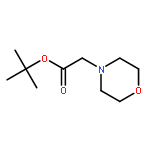
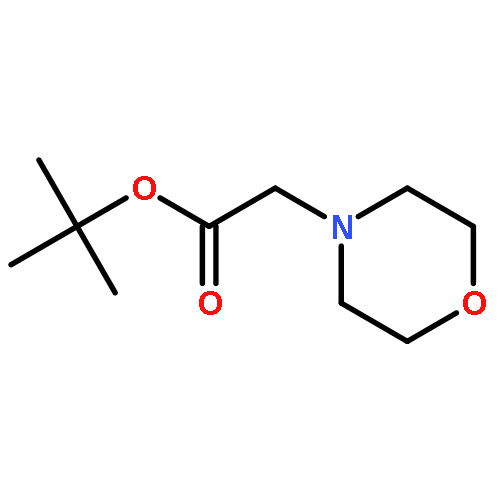
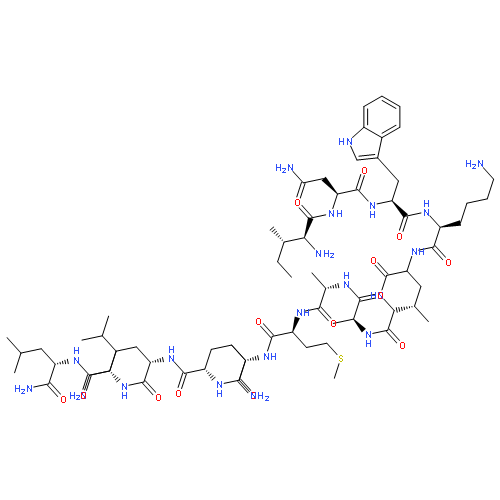
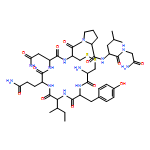
Co-reporter:Youhei Sohma, Yuta Hirayama, Atsuhiko Taniguchi, Hidehito Mukai, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 5) pp:1729-1733
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmc.2011.01.021
The O-acyl isopeptide of Aβ1–42 (1), possessing an ester bond at the Gly25-Ser26 sequence, is a water-soluble and non-aggregative precursor molecule and is capable of production of monomer Aβ1–42. The SDS–PAGE result showed that the Aβ1–42, produced from 1, adopted monomeric state at first and then self-assembled to oligomer. The oligomeric state was stabilized by nordihydroguaiaretic acid. The Thioflavin-T (ThT) fluorescence intensity derived from Aβ1–42 (generated from 1) was suppressed by various aggregation inhibitors. Finally, 1 could generate Aβ1–42 via the O-to-N acyl migration under cellular medium conditions and the produced Aβ1–42 exhibited cytotoxicity against PC12 cells. These results suggest that the click peptide system, which enables us to predominantly produce monomer Aβ1–42 under physiological conditions, would be adoptable to various biochemical and biophysical experiments including cellular system to investigate the functions of Aβ1–42.
Co-reporter:Jeffrey-Tri Nguyen, Keiko Kato, Koushi Hidaka, Henri-Obadja Kumada, Tooru Kimura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 8) pp:2425-2429
Publication Date(Web):15 April 2011
DOI:10.1016/j.bmcl.2011.02.066
The human T cell leukemia/lymphotropic virus type 1 (HTLV-I) is clinically associated with adult T cell leukemia/lymphoma, HTLV-I associated myelopathy/tropical spastic paraparesis, and a number of other chronic inflammatory diseases. To stop the replication of the virus, we developed highly potent tetrapeptidic HTLV-I protease inhibitors. In a recent X-ray crystallography study, several of our inhibitors could not form co-crystal complexes with the protease due to their high hydrophobicity. In the current study, we designed, synthesized and evaluated the HTLV-I protease inhibition potency of compounds with hydrophilic end-capping moieties with the aim of improving pharmaceutic and pharmacokinetic properties.
Co-reporter:Jeffrey-Tri Nguyen, Keiko Kato, Henri-Obadja Kumada, Koushi Hidaka, Tooru Kimura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 6) pp:1832-1837
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmcl.2011.01.048
The human T cell lymphotropic/leukemia virus type 1 (HTLV-I) causes adult T cell lymphoma/leukemia. The virus is also responsible for chronic progressive myelopathy and several inflammatory diseases. To stop the manufacturing of new viral components, in our previous reports, we derived small tetrapeptidic HTLV-I protease inhibitors with an important amide-capping moiety at the P3 residue. In the current study, we removed the P3-cap moiety and, with great difficulty, optimized the P3 residue for HTLV-I protease inhibition potency. We discovered a very potent and small tetrapeptidic HTLV-I protease inhibitor (KNI-10774a, IC50 = 13 nM).
Co-reporter:Dr. Taku Yoshiya;Ayano Higa;Naoko Abe;Fukue Fukao;Tomomi Kuruma;Yuki Toda; Dr. Youhei Sohma; Dr. Yoshiaki Kiso
ChemBioChem 2011 Volume 12( Issue 8) pp:1216-1222
Publication Date(Web):
DOI:10.1002/cbic.201100025
Abstract
The O-acyl isopeptide (1) of islet amyloid polypeptide (IAPP), which contains an ester moiety at both Ala8-Thr9 and Ser19-Ser20, was prepared by sequential segment condensation based on the O-acyl isopeptide method. Isopeptide 1 possessed nonaggregative properties, retaining its random coil structure under the acidic conditions; this suggests that the insertion of the O-acyl isopeptide structures in IAPP suppressed aggregation of the molecule. As a result of the rapid O-to-N acyl shift of 1 under neutral pH, in situ-formed IAPP adopted a random-coil structure at the start of the experiment, and then underwent conformational change to α-helix/β-sheet mixed structures as well as aggregation. The click peptide strategy with the nonaggregative precursor molecule 1 could be a useful experimental tool to identify the functions of IAPP, by overcoming the handling difficulties that arise from IAPP's intense and uncontrollable self-assembling nature.
Co-reporter:Harichandra D. Tagad, Yoshio Hamada, Jeffrey-Tri Nguyen, Takashi Hamada, Hamdy Abdel-Rahman, Abdellah Yamani, Ayaka Nagamine, Hayato Ikari, Naoto Igawa, Koushi Hidaka, Youhei Sohma, Tooru Kimura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 9) pp:3175-3186
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmc.2010.03.032
We previously reported potent BACE1 inhibitors KMI-420 and KMI-570 possessing a hydroxymethylcarbonyl isostere as a substrate transition-state mimic. Acidic moieties at the P1′ and P4 positions of KMI inhibitors are thought to be unfavorable in terms of membrane permeability across the blood–brain barrier. Herein, we replaced acidic moieties at the P4 position with hydrogen bond accepting groups and acidic moieties at the P1′ position with less acidic and similar molecular-size moieties (carboxylic acid or tetrazole bioisosteres). These inhibitors exhibited improved BACE1 inhibitory activities and a thorough quantitative structure–activity relationship study was performed.
Co-reporter:Takuya Miura, Koushi Hidaka, Tsuyoshi Uemura, Keisuke Kashimoto, Yuto Hori, Yuko Kawasaki, Adam J. Ruben, Ernesto Freire, Tooru Kimura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 16) pp:4836-4839
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmcl.2010.06.099
We attached 2-aminoethylamino groups to allophenylnorstatine-containing plasmepsin (Plm) inhibitors and investigated SAR of the methyl or ethyl substitutions on the amino groups. Unexpectedly, compounds 22 (KNI-10743) and 25 (KNI-10742) exhibited extremely potent Plm II inhibitory activities (Ki <0.1 nM). Moreover, among our peptidomimetic Plm inhibitors, we identified the compounds with the highest antimalarial activity using a SYBR Green I-based fluorescence assay.Attachments of 2-aminoethylamino substituents to an allophenylnorstatine-containing plasmepsin inhibitor enhanced both plasmepsin inhibitory and antimalarial activities.
Co-reporter:Koushi Hidaka ; Tooru Kimura ; Hamdy M. Abdel-Rahman ; Jeffrey-Tri Nguyen ; Keith F. McDaniel ; William E. Kohlbrenner ; Akhteruzzaman Molla ; Motoyasu Adachi ; Taro Tamada ; Ryota Kuroki ; Noriko Katsuki ; Yoshiaki Tanaka ; Hikaru Matsumoto ; Jun Wang ; Yoshio Hayashi ; Dale J. Kempf
Journal of Medicinal Chemistry 2009 Volume 52(Issue 23) pp:7604-7617
Publication Date(Web):July 17, 2009
DOI:10.1021/jm9005115
A series of HIV protease inhibitor based on the allophenylnorstatine structure with various P2′ moieties were synthesized. Among these analogues, we discovered that a small allyl group would maintain potent enzyme inhibitory activity compared to the o-methylbenzyl moiety in clinical candidate 1 (KNI-764, also known as JE-2147, AG-1776, or SM-319777). Introduction of an anilinic amino group to 2 (KNI-727) improved water-solubility and anti-HIV-1 activity. X-ray crystallographic analysis of 13k (KNI-1689) with a β-methallyl group at P2′ position revealed hydrophobic interactions with Ala28, Ile84, and Ile50′ similar to that of 1. The presence of an additional methyl group on the allyl group in compound 13k significantly increased anti-HIV activity over 1 while providing a rational drug design for structural minimization and improving membrane permeability.
Co-reporter:Taku Yoshiya, Hiroyuki Kawashima, Youhei Sohma, Tooru Kimura and Yoshiaki Kiso
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 14) pp:2894-2904
Publication Date(Web):18 May 2009
DOI:10.1039/B903624E
We report the establishment of the O-acyl isopeptide method-based racemization-free segment condensation reaction toward future chemical protein synthesis. Peptide segments containing C-terminal O-acyl Ser/Thr residues were successfully synthesized by use of a lower nucleophilic base cocktail for Fmoc removal, and then coupled to an amino group of a peptide-resin without side reactions or epimerization. We also succeeded in performing the segment condensation in a sequential manner and in solution phase conditions as well.
Co-reporter:Hui Wang, Taeko Kakizawa, Atsuhiko Taniguchi, Takaaki Mizuguchi, Tooru Kimura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 14) pp:4881-4887
Publication Date(Web):15 July 2009
DOI:10.1016/j.bmc.2009.06.017
Amyloid β peptide (Aβ) 1–42 is known to be involved in the onset of Alzheimer’s disease (AD). We developed a click peptide of Aβ1–42 as a useful tool for AD research on the basis of an O-acyl isopeptide method. The click peptide quickly produced intact Aβ1–42 via a pH-dependent O-to-N intramolecular acyl migration (pH-click). Herein, a click peptide (26-O-acyl isoAβ1–42 (E22Δ)) of a new mutant Aβ1–42 (E22Δ) was synthesized. The mutant click peptide was more water-soluble than Aβ1–42 (E22Δ). Moreover it quantitatively converted to the native peptide under physiological conditions (pH 7.4, 37 °C). CD analyses showed a conformational change from a random-coil structure of the click peptide to a β-sheet structure of the in situ produced Aβ1–42 (E22Δ). This click peptide is a useful precursor of a mutant Aβ1–42 to establish an experiment system for investigating the properties of the mutant.
Co-reporter:Koushi Hidaka, Tooru Kimura, Adam J. Ruben, Tsuyoshi Uemura, Mami Kamiya, Aiko Kiso, Tetsuya Okamoto, Yumi Tsuchiya, Yoshio Hayashi, Ernesto Freire, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 4) pp:1772
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmc.2008.10.094
Co-reporter:Thomas Regnier, Diganta Sarma, Koushi Hidaka, Usman Bacha, Ernesto Freire, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2722-2727
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.03.118
A series of trifluoromethyl, benzothiazolyl or thiazolyl ketone-containing peptidic compounds as SARS-CoV 3CL protease inhibitors were developed and their potency was evaluated by in vitro protease inhibitory assays. Three candidates had encouraging results for the development of new anti-SARS compounds.A series of trifluoromethyl, benzothiazolyl and thiazolyl ketone-containing peptidic compounds were synthesized and evaluated in vitro against SARS-CoV 3CLpro.
Co-reporter:Yoshio Hamada, Hiroko Ohta, Naoko Miyamoto, Diganta Sarma, Takashi Hamada, Tomoya Nakanishi, Moe Yamasaki, Abdellah Yamani, Shoichi Ishiura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 9) pp:2435-2439
Publication Date(Web):1 May 2009
DOI:10.1016/j.bmcl.2009.03.049
Recently, we reported potent substrate-based pentapeptidic BACE1 inhibitors possessing a hydroxymethylcarbonyl isostere as a substrate transition-state mimic. Because these inhibitors contained some natural amino acids, we would need to improve their enzymatic stability in vivo and permeability across the blood–brain barrier, so that they become practically useful. Subsequently, non-peptidic and small-sized BACE1 inhibitors possessing a heterocyclic scaffold, 2,6-pyridenedicarboxylic, chelidamic or chelidonic moiety, at the P2 position were reported. These inhibitors were designed based on the conformer of docked inhibitor in BACE1. In this study, we discuss the role and significance of interactions between Arg235 of BACE1 and its inhibitor in BACE1 inhibitory mechanism. Moreover, we designed more potent small-sized BACE1 inhibitors with a 2,6-pyridinedicarboxylic scaffold at the P2 position, that were optimized for the interactions with Arg235 of BACE1.Following our hypothesis concerning the role of interaction of BACE1–Arg235 with its substrates/inhibitors, we designed a series of small-sized potent BACE1 inhibitors.
Co-reporter:Atsuhiko Taniguchi ;Youhei Sohma Dr.;Yuta Hirayama;Hidehito Mukai Dr.;Tooru Kimura Dr.;Yoshio Hayashi Dr.;Katsumi Matsuzaki Dr. Dr.
ChemBioChem 2009 Volume 10( Issue 4) pp:710-715
Publication Date(Web):
DOI:10.1002/cbic.200800765
Co-reporter:Shingo Nakatani ; Koushi Hidaka ; Ei’ichi Ami ; Koichiro Nakahara ; Akihiko Sato ; Jeffrey-Tri Nguyen ; Yoshio Hamada ; Yasuko Hori ; Nobuyuki Ohnishi ; Akinori Nagai ; Tooru Kimura ; Yoshio Hayashi
Journal of Medicinal Chemistry 2008 Volume 51(Issue 10) pp:2992-3004
Publication Date(Web):April 22, 2008
DOI:10.1021/jm701555p
Several non-natural d-amino acid derivatives were introduced as P2/P3 residues in allophenylnorstatine-containing (Apns; (2S,3S)-3-amino-2-hydroxy-4-phenylbutyric acid) HIV protease inhibitors. The synthetic analogues exhibited potent inhibitory activity against HIV-1 protease enzyme and HIV-1 replication in MT-4 cells. Structure–activity relationships revealed that d-cysteine or serine derivatives contributed to highly potent anti-HIV activities. Interestingly, anti-HIV activity of all the d-amino acid-introduced inhibitors was remarkably enhanced in their anti-HIV activities against a Nelfinavir-resistant clone, which has a D30N mutation in the protease, over that of the wild-type strain. HIV inhibitory activity of several analogues was moderately affected by an inclusion of α1-acid glycoprotein in the test medium.
Co-reporter:Meihui Zhang, Jeffrey-Tri Nguyen, Henri-Obadja Kumada, Tooru Kimura, Maosheng Cheng, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 10) pp:5795-5802
Publication Date(Web):15 May 2008
DOI:10.1016/j.bmc.2008.03.055
The causative agent behind adult T-cell leukemia and tropical spastic paraparesis/HTLV-I-associated myelopathy is the human T-cell leukemia virus type 1 (HTLV-I). Tetrapeptidic HTLV-I protease inhibitors were designed on a previously reported potent inhibitor KNI-10516, with modifications at the P3-cap moieties. All the inhibitors showed high HIV-1 protease inhibitory activity (over 98% inhibition at 50 nM) and most exhibited highly potent inhibition against HTLV-I protease (IC50 values were less than 100 nM).A series of novel tetrapeptidic HTLV-I protease inhibitors were prepared using an hydroxymethylcarbonyl isostere. Most inhibitors showed high HTLV-I and HIV-1 proteases inhibitory activity.
Co-reporter:Koushi Hidaka, Tooru Kimura, Adam J. Ruben, Tsuyoshi Uemura, Mami Kamiya, Aiko Kiso, Tetsuya Okamoto, Yumi Tsuchiya, Yoshio Hayashi, Ernesto Freire, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 23) pp:10049-10060
Publication Date(Web):1 December 2008
DOI:10.1016/j.bmc.2008.10.011
Plasmepsin (Plm) is a potential target for new antimalarial drugs, but most reported Plm inhibitors have relatively low antimalarial activities. We synthesized a series of dipeptide-type HIV protease inhibitors, which contain an allophenylnorstatine-dimethylthioproline scaffold to exhibit potent inhibitory activities against Plm II. Their activities against Plasmodium falciparum in the infected erythrocyte assay were largely different from those against the target enzyme. To improve the antimalarial activity of peptidomimetic Plm inhibitors, we attached substituents on a structure of the highly potent Plm inhibitor KNI-10006. Among the derivatives, we identified alkylamino compounds such as 44 (KNI-10283) and 47 (KNI-10538) with more than 15-fold enhanced antimalarial activity, to the sub-micromolar level, maintaining their potent Plm II inhibitory activity and low cytotoxicity. These results suggest that auxiliary substituents on a specific basic group contribute to deliver the inhibitors to the target Plm.We identified mono-substituted amino derivatives of an allophenylnorstatine-containing plasmepsin inhibitor with promising anti-malarial activity and low cytotoxicity.
Co-reporter:Meihui Zhang, Jeffrey-Tri Nguyen, Henri-Obadja Kumada, Tooru Kimura, Maosheng Cheng, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 14) pp:6880-6890
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmc.2008.05.052
Adult T-cell leukemia and tropical spastic paraparesis/HTLV-I-associated myelopathy are only some of the more common end results of an infection with a human T-cell leukemia virus type 1 (HTLV-I). Expanding from our previous reports, we synthesized all different permutations of tetrapeptidic HTLV-I protease inhibitors using at least eight P3-cap and five P1′-cap moieties. The inhibitors exhibited over 97% inhibition against HIV-1 protease and a wide range of inhibitory activity against HTLV-I protease.Structure–activity relationship studies were performed on 44 novel tetrapeptidic HTLV-I protease inhibitors possessing different hydrophobic P3-cap and P1′-cap moieties.
Co-reporter:Mayo Noguchi, Mariusz Skwarczynski, Halan Prakash, Shun Hirota, Tooru Kimura, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 10) pp:5389-5397
Publication Date(Web):15 May 2008
DOI:10.1016/j.bmc.2008.04.022
A novel coumarin-based highly water-soluble photocleavable protective group was designed and synthesized, and then this photosensitive protecting group was used to design paclitaxel prodrugs. These novel paclitaxel conjugates demonstrated excellent water solubility, over 100 mg mL−1. Thus, the use of a detergent in the formulation can be omitted completely, even at high doses. Phototaxel 11 released the parent drug, paclitaxel, quickly and efficiently by minimal tissue-damaging 365 nm UV light irradiation at low power, while laser activation at 355 nm led to extensive decomposition of the prodrug. The carbamate-type prodrug, phototaxel 11, was stable in the dark prior to activation, whereas carbonate-type phototaxel 9 demonstrated poor stability under aqueous conditions. For such prodrugs, tumor-tissue targeting after administration could be achieved by selective light delivery, similar to that used in photodynamic therapy. In addition, newly designed coumarin derivative 8 can be applied in organic chemistry as a photosensitive protective group and for the design of caged compounds.
Co-reporter:Yoshio Hamada, Hiroko Ohta, Naoko Miyamoto, Ryoji Yamaguchi, Abdellah Yamani, Koushi Hidaka, Tooru Kimura, Kazuki Saito, Yoshio Hayashi, Shoichi Ishiura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 5) pp:1654-1658
Publication Date(Web):1 March 2008
DOI:10.1016/j.bmcl.2008.01.056
Recently, we reported substrate-based β-secretase (BACE1) inhibitors with a hydroxymethylcarbonyl (HMC) isostere as a substrate transition-state mimic. These inhibitors showed potent BACE1 inhibitory activities (∼1.2 nM IC50). In order to improve in vivo enzymatic stability and permeability across the blood–brain barrier, these penta-peptidic inhibitors would need to be further optimized. On the other hand, non-peptidic inhibitors possessing isophthalic residue at the P2 position were reported from other research groups. We selected isophthalic-type aromatic residues at the P2 position and an HMC isostere at the P1 position as lead compounds. On the basis of the design approach focused on the conformer of docked inhibitor in BACE1, we found novel non-peptidic and small-sized BACE1 inhibitors possessing a 2,6-pyridinedicarboxylic, chelidamic or chelidonic residue at the P2 position.
Co-reporter:Yoshio Hamada, Hamdy Abdel-Rahman, Abdellah Yamani, Jeffrey-Tri Nguyen, Monika Stochaj, Koushi Hidaka, Tooru Kimura, Yoshio Hayashi, Kazuki Saito, Shoichi Ishiura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 5) pp:1649-1653
Publication Date(Web):1 March 2008
DOI:10.1016/j.bmcl.2008.01.058
Recently, we reported potent BACE1 inhibitors KMI-429, -684, and -574 possessing a hydroxymethylcarbonyl isostere as a substrate transition-state mimic. These inhibitors showed potent inhibitory activities in enzymatic and cell assays, especially, KMI-429 was confirmed to significantly inhibit Aβ production in vivo. However, acidic moieties at the P4 and P1′ positions of KMI-compounds were thought to be unfavorable for membrane permeability across the blood–brain barrier. Herein, we replaced acidic moieties at the P4 position with other hydrogen bond acceptor groups, and these inhibitors exhibited improved BACE1 inhibitory activities in cultured cells. In this study, we replaced the acidic moieties at the P1′ position with non-acidic and low molecular sized moieties.
Co-reporter:Jeffrey-Tri Nguyen;Yoshio Hamada;Tooru Kimura
Archiv der Pharmazie 2008 Volume 341( Issue 9) pp:523-535
Publication Date(Web):
DOI:10.1002/ardp.200700267
Abstract
In this retrospective, personal review covering our research from the late 1980s until 2007, we outline nearly two-decade worth of our own work on several aspartic protease inhibitors including those affecting renin, HIV-1 protease, plasmepsins, β-secretase, and HTLV-I protease and we report on aspartic protease inhibitors as potential drugs to treat hypertension, AIDS, malaria, Alzheimer's disease and adult T-cell leukemia, HTLV-I associated myelopathy / tropical spastic paraparesis, and various, respectively, associated diseases. Herein, we describe our methods for rational substrate-based drug design of peptidomimetics that potently inhibit the activity of renin, HIV-1 protease, plasmepsins, β-secretase, and HTLV-I protease accordingly, using an appropriately selected inhibitory residue that contained a hydroxymethylcarbonyl isostere. Although this non-hydrolyzable isostere mimics the transition state that is formed during protein cleavage of a substrate, the isostere-containing inhibitor is not cleaved. We highlight our optimization studies in which we used various techniques and tools such as truncation studies, natural and non-natural amino acid substitution studies, various moieties to promote chemical and pharmacological stability, X-ray crystallography, computer-assisted docking and dynamic simulations, quantitative structure-activity relationship studies, and various other methods that this review can barely mention.
Co-reporter:Jeffrey-Tri Nguyen, Meihui Zhang, Henri-Obadja Kumada, Ayako Itami, Keiji Nishiyama, Tooru Kimura, Maosheng Cheng, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 1) pp:366-370
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmcl.2007.10.066
The culprit behind adult T-cell leukemia, myelopathy/tropical paraparesis, and a plethora of inflammatory diseases is the human T-cell leukemia virus type 1 (HTLV-I). We recently unveiled a potent hexapeptidic HTLV-I protease inhibitor, KNI-10166, composed mostly of natural amino acid residues. Herein, we report the derivation of potent tetrapeptidic inhibitor KNI-10516, possessing only non-natural amino acid residues.
Co-reporter:Atsuhiko Taniguchi;Mariusz Skwarczynski Dr.;Youhei Sohma Dr.;Takuma Okada Dr.;Keisuke Ikeda;Halan Prakash Dr.;Hidehito Mukai Dr.;Yoshio Hayashi Dr.;Tooru Kimura Dr.;Shun Hirota Dr.;Katsumi Matsuzaki Dr. Dr.
ChemBioChem 2008 Volume 9( Issue 18) pp:3055-3065
Publication Date(Web):
DOI:10.1002/cbic.200800503
Abstract
In biological experiments, poor solubility and uncontrolled assembly of amyloid β peptide (Aβ) 1–42 pose significant obstacles to establish an experiment system that clarifies the function of Aβ1–42 in Alzheimer's disease (AD). Herein, as an experimental tool to overcome these problems, we developed a water-soluble photo-“click peptide” with a coumarin-derived photocleavable protective group that is based on an O-acyl isopeptide method. The click peptide had nearly 100-fold higher water solubility than Aβ1–42 and did not self-assemble, as the isomerized structure in its peptide backbone drastically changed the conformation that was derived from Aβ1–42. Moreover, the click peptide afforded Aβ1–42 quickly under physiological conditions (pH 7.4, 37 °C) by photoirradiation followed by an O–N intramolecular acyl migration. Because the in situ production of intact Aβ1–42 from the click peptide could improve the difficulties in handling Aβ1–42 caused by its poor solubility and highly aggregative nature, this click peptide strategy would provide a reliable experiment system for investigating the pathological function of Aβ1–42 in AD.
Co-reporter:Koushi Hidaka, Tooru Kimura, Yumi Tsuchiya, Mami Kamiya, Adam J. Ruben, Ernesto Freire, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 11) pp:3048-3052
Publication Date(Web):1 June 2007
DOI:10.1016/j.bmcl.2007.03.052
Based on a highly potent allophenylnorstatine-containing inhibitor, KNI-10006, against the plasmepsins of Plasmodium falciparum, we synthesized a series of tripeptide-type compounds with various N-terminal moieties and evaluated their inhibitory activities against plasmepsin II. Certain phenylacetyl derivatives exhibited extremely high affinity with Ki values of less than 0.1 nM suggesting successful hydrophobic interactions.A series of allophenylnorstatine-containing tripeptidic compounds were synthesized with various N-terminal phenylacetyl moieties. Certain derivatives exhibited extremely potent inhibitory activity against plasmepsin II.
Co-reporter:Ei’ichi Ami, Koichiro Nakahara, Akihiko Sato, Jeffrey-Tri Nguyen, Koushi Hidaka, Yoshio Hamada, Shingo Nakatani, Tooru Kimura, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 15) pp:4213-4217
Publication Date(Web):1 August 2007
DOI:10.1016/j.bmcl.2007.05.039
We designed several HIV protease inhibitors with various d-cysteine derivatives as P2/P3 moieties based on the structure of clinical drug candidate, KNI-764. Herein, we report their synthesis, HIV protease inhibitory activity, HIV IIIB cell inhibitory activity, cellular toxicity, and inhibitory activity against drug-resistant HIV strains. KNI-1931 showed distinct selectivity against HIV proteases and high potency against drug-resistant strains, surpassing those of Ritonavir and Nelfinavir.
Co-reporter:Yoshio Hamada, Naoto Igawa, Hayato Ikari, Zyta Ziora, Jeffrey-Tri Nguyen, Abdellah Yamani, Koushi Hidaka, Tooru Kimura, Kazuki Saito, Yoshio Hayashi, Maiko Ebina, Shoichi Ishiura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 16) pp:4354-4359
Publication Date(Web):15 August 2006
DOI:10.1016/j.bmcl.2006.05.046
Recently, we reported potent and small-sized β-secretase (BACE1) inhibitors KMI-570 and KMI-684 in which we replaced carboxylic acid groups at the P1′ position of KMI-420 and KMI-429, respectively, with tetrazole derivatives as carboxylic acid bioisosteres. These modifications improved significantly BACE1 inhibitory activity and chemical stability. In this study, the acidic tetrazole ring of the P4 position of KMI-420 and KMI-570, respectively, was replaced with various hydrogen bond acceptor groups. We found BACE1 inhibitor KMI-574 that exhibited potent inhibitory activity in cultured cells as well as in vitro enzymatic assay.
Co-reporter:Tooru Kimura, Yoshio Hamada, Monika Stochaj, Hayato Ikari, Ayaka Nagamine, Hamdy Abdel-Rahman, Naoto Igawa, Koushi Hidaka, Jeffrey-Tri Nguyen, Kazuki Saito, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 9) pp:2380-2386
Publication Date(Web):1 May 2006
DOI:10.1016/j.bmcl.2006.01.108
Recently, we reported potent and small-sized β-secretase (BACE1) inhibitors KMI-420 and KMI-429 in which we replaced the Glu residue at the P4 position of KMI-260 and KMI-360, respectively, with a 1H-tetrazole-5-carbonyl DAP (l-α,β-diaminopropionic acid) residue. At the P1′ position, these compounds contain one or two carboxylic acid groups, which are unfavorable for crossing the blood–brain barrier. Herein, we report BACE1 inhibitors with P1′ carboxylic acid bioisosteres in order to develop practical anti-Alzheimer’s disease drugs. Among them, tetrazole ring-containing compounds, KMI-570 (IC50 = 4.8 nM) and KMI-684 (IC50 = 1.2 nM), exhibited significantly potent BACE1 inhibitory activities.
Co-reporter:Mariusz Skwarczynski, Mayo Noguchi, Shun Hirota, Youhei Sohma, Tooru Kimura, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 17) pp:4492-4496
Publication Date(Web):1 September 2006
DOI:10.1016/j.bmcl.2006.06.030
A prodrug of paclitaxel which has a coumarin derivative conjugated to the amino acid moiety of isotaxel (O-acyl isoform of paclitaxel) has been synthesized. The prodrug was selectively converted to isotaxel by visible light irradiation (430 nm) with the cleavage of coumarin. Finally, paclitaxel was released by subsequent spontaneous O–N intramolecular acyl migration.
Co-reporter:Tooru Kimura, Daisuke Shuto, Yoshio Hamada, Naoto Igawa, Soko Kasai, Ping Liu, Koushi Hidaka, Takashi Hamada, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 1) pp:211-215
Publication Date(Web):3 January 2005
DOI:10.1016/j.bmcl.2004.09.090
Recently, we reported potent and small-sized BACE1 inhibitors KMI-358 and KMI-370 in which the Glu residue is replaced by a β-N-oxalyl-DAP (l-α,β-diaminopropionyl) residue at the P4 position. The β-N-oxalyl-DAP group is important for enhancing BACE1 inhibitory activity, but these inhibitors isomerized to α-N-oxalyl-DAP derivatives in solvents. Hence, we used a tetrazole moiety as a bioisostere of the free carboxylic acid of the oxalyl group. KMI-420 and KMI-429, containing a tetrazole ring, showed improved stability and potent enzyme inhibitory activity.
Co-reporter:Youhei Sohma, Masato Sasaki, Yoshio Hayashi, Tooru Kimura and Yoshiaki Kiso
Chemical Communications 2004 (Issue 1) pp:124-125
Publication Date(Web):21 Nov 2003
DOI:10.1039/B312129A
A novel and efficient method for the synthesis of difficult sequence-containing peptides has been developed based on the synthesis of O-acyl isopeptides followed by an O–N intramolecular acyl migration reaction, resulting in a remarkable improvement of the yields.
Co-reporter:Daisuke Shuto, Soko Kasai, Tooru Kimura, Ping Liu, Koushi Hidaka, Takashi Hamada, Saeko Shibakawa, Yoshio Hayashi, Chinatsu Hattori, Beata Szabo, Shoichi Ishiura, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 24) pp:4273-4276
Publication Date(Web):15 December 2003
DOI:10.1016/j.bmcl.2003.09.053
A novel class of substrate-based β-secretase (BACE1) inhibitors containing a hydroxymethylcarbonyl (HMC) isostere was designed and synthesized. Phenylnorstatine [(2R,3S)-3-amino-2-hydroxy-4-phenylbutyric acid; Pns] was an effective transition-state mimic at the P1 position. Structure–activity relationships (SARs) of the P3–P3′ positions of BACE1 inhibitors were studied.Graphic
Co-reporter:Koushi Hidaka, Tooru Kimura, Yoshio Hayashi, Keith F McDaniel, Tatyana Dekhtyar, Lynn Colletti, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2003 Volume 13(Issue 1) pp:93-96
Publication Date(Web):January 2003
DOI:10.1016/S0960-894X(02)00848-X
Pseudo-symmetric HIV-1 protease inhibitors containing a novel HMC-hydrazide isostere as the transition-state mimic were designed and synthesized. Most of the synthetic compounds with varied structures at the P and P′ sites around this core unit showed potent inhibitory activity against HIV-1 protease with nanomolar Ki values.Graphic
Co-reporter:Hikaru Matsumoto, Tooru Kimura, Tomonori Hamawaki, Akira Kumagai, Toshiyuki Goto, Kouichi Sano, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry 2001 Volume 9(Issue 6) pp:1589-1600
Publication Date(Web):June 2001
DOI:10.1016/S0968-0896(01)00045-1
Based on the prodrug concept as well as the combination of two different classes of anti-HIV agents, we designed and synthesized a series of anti-HIV double-drugs consisting of HIV protease inhibitors conjugated with a nucleoside reverse transcriptase inhibitor in an effort to enhance the antiviral activity. For the conjugation, a series of linkers that conjoins the two different classes of inhibitors has been investigated. Double-drugs using a succinyl amino acid linker were shown to release the parent drugs via spontaneous imide formation at a faster rate compared to compounds using a glutaryl amino acid linker, as expected from the energetically favorable cyclization to the five-membered ring. Among the double-drugs, KNI-1039 (3b) with a glutarylglycine linker exhibited extremely potent anti-HIV activity compared with that of the individual components. Double-drug 3b was relatively stable in culture medium, whereas it regenerated active species in cell homogenate. These results suggested that the synergistic enhancement of anti-HIV activities of 3b may be due to their ability to penetrate into the target cell and subsequent regeneration of two different classes of anti-HIV agents in the cytoplasm.KNI-1039, a double-drug conjugating an HIV potease inhibitor with AZT by a glutarylglycine linker, exhibited excellent anti-HIV activity.
Co-reporter:Hikaru Matsumoto, Tomonori Hamawaki, Hisashi Ota, Tooru Kimura, Toshiyuki Goto, Kouichi Sano, Yoshio Hayashi, Yoshiaki Kiso
Bioorganic & Medicinal Chemistry Letters 2000 Volume 10(Issue 11) pp:1227-1231
Publication Date(Web):5 June 2000
DOI:10.1016/S0960-894X(00)00202-X
We designed and synthesized a new series of prodrug-type anti-HIV agents consisting of a peptidomimetic HIV protease inhibitor conjugated with a nucleoside reverse transcriptase inhibitor in an effort to enhance the antiviral activity. For the conjugation, a series of linkers that conjoin the two different classes of inhibitors have been investigated. Conjugates using a succinyl amino acid linker were shown to release the parent components via the spontaneous imide formation at a faster rate compared to conjugates using a glutaryl amino acid linker, as expected from the energetically favorable cyclization to the five-membered ring. Herein, we report a new ‘double-drug’ 4b (KNI-1039) with a glutarylglycine linker, which exhibited extremely potent anti-HIV activity compared with that of the individual components.
Co-reporter:Taku Yoshiya, Hiroyuki Kawashima, Youhei Sohma, Tooru Kimura and Yoshiaki Kiso
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 14) pp:NaN2904-2904
Publication Date(Web):2009/05/18
DOI:10.1039/B903624E
We report the establishment of the O-acyl isopeptide method-based racemization-free segment condensation reaction toward future chemical protein synthesis. Peptide segments containing C-terminal O-acyl Ser/Thr residues were successfully synthesized by use of a lower nucleophilic base cocktail for Fmoc removal, and then coupled to an amino group of a peptide-resin without side reactions or epimerization. We also succeeded in performing the segment condensation in a sequential manner and in solution phase conditions as well.