interaction of 5-LO with coactosin-like protein and abrogated 5-LO nuclear membrane translocation in ionomycin-stimulated neutrophils, processes that are typically mediated via the regulatory 5-LO C2-like domain. Together, hyperforin is a novel type of 5-LO inhibitor apparently acting by interference with the C2-like domain, with high effectiveness in vivo.
Co-reporter:Oliver Werz ; Christine Greiner ; Andreas Koeberle ; Christina Hoernig ; Sven George ; Laura Popescu ; Ivonne Syha ; Manfred Schubert-Zsilavecz ;Dieter Steinhilber
Journal of Medicinal Chemistry 2008 Volume 51(Issue 17) pp:5449-5453
Publication Date(Web):August 19, 2008
DOI:10.1021/jm800588x
A novel class of potent 5-lipoxygenase (5-LO) product synthesis inhibitors based on the structure of pirinixic acid (4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid, compound 1) is presented. Systematic profiling of 1, i.e., esterification of the carboxylic acid, α-substitution, and replacement of the o-dimethylaniline by 6-aminoquinoline, leads to potent suppressors of 5-LO product formation in activated polymorphonuclear leukocytes, exemplified by ethyl 2-[4-chloro-6-(quinoline-6-ylamino)-pyrimidin-2-ylsulfanyl]octane-1-carboxylate (6d, IC50 = 0.6 μM). These derivatives may possess potential for intervention with inflammatory and allergic diseases.
Co-reporter:Andreas Koeberle ; Heiko Zettl ; Christine Greiner ; Mario Wurglics ; Manfred Schubert-Zsilavecz
Journal of Medicinal Chemistry 2008 Volume 51(Issue 24) pp:8068-8076
Publication Date(Web):November 19, 2008
DOI:10.1021/jm801085s
Dual inhibition of the prostaglandin (PG) and leukotriene (LT) biosynthetic pathway is supposed to be superior over single interference, both in terms of efficacy and side effects. Here, we present a novel class of dual microsomal PGE2 synthase-1/5-lipoxygenase (5-LO) inhibitors based on the structure of pirinixic acid [PA, 2-(4-chloro-6-(2,3-dimethylphenylamino)pyrimidin-2-ylthio)acetic acid, compound 1]. Target-oriented structural modification of 1, particularly α substitution with extended n-alkyl or bulky aryl substituents and concomitant replacement of the 2,3-dimethylaniline by a biphenyl-4-yl-methane-amino residue, resulted in potent suppression of mPGES-1 and 5-LO activity, exemplified by 2-(4-(biphenyl-4-ylmethylamino)-6-chloropyrimidin-2-ylthio)octanoic acid (7b, IC50 = 1.3 and 1 μM, respectively). Select compounds also potently reduced PGE2 and 5-LO product formation in intact cells. Importantly, inhibition of cyclooxygenases-1/2 was significantly less pronounced. Taken together, these pirinixic acid derivatives constitute a novel class of dual mPGES-1/5-LO inhibitors with a promising pharmacologial profile and a potential for therapeutic use.
Co-reporter:Irina Tretiakova;Dagmar Blaesius;Lucia Maxia;Sebastian Wesselborg
Apoptosis 2008 Volume 13( Issue 1) pp:119-131
Publication Date(Web):2008 January
DOI:10.1007/s10495-007-0150-0
Myrtucommulone (MC) is a unique, nonprenylated acylphloroglucinol contained in the leaves of myrtle (Myrtus communis). Here, we addressed the potential of MC to induce apoptosis of cancer cells. MC potently induced cell death of different cancer cell lines (EC50 3–8 μM) with characteristics of apoptosis, visualized by the activation of caspase-3, -8 and -9, cleavage of poly(ADP-ribose)polymerase (PARP), release of nucleosomes into the cytosol, and DNA fragmentation. MC was much less cytotoxic for non-transformed human peripheral blood mononuclear cells (PBMC) or foreskin fibroblasts (EC50 cell death = 20–50 μM), and MC up to 30 μM hardly caused processing of PARP, caspase-3, -8 and -9 in human PBMC. MC-induced apoptosis was mediated by the intrinsic rather than the extrinsic death pathway. Thus, MC caused loss of the mitochondrial membrane potential in MM6 cells and evoked release of cytochrome c from mitochondria. Interestingly, Jurkat cells deficient in caspase-9 were resistant to MC-induced cell death and no processing of PARP or caspase-8 was evident. In cell lines deficient in either CD95 (Fas, APO-1) signalling, FADD or caspase-8, MC was still able to potently induce cell death and PARP cleavage. Conclusively, MC induces apoptosis in cancer cell lines, with marginal cytotoxicity for non-transformed cells, via the mitochondrial cytochrome c/Apaf-1/caspase-9 pathway.
Co-reporter:Carlo Pergola;Gabriele Dodt;Antonietta Rossi;Eva Neunhoeffer;Barbara Lawrenz;Hinnak Northoff;Bengt Samuelsson;Olof Rådmark;Lidia Sautebin
PNAS 2008 Volume 105 (Issue 50 ) pp:19881-19886
Publication Date(Web):2008-12-16
DOI:10.1073/pnas.0809120105
5-Lipoxygenase initiates the biosynthesis of leukotrienes, lipid mediators involved in normal host defense and in inflammatory
and allergic disorders. Despite an obvious gender bias in leukotriene-related diseases (e.g., asthma), gender aspects have
been neglected in studies on leukotrienes and 5-lipoxygenase. Here, we show that leukotriene formation in stimulated whole
blood or neutrophils from males is substantially lower compared with females, accompanied by changed 5-lipoxygenase trafficking.
This is due to gender-specific differential activation of extracellular signal-regulated kinases (ERKs). The differences are
directly related to variant male/female testosterone plus 5α-dihydrotestosterone levels, and addition of 5α-dihydrotestosterone
to female blood or neutrophils reduced the high (female) LT biosynthesis capacity to low (male) levels. In conclusion, regulation
of ERKs and leukotriene formation by androgens constitutes a molecular basis for gender differences in the inflammatory response,
and in inflammatory diseases such as asthma.
Co-reporter:L Fischer;M Hornig;C Pergola;N Meindl;L Franke;Y Tanrikulu;G Dodt;G Schneider;D Steinhilber;O Werz
British Journal of Pharmacology 2007 Volume 152(Issue 4) pp:
Publication Date(Web):29 JAN 2009
DOI:10.1038/sj.bjp.0707416
Licofelone is a dual inhibitor of the cyclooxygenase and 5-lipoxygenase (5-LO) pathway, and has been developed for the treatment of inflammatory diseases. Here, we investigated the molecular mechanisms underlying the inhibition by licofelone of the formation of 5-LO products.
The efficacy of licofelone to inhibit the formation of 5-LO products was analysed in human isolated polymorphonuclear leukocytes (PMNL) or transfected HeLa cells, as well as in cell-free assays using respective cell homogenates or purified recombinant 5-LO. Moreover, the effects of licofelone on the subcellular redistribution of 5-LO were studied.
Licofelone potently blocked synthesis of 5-LO products in Ca2+-ionophore-activated PMNL (IC50=1.7 μM) but was a weak inhibitor of 5-LO activity in cell-free assays (IC50≫10 μM). The structures of licofelone and MK-886, an inhibitor of the 5-LO-activating protein (FLAP), were superimposable. The potencies of both licofelone and MK-886 in ionophore-activated PMNL were impaired upon increasing the concentration of arachidonic acid, or under conditions where 5-LO product formation was evoked by genotoxic, oxidative or hyperosmotic stress. Furthermore, licofelone prevented nuclear redistribution of 5-LO in ionophore-activated PMNL, as had been observed for FLAP inhibitors. Finally, licofelone as well as MK-886 caused only moderate inhibition of the synthesis of 5-LO products in HeLa cells, unless FLAP was co-transfected.
Our data suggest that the potent inhibition of the biosynthesis of 5-LO products by licofelone requires an intact cellular environment and appears to be due to interference with FLAP.
British Journal of Pharmacology (2007) 152, 471–480; doi:10.1038/sj.bjp.0707416; published online 20 August 2007
Co-reporter:Angela A.Y. Michel, Dieter Steinhilber, Oliver Werz
Protein Expression and Purification (May 2008) Volume 59(Issue 1) pp:110-116
Publication Date(Web):1 May 2008
DOI:10.1016/j.pep.2008.01.010
5-Lipoxygenase (5-LO), the key enzyme in leukotriene biosynthesis, is built of a catalytic C-terminal domain and a regulatory N-terminal C2-like domain. The C2-like domain is the target of many regulatory factors or proteins including Ca2+, phospholipids, glycerides, coactosin-like protein and presumably other components that modulate the catalytic activity of 5-LO by acting at this domain, but the detailed underlying molecular mechanisms of these interactions are still unclear. In order to obtain the 5-LO C2-like domain as purified protein in good yields for further mechanistic studies and structure elucidation, a novel expression and purification approach has been applied. A plasmid was constructed expressing a fusion protein of maltose-binding protein (MBP) and the regulatory C2-like domain of 5-LO (AS 1–128), separated by a tobacco etch virus (TEV) protease-cleavage site. The fusion protein MBP-5LO1-128 could be essentially expressed as a soluble protein in Escherichia coli and was efficiently purified by amylose affinity chromatography. By means of this procedure, approximately 80 mg purified fusion protein out of 1 L E. coli culture were obtained. Digestion with TEV protease yielded the C2-like domain that was further purified using hydrophobic interaction chromatography. Alternatively, the uncleaved fusion protein MBP-5LO1-128 may be suitable to immobilize the C2-like domain on an amylose resin for co-factor interaction studies. Together, we present a convenient expression and purification strategy of the 5-LO C2-like domain that opens many possibilities for structural determination and mechanistic studies, aiming to reveal the precise role and function of this regulatory domain.
Co-reporter:Marika Hoffmann, Jakob J. Lopez, Carlo Pergola, Christian Feisst, Sven Pawelczik, Per-Johan Jakobsson, Bernd L. Sorg, Clemens Glaubitz, Dieter Steinhilber, Oliver Werz
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids (April 2010) Volume 1801(Issue 4) pp:
Publication Date(Web):April 2010
DOI:10.1016/j.bbalip.2009.12.007
Here, we investigated the modulation of cytosolic phospholipase A2 (cPLA2)-mediated arachidonic acid (AA) release by the polyprenylated acylphloroglucinol hyperforin. Hyperforin increased AA release from human platelets up to 2.6 fold (maximal effect at 10 µM) versus unstimulated cells, which was blocked by cPLA2α-inhibition, and induced translocation of cPLA2 to a membrane compartment. Interestingly, these stimulatory effects of hyperforin were even more pronounced after depletion of intracellular Ca2+ by EDTA plus BAPTA/AM. Hyperforin induced phosphorylation of cPLA2 at Ser505 and activated p38 mitogen-activated protein kinase (MAPK), and inhibition of p38 MAPK by SB203580 prevented cPLA2 phosphorylation. However, neither AA release nor translocation of cPLA2 was abrogated by SB203580. In cell-free assays using liposomes prepared from different lipids, hyperforin failed to stimulate phospholipid hydrolysis by isolated cPLA2 in the presence of Ca2+. However, when Ca2+ was omitted, hyperforin caused a prominent increase in cPLA2 activity using liposomes composed of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphoethanolamine but not of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine (PAPC) unless the PAPC liposomes were enriched in cholesterol (20 to 50%). Finally, two-dimensional 1H-MAS-NMR analysis visualized the directed insertion of hyperforin into POPC liposomes. Together, hyperforin, through insertion into phospholipids, may facilitate cPLA2 activation by enabling its access towards select lipid membranes independent of Ca2+ ions. Such Ca2+- and phosphorylation-independent mechanism of cPLA2 activation may apply also to other membrane-interfering molecules.