Co-reporter:Yukihiro Fukata, Koichi Yao, Ryota Miyaji, Keisuke Asano, and Seijiro Matsubara
The Journal of Organic Chemistry December 1, 2017 Volume 82(Issue 23) pp:12655-12655
Publication Date(Web):November 2, 2017
DOI:10.1021/acs.joc.7b02451
In this study, the isothiourea-catalyzed enantioselective formal [4+3] cycloaddition of various α,β-unsaturated carboxylic acid derivatives with 2-aminothiophenols was developed. Mechanistic studies suggested that the reaction proceeds via a reversible sulfa-Michael addition to α,β-unsaturated acylammonium intermediates, followed by the enantioselective formation of a seven-membered ring, enabling the facile and divergent synthesis of optically active 2- and 3-substituted 1,5-benzothiazepines. This process was demonstrated to be highly versatile, affording the corresponding products in excellent regioselectivities and high enantioselectivities. Furthermore, this method enabled the synthesis of chiral 2,3-disubstituted 1,5-benzothiazepines in high regio-, enantio-, and diastereoselectivities. Hence, this protocol can be applied for the construction of a library of useful pharmaceutical candidates.
Co-reporter:Takuya Kurahashi and Seijiro Matsubara
Accounts of Chemical Research 2015 Volume 48(Issue 6) pp:1703
Publication Date(Web):May 19, 2015
DOI:10.1021/acs.accounts.5b00041
Heterocycles have garnered significant attention because they are important functional building blocks in various useful molecules, such as pharmaceuticals, agricultural chemicals, pesticides, and materials. Several studies have been conducted regarding the preparation of heterocyclic skeletons with an emphasis on selectivity and efficiency. Three strategies are typically employed to construct cyclic molecules, namely, cyclization, cycloaddition, and ring-size alterations. Although each method has certain advantages, cycloaddition may be superior from the viewpoint of divergence. Specifically, cycloadditions enable the construction of rings from several pieces. However, the construction of heterocycles via cycloadditions is more challenging than the construction of carbocycles. For heterocycle construction, simple pericyclic reactions rarely work smoothly because of the large HOMO–LUMO gap unless well-designed combinations, such as electron-rich dienes and aldehydes, are utilized. Thus, a different approach should be employed to prepare heterocycles via cycloadditions. To this end, the use of metallacycles containing heteroatoms is expected to serve as a promising solution. In this study, we focused on the preparation of heteroatom-containing nickelacycles. Because nickel possesses a relatively high redox potential and an affinity for heteroatoms, several methods were developed to synthesize heteronickelacycles from various starting materials. The prepared nickelacycles were demonstrated to be reasonable intermediates in cycloaddition reactions, which were used to prepare various heterocycles. In this Account, we introduce the following four methods to prepare heterocycles via heteronickelacycles. (1) Direct oxidative insertion of Ni(0) to α,β-unsaturated enone derivatives: treatment of 3-ethoxycarbonyl-4-phenyl-3-buten-2-one with Ni(0) afforded an oxa-nickelacycle, which reacted with alkynes to give pyrans. (2) Substitution of a part of a cyclic compound with low-valent nickel, accompanied by elimination of small molecules such as CO, CO2, and acetophenone: treatment of phthalic anhydride with Ni(0) in the presence of ZnCl2 afforded the oxanickelacycle, which was formed via decarbonylative insertion of Ni(0) and reacted with alkynes to give isocumarins. (3) Cyclization to a nickelacycle, accompanied by two C–C σ-bond activations: insertion of Ni(0) into an arylnitrile, followed by aryl cyanation of an alkyne, gave alkenylnickel as an intermediate. The alkenylnickel species subsequently underwent an intramolecular nucleophilic attack with an arylcarbonyl group to form a cyclized product with concomitant cleavage of the C–C σ-bond between the carbonyl and aryl groups. (4) Assembly of several components to form a heteroatom-containing nickelacycle via cycloaddition: a new [2 + 2 + 1] cyclization reaction was carried out using an α,β-unsaturated ester, isocyanate, and alkyne via a nickelacycle. On the basis of these four strategies, we developed new methods to prepare heterocyclic compounds using nickelacycles as the key active species.
Co-reporter:Yukihiro Fukata; Keisuke Asano
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5320-5323
Publication Date(Web):April 9, 2015
DOI:10.1021/jacs.5b02537
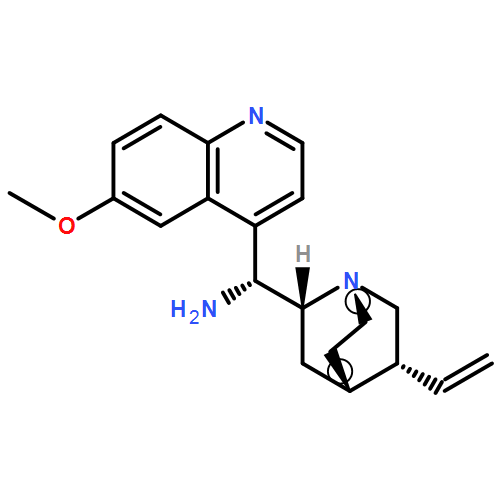
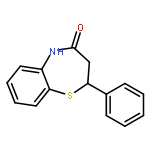
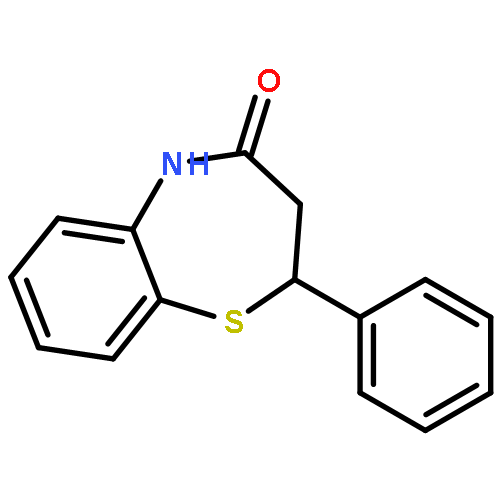
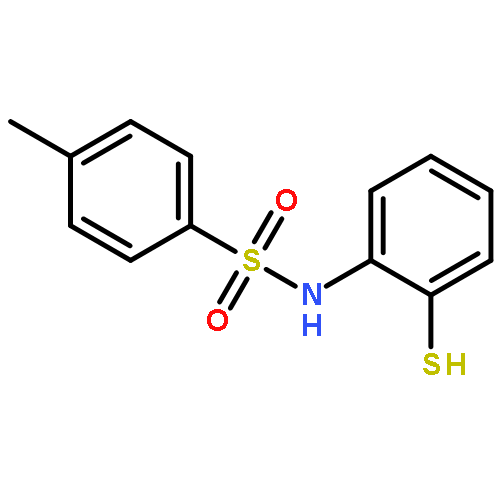
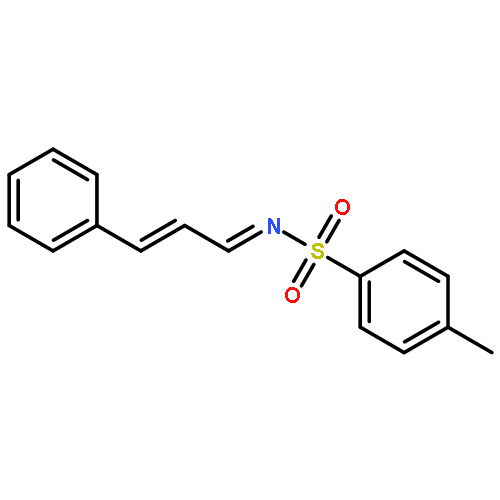
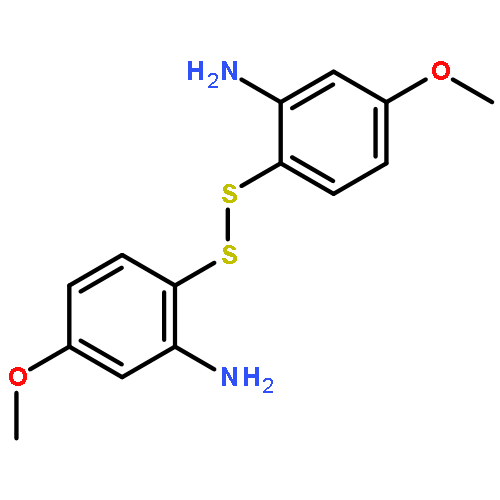
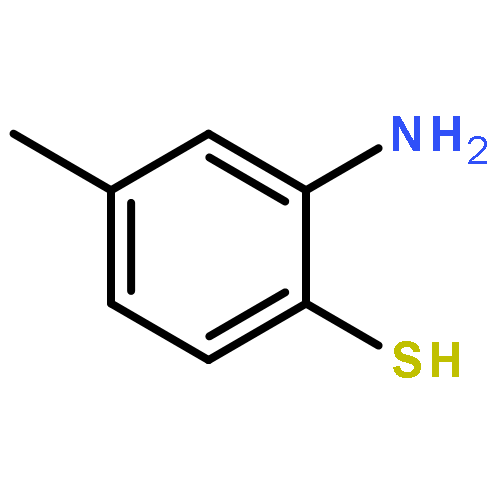
The 1,5-benzothiazepine moiety is well-known as a versatile pharmacophore, and its derivatives are expected to have antagonism against numerous diseases. Thus, it is desirable to develop a synthetic route that enables facile enantioselective preparation of a wide range of such derivatives. Although the cycloaddition approach could be considered a possible route to these compounds, to date, there has been no precedent of such a protocol. We therefore present the first example of a highly enantioselective net [4 + 3] cycloaddition to afford 1,5-benzothiazepines by utilizing α,β-unsaturated acylammonium intermediates generated by chiral isothiourea catalysts, which undergo two sequential chemoselective nucleophilic attacks by 2-aminothiophenols. This protocol provided cycloadducts in extremely high regioselectivity, with a good-to-excellent stereoselectivity being achieved regardless of the steric and electronic properties of the substrates. This method therefore offers promising synthetic routes for the construction of a library of optically active 1,5-benzothiazepines for assay evaluation.
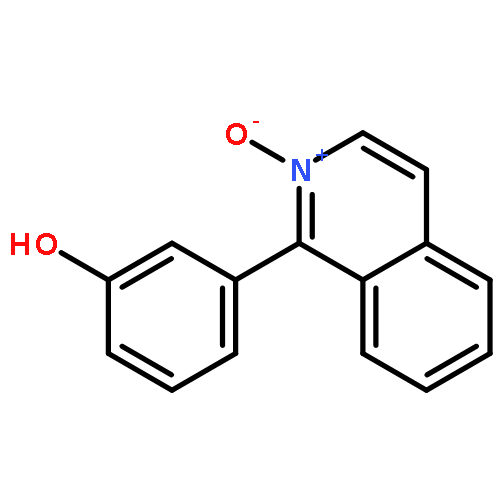
Co-reporter:Ryota Miyaji; Keisuke Asano
Journal of the American Chemical Society 2015 Volume 137(Issue 21) pp:6766-6769
Publication Date(Web):May 16, 2015
DOI:10.1021/jacs.5b04151
Bifunctional catalysts bearing amino and urea functional groups have been applied for a novel, highly enantioselective synthesis of axially chiral isoquinoline N-oxides, which are promising chiral ligands or organocatalysts in organic synthesis. This is the first example of highly enantioselective synthesis of axially chiral biaryls by bifunctional organocatalysts. Good-to-excellent enantioselectivities were obtained with a range of substrates.
Co-reporter:Kazuki Maeda, Takuma Terada, Takahiro Iwamoto, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2015 Volume 17(Issue 21) pp:5284-5287
Publication Date(Web):October 22, 2015
DOI:10.1021/acs.orglett.5b02654
A new efficient synthetic route to unsymmetrically substituted dihydropyridine scaffolds via dehydrative [4 + 2] cycloaddition of N-tosylated α,β-unsaturated imines with aldehydes has been developed. This transformation is enabled by (i) the remarkable catalytic ability of the cationic Ru(IV) porphyrin complex to activate both the imino and carbonyl groups and (ii) the hydrophobic nature of the porphyrin ligand, which helps realize robust Lewis acidity in the dehydrative cycloaddition.
Co-reporter:Tasuku Inami, Takuya Kurahashi and Seijiro Matsubara
Chemical Communications 2015 vol. 51(Issue 7) pp:1285-1288
Publication Date(Web):05 Dec 2014
DOI:10.1039/C4CC09123J
The thiocarbamoylation of internal alkynes to produce tetrasubstituted β-aminocarbonyl vinyl sulfides was demonstrated using a nickel catalyst. This reaction was successful with a wide variety of substituents, and gave the syn-adducts exclusively.
Co-reporter:Akira Matsumoto, Keisuke Asano and Seijiro Matsubara
Chemical Communications 2015 vol. 51(Issue 58) pp:11693-11696
Publication Date(Web):12 Jun 2015
DOI:10.1039/C5CC04124D
A novel method of enantioselective 1,3-dioxane construction via a hemiacetalization/intramolecular oxy-Michael addition cascade by a chiral phosphoric acid catalyst was developed. The product was successfully transformed into an optically active 1,3-polyol motif, indicating that the proposed reaction can provide useful chiral building blocks for the de novo synthesis of polyketides.
Co-reporter:Ryosuke Haraguchi, Yoshiaki Takada and Seijiro Matsubara
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 1) pp:241-247
Publication Date(Web):2014/10/20
DOI:10.1039/C4OB01474J
The nucleophilic cyclopropanation of hexa-1,5-diene-3,4-diones with bis(iodozincio)methane afforded the Zn alkoxides of cis-dialkenylcyclopropane-1,2-diols stereoselectively. The subsequent oxy-Cope rearrangement afforded the corresponding Zn alkoxides of 5,6-dialkylcyclohepta-3,7-diene-1,3-diols.
Co-reporter:Ryosuke Haraguchi, Zenichi Ikeda, Akihiro Ooguri, Seijiro Matsubara
Tetrahedron 2015 Volume 71(Issue 46) pp:8830-8837
Publication Date(Web):18 November 2015
DOI:10.1016/j.tet.2015.08.060
The palladium catalyzed cross-coupling reaction of thiol esters with bis(iodozincio)methane or 1,1-bis(iodozincio)ethane gave Reformatsky-type enolates. They can react with some electrophiles to give the corresponding adducts and were also trapped by silylation reagents to afford silyl enol ethers. As the method applicable to the thiol ester carrying ketone moiety, it afforded zinc enolates carrying ketone in the same molecule.
Co-reporter:Kenichiro Nakai, Takuya Kurahashi, Seijiro Matsubara
Tetrahedron 2015 Volume 71(26–27) pp:4512-4517
Publication Date(Web):1 July 2015
DOI:10.1016/j.tet.2015.02.064
A nickel/Lewis acid catalyzed intermolecular cycloaddition reaction of o-cyanobenzylarylketones with alkynes to form naphthalenones is developed. This reaction is promoted by the nickel/Lewis Acid catalyst pair and involves the cleavage of two C–C σ bonds to eliminate arylcyanide and the formation of different two C–C σ bonds with alkyne insertion.
Co-reporter:Kenichiro Nakai ; Yuji Yoshida ; Takuya Kurahashi
Journal of the American Chemical Society 2014 Volume 136(Issue 22) pp:7797-7800
Publication Date(Web):May 14, 2014
DOI:10.1021/ja500666h
We have developed a redox-economical coupling reaction of alcohols and alkynes to form allylic alcohols under mild conditions. The reaction is redox-neutral as well as redox-economical and thus free from any additives such as a reductant or an oxidant. This atom-economical coupling can be applied for the conversion of both aliphatic and benzylic alcohols to the corresponding substituted allylic alcohols in a single synthetic operation.
Co-reporter:Yukihiro Fukata, Takaaki Okamura, Keisuke Asano, and Seijiro Matsubara
Organic Letters 2014 Volume 16(Issue 8) pp:2184-2187
Publication Date(Web):March 31, 2014
DOI:10.1021/ol500637x
We present a novel methodology for the asymmetric synthesis of β-mercaptolactones via isomerization of ω-hydroxy-α,β-unsaturated thioesters by means of a bifunctional aminothiourea catalyst. The catalyst interacts with the substrate through the cooperative action of both a covalent bond at the amino group and noncovalent bonding at the thiourea group. The potential for an enantiodivergent synthesis could also be demonstrated by carrying out the reaction in a different solvent system.
Co-reporter:Takuma Terada, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2014 Volume 16(Issue 10) pp:2594-2597
Publication Date(Web):April 30, 2014
DOI:10.1021/ol500625r
Lewis acid catalyzed cycloaddition of cyclohexenone and butadiene affords trans-fused octalone with high regio- and diastereoselectivity. The use of the ruthenium porphyrin complex as the Lewis acid catalyst is key to the reaction. The cycloaddition proceeds in toluene with 1 mol % of the ruthenium catalyst at 25 °C.
Co-reporter:Tasuku Inami, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2014 Volume 16(Issue 21) pp:5660-5662
Publication Date(Web):October 27, 2014
DOI:10.1021/ol5026102
A new synthetic method for thiochromones was developed by using nickel-catalyzed decarbonylative cycloaddition of readily available thioisatins with alkynes. This reaction proceeded under very mild conditions and has quite high functional group compatibility.
Co-reporter:Naoki Yoneda, Ayano Hotta, Keisuke Asano, and Seijiro Matsubara
Organic Letters 2014 Volume 16(Issue 23) pp:6264-6266
Publication Date(Web):November 10, 2014
DOI:10.1021/ol503104b
Formaldehyde was utilized as an oxygen-centered nucleophile in an asymmetric oxy-Michael addition to γ-hydroxy-α,β-unsaturated carbonyl compounds using bifunctional organocatalysts through hemiacetal intermediates. The cyclic acetal product could be further transformed into β-hydroxycarbonyl compounds, useful synthetic intermediates leading to various important target molecules. As such, this method is an example of a novel formal asymmetric hydration of α,β-unsaturated carbonyl compounds.
Co-reporter:Ryota Miyaji, Keisuke Asano and Seijiro Matsubara
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 1) pp:119-122
Publication Date(Web):29 Oct 2013
DOI:10.1039/C3OB41938J
Cinchona-alkaloid-urea-based bifunctional organocatalysts facilitate the catalytic asymmetric synthesis of chroman derivatives via an intramolecular oxy-Michael addition reaction. Phenol derivatives bearing an easily available (E)-α,β-unsaturated ketone or a thioester moiety are useful substrates for the title transformation. This method represents a facile synthesis of various optically active 2-substituted chromans in high yield.
Co-reporter:Yukihiro Fukata ; Keisuke Asano
Journal of the American Chemical Society 2013 Volume 135(Issue 33) pp:12160-12163
Publication Date(Web):August 5, 2013
DOI:10.1021/ja407027e
In a novel organocatalytic formal [3 + 2] cycloaddition to afford chiral 2-oxazolidinones, an enantioselectivity switch could be induced by changing the manner of addition of the reactants, even when the reaction components (cinchona-alkaloid-derived aminothiourea catalyst, substrates, and solvent) were the same.
Co-reporter:Takahiro Shiba ; Takuya Kurahashi
Journal of the American Chemical Society 2013 Volume 135(Issue 37) pp:13636-13639
Publication Date(Web):September 5, 2013
DOI:10.1021/ja4068172
We have developed a nickel-catalyzed transformation, in which phthalimides react with trimethylsilyl-substituted alkynes in the presence of Ni(0)/PMe3/MAD catalyst to provide isoindolinones. The reaction process displays an unusual mechanistic feature—decarbonylation and alkylidenation. The use of both trimethylsilyl-substiuted alkynes and MAD was found to be essential for the transformation with high selectivities.
Co-reporter:Kenichiro Nakai, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2013 Volume 15(Issue 4) pp:856-859
Publication Date(Web):January 25, 2013
DOI:10.1021/ol303546p
Substituted quinolones were efficiently synthesized via the nickel-catalyzed cycloaddition of o-cyanophenylbenzamide derivatives with alkynes. The reaction involves elimination of a nitrile group by cleavage of the two independent aryl–cyano and aryl–carbonyl C–C bonds of the amides.
Co-reporter:Rihoko Tombe, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2013 Volume 15(Issue 8) pp:1791-1793
Publication Date(Web):April 8, 2013
DOI:10.1021/ol4005068
Nickel-catalyzed intermolecular [3 + 2] cycloaddition of vinylcyclopropanes to imines has been developed. This transformation generates substituted pyrrolidines in high yields, with good regio- and diastereo- selectivity under mild reaction conditions. A variety of imines can be used in this reaction. An asymmetric variant of the reaction has also been demonstrated.
Co-reporter:Ryota Miyaji, Keisuke Asano, and Seijiro Matsubara
Organic Letters 2013 Volume 15(Issue 14) pp:3658-3661
Publication Date(Web):July 11, 2013
DOI:10.1021/ol401538b
A novel method for the asymmetric synthesis of 2-substituted indolines, employing bifunctional amino(thio)urea catalysts, was developed. The reaction proceeded via an intramolecular aza-Michael addition mediated by activation through hydrogen bonding. The catalytic process was shown to be highly versatile and applicable to a wide range of substrates due to the flexible catalytic mechanism utilizing a noncovalent interaction.
Co-reporter:Ryosuke Haraguchi and Seijiro Matsubara
Organic Letters 2013 Volume 15(Issue 13) pp:3378-3380
Publication Date(Web):June 11, 2013
DOI:10.1021/ol401425s
Treatment of phenyl isocyanate with bis(iodozincio)methane gave a zinciomethylenated product, which acts as an amide-enoate equivalent. It did not react with an aldehyde efficiently, but gave the corresponding adduct in good yield in the presence of an aminoalcohol. Use of a catalytic amount of chiral aminoalcohol led the process to the catalytic asymmetric Aldol-type reaction.
Co-reporter:Makoto Hasegawa, Takuya Kurahashi, Seijiro Matsubara
Tetrahedron Letters 2013 Volume 54(Issue 46) pp:6196-6198
Publication Date(Web):13 November 2013
DOI:10.1016/j.tetlet.2013.08.128
Cycloisomerization of 1,6-enynes is successfully carried out in the presence of a dicationic platinum(IV) catalyst to afford five-membered ring systems. The use of a weakly coordinating axial ligand is the key to bringing out the catalytic activity of platinum porphyrin for the reaction.
Co-reporter:Ryota Wakabayashi, Takuya Kurahashi, and Seijiro Matsubara
Organic Letters 2012 Volume 14(Issue 18) pp:4794-4797
Publication Date(Web):September 4, 2012
DOI:10.1021/ol3020946
An efficient protocol for the aza-Diels–Alder reaction of electron-deficient 1,3-dienes with unactivated imines in the presence of a cationic cobalt(III) porphyrin complex was developed. The transformation proceeded smoothly to afford the desired piperidine scaffold within 2 h at ambient temperature. Highly chemoselective cycloaddition of imines with dienes in the presence of a variety of carbonyl compounds was also demonstrated.
Co-reporter:Tasuku Inami, Takuya Kurahashi and Seijiro Matsubara
Chemical Communications 2015 - vol. 51(Issue 7) pp:NaN1288-1288
Publication Date(Web):2014/12/05
DOI:10.1039/C4CC09123J
The thiocarbamoylation of internal alkynes to produce tetrasubstituted β-aminocarbonyl vinyl sulfides was demonstrated using a nickel catalyst. This reaction was successful with a wide variety of substituents, and gave the syn-adducts exclusively.
Co-reporter:Ryota Miyaji, Keisuke Asano and Seijiro Matsubara
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 1) pp:NaN122-122
Publication Date(Web):2013/10/29
DOI:10.1039/C3OB41938J
Cinchona-alkaloid-urea-based bifunctional organocatalysts facilitate the catalytic asymmetric synthesis of chroman derivatives via an intramolecular oxy-Michael addition reaction. Phenol derivatives bearing an easily available (E)-α,β-unsaturated ketone or a thioester moiety are useful substrates for the title transformation. This method represents a facile synthesis of various optically active 2-substituted chromans in high yield.
Co-reporter:Ryosuke Haraguchi, Yoshiaki Takada and Seijiro Matsubara
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 1) pp:NaN247-247
Publication Date(Web):2014/10/20
DOI:10.1039/C4OB01474J
The nucleophilic cyclopropanation of hexa-1,5-diene-3,4-diones with bis(iodozincio)methane afforded the Zn alkoxides of cis-dialkenylcyclopropane-1,2-diols stereoselectively. The subsequent oxy-Cope rearrangement afforded the corresponding Zn alkoxides of 5,6-dialkylcyclohepta-3,7-diene-1,3-diols.
Co-reporter:Akira Matsumoto, Keisuke Asano and Seijiro Matsubara
Chemical Communications 2015 - vol. 51(Issue 58) pp:NaN11696-11696
Publication Date(Web):2015/06/12
DOI:10.1039/C5CC04124D
A novel method of enantioselective 1,3-dioxane construction via a hemiacetalization/intramolecular oxy-Michael addition cascade by a chiral phosphoric acid catalyst was developed. The product was successfully transformed into an optically active 1,3-polyol motif, indicating that the proposed reaction can provide useful chiral building blocks for the de novo synthesis of polyketides.

![Bicyclo[2.2.2]octanone, 3-methyl-](http://img.cochemist.com/ccimg/26100/26051-25-2.png)
![Bicyclo[2.2.2]octanone, 3-methyl-](http://img.cochemist.com/ccimg/26100/26051-25-2_b.png)





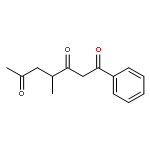
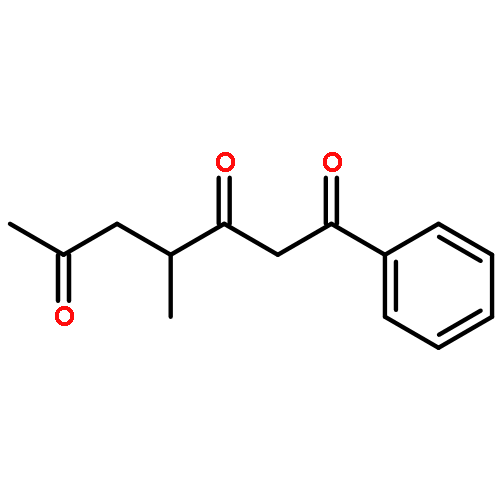
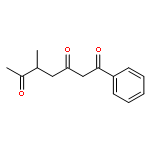
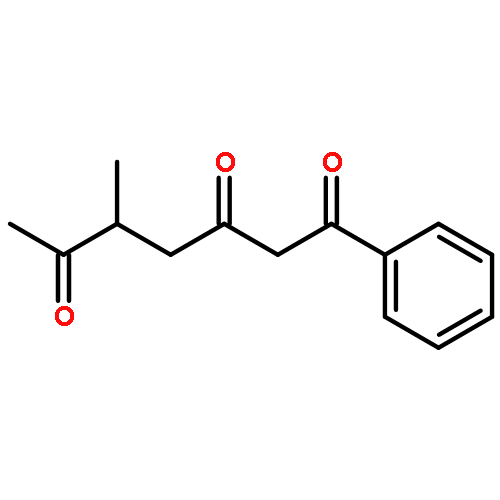
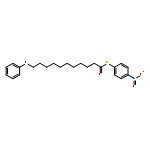
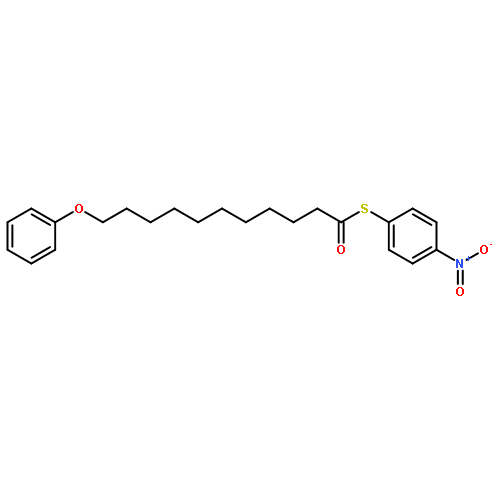
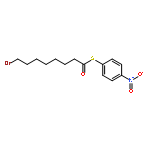
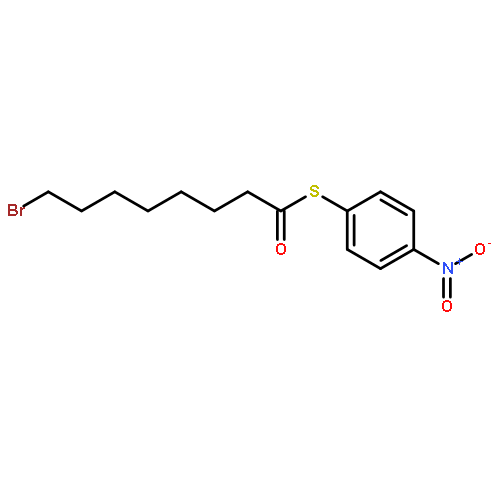
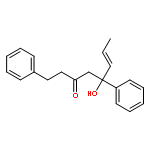
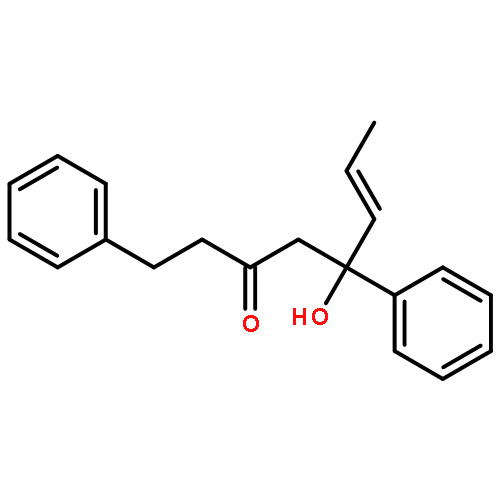
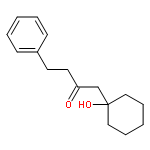
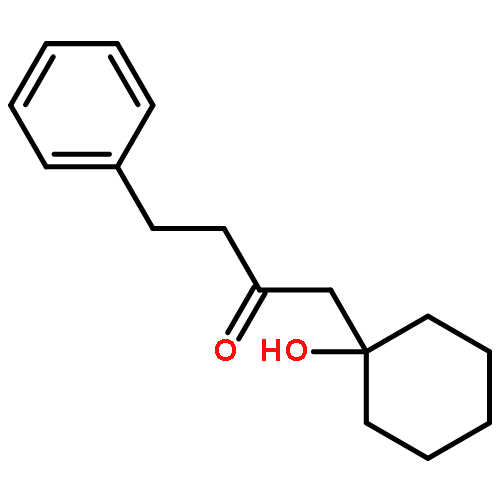
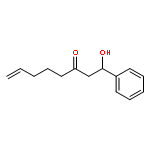
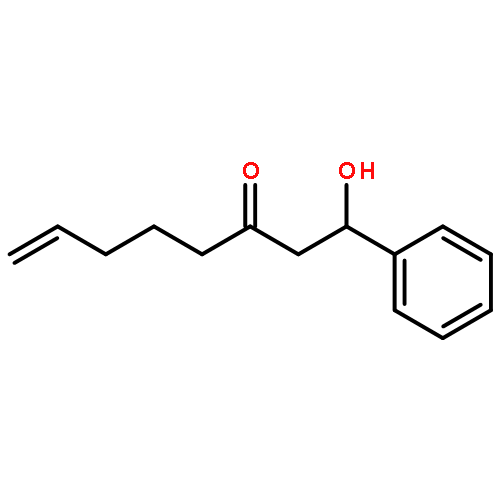
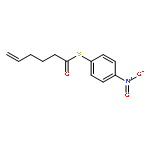
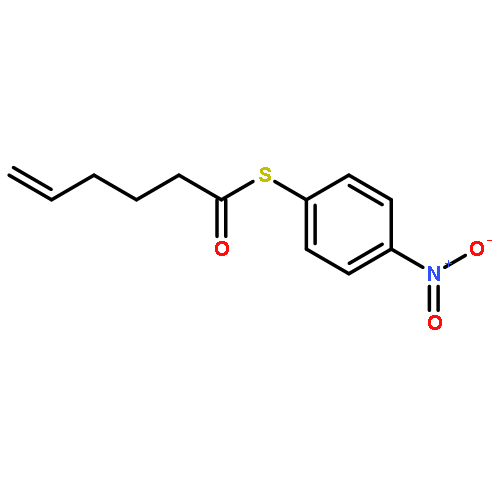
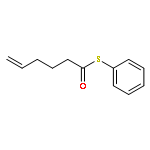
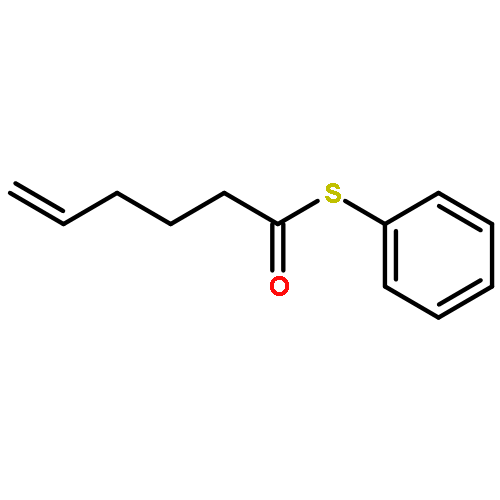
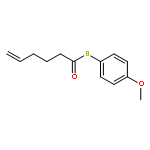
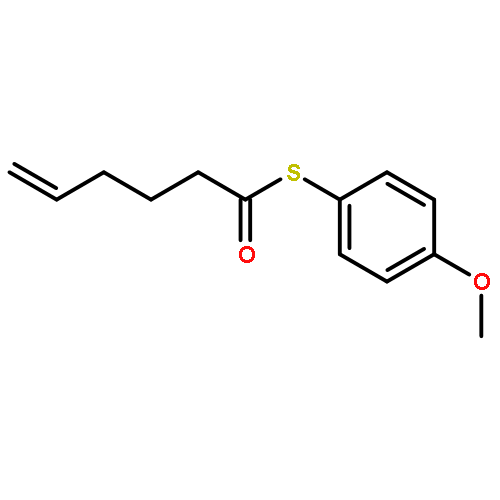


![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)



![N-[2-(4-fluorophenyl)ethyl]-3-methoxybenzamide](http://img.cochemist.com/ccimg/423800/423739-40-6.png)
![N-[2-(4-fluorophenyl)ethyl]-3-methoxybenzamide](http://img.cochemist.com/ccimg/423800/423739-40-6_b.png)










![2-Penten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-, (2E)-](http://img.cochemist.com/ccimg/199000/198977-95-6.png)
![2-Penten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-, (2E)-](http://img.cochemist.com/ccimg/199000/198977-95-6_b.png)


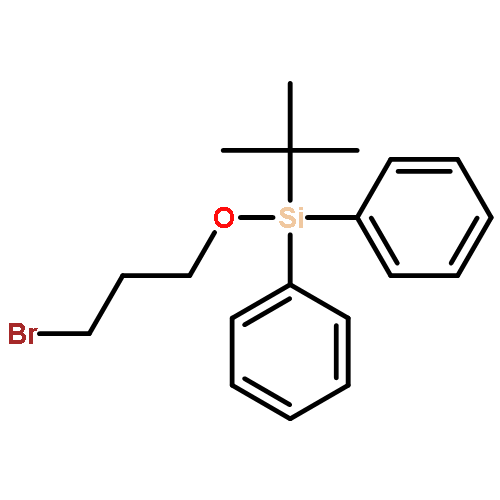
![2-Methoxy-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/175200/175153-20-5.png)
![2-Methoxy-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/175200/175153-20-5_b.png)


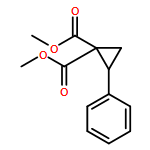

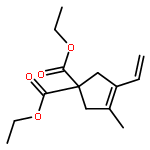

![Propanedioic acid, [(2E)-3-phenyl-2-propenyl]-2-propynyl-, diethyl ester](http://img.cochemist.com/ccimg/158400/158389-22-1.png)
![Propanedioic acid, [(2E)-3-phenyl-2-propenyl]-2-propynyl-, diethyl ester](http://img.cochemist.com/ccimg/158400/158389-22-1_b.png)






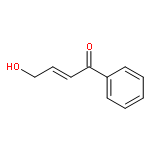



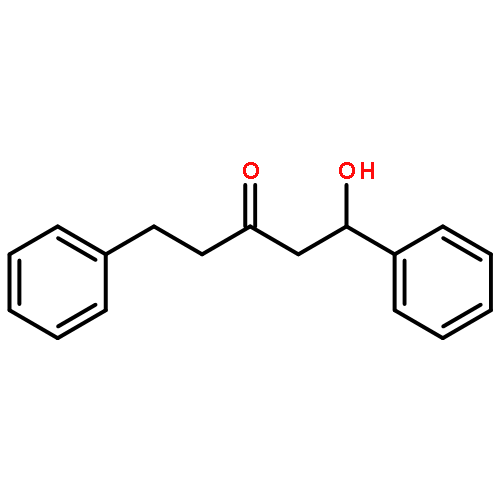
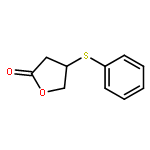
![2(3H)-Furanone, dihydro-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/141300/141248-79-5.png)
![2(3H)-Furanone, dihydro-4-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/141300/141248-79-5_b.png)




![1,3-Dithiane, 2-[3-[(tetrahydro-2H-pyran-2-yl)oxy]propyl]-](http://img.cochemist.com/ccimg/139300/139230-31-2.png)
![1,3-Dithiane, 2-[3-[(tetrahydro-2H-pyran-2-yl)oxy]propyl]-](http://img.cochemist.com/ccimg/139300/139230-31-2_b.png)







![Acetaldehyde, [[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-83-5.png)
![Acetaldehyde, [[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/112000/111998-83-5_b.png)


![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7.png)
![21H,23H-Porphine, 5,10,15,20-tetrakis[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/133300/110452-48-7_b.png)



![Silane, trimethyl[[1-(3-thienyl)ethenyl]oxy]-](http://img.cochemist.com/ccimg/101400/101306-15-4.png)
![Silane, trimethyl[[1-(3-thienyl)ethenyl]oxy]-](http://img.cochemist.com/ccimg/101400/101306-15-4_b.png)




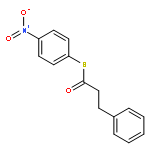
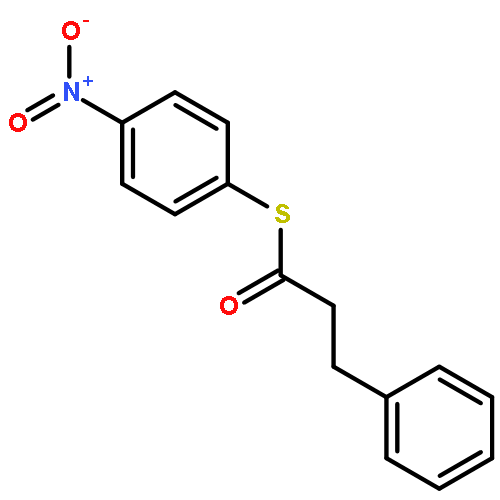
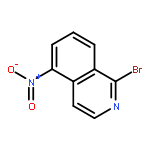



![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/2246300/92669-43-7.png)
![Ruthenium, carbonyl[5,10,15,20-tetrakis(2,4,6-trimethylphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-31)-](/data/chemimg/2246300/92669-43-7_b.png)
![Benzene, [2-(1,1-dimethylethyl)-1-methoxy-2,3-butadienyl]-](http://img.cochemist.com/ccimg/92600/92544-49-5.png)
![Benzene, [2-(1,1-dimethylethyl)-1-methoxy-2,3-butadienyl]-](http://img.cochemist.com/ccimg/92600/92544-49-5_b.png)




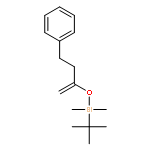


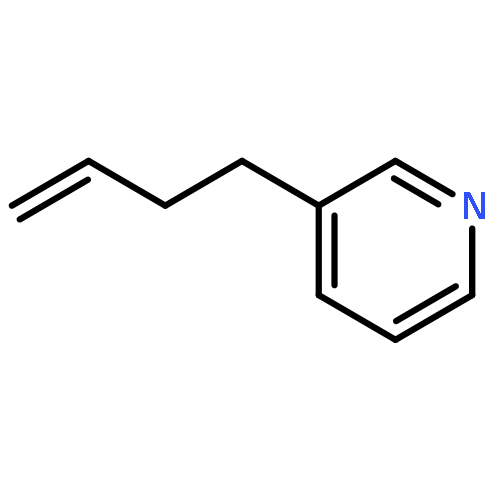
![Imidazo[2,1-b]benzothiazole, 2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/78300/78291-80-2.png)
![Imidazo[2,1-b]benzothiazole, 2,3-dihydro-2-phenyl-](http://img.cochemist.com/ccimg/78300/78291-80-2_b.png)
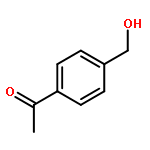



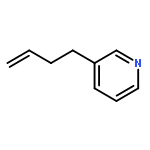
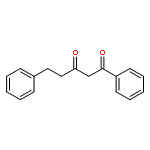
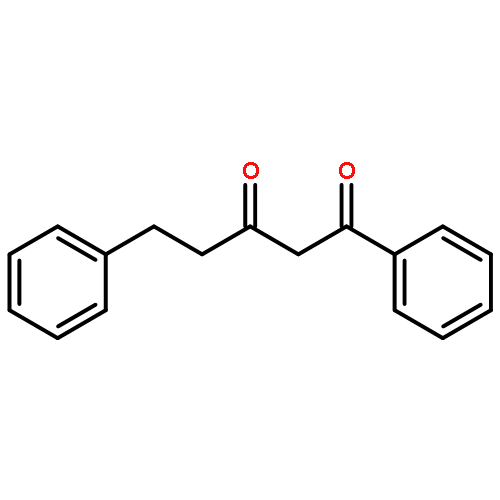
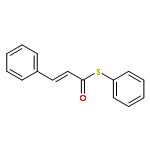

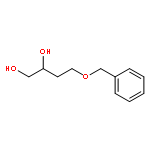
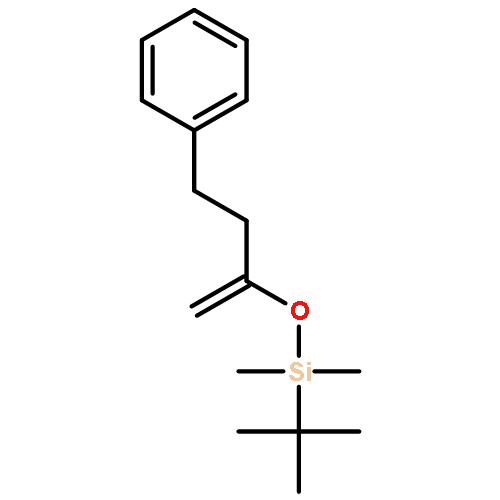
![6-Methoxybenzo[b]thiophene-2,3-dione](http://img.cochemist.com/ccimg/63700/63675-77-4.png)
![6-Methoxybenzo[b]thiophene-2,3-dione](http://img.cochemist.com/ccimg/63700/63675-77-4_b.png)


![Cobalt, chloro[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/2247200/60166-10-1.png)
![Cobalt, chloro[5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/2247200/60166-10-1_b.png)














![Benzo[b]thiophene-2,3-dione, 5-methyl-](http://img.cochemist.com/ccimg/50900/50891-89-9.png)
![Benzo[b]thiophene-2,3-dione, 5-methyl-](http://img.cochemist.com/ccimg/50900/50891-89-9_b.png)




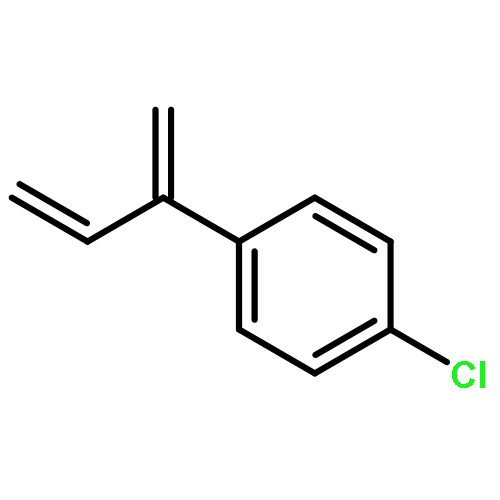
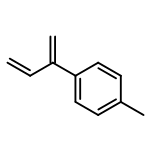
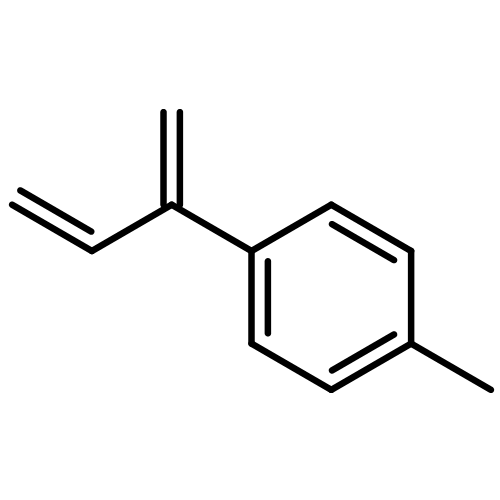
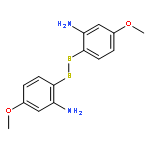


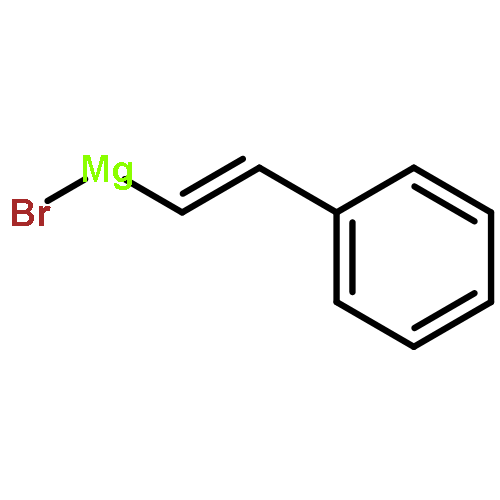
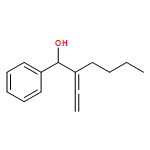
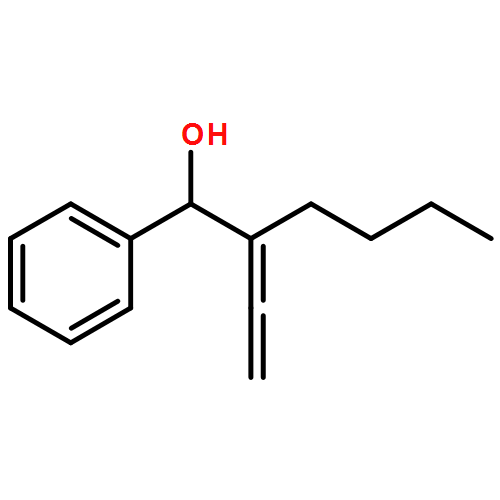








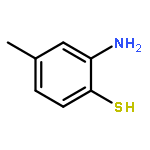
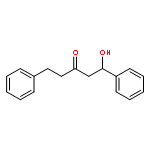
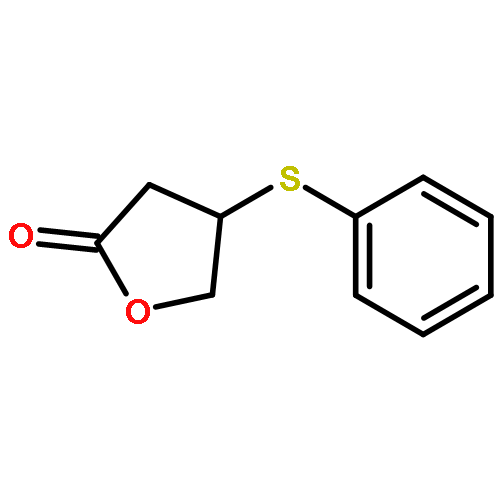
![2(3H)-Furanone, dihydro-4-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/30600/30544-85-5.png)
![2(3H)-Furanone, dihydro-4-[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/30600/30544-85-5_b.png)


![Benzene, 1-methoxy-4-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/22900/22865-60-7.png)
![Benzene, 1-methoxy-4-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/22900/22865-60-7_b.png)







































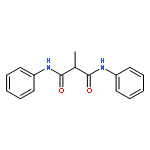
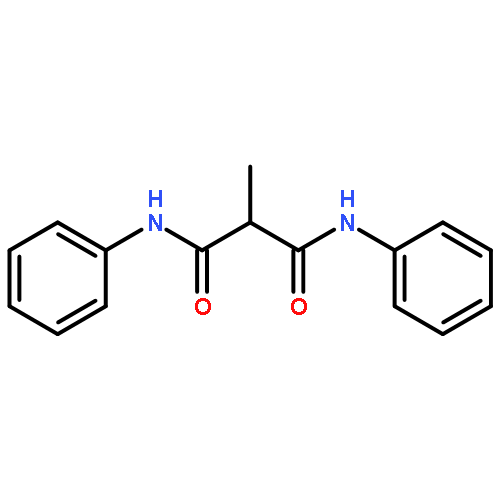
![N,N'-BIS[4-(TRIFLUOROMETHYL)PHENYL]PROPANEDIAMIDE](http://img.cochemist.com/ccimg/7600/7574-50-7.png)
![N,N'-BIS[4-(TRIFLUOROMETHYL)PHENYL]PROPANEDIAMIDE](http://img.cochemist.com/ccimg/7600/7574-50-7_b.png)





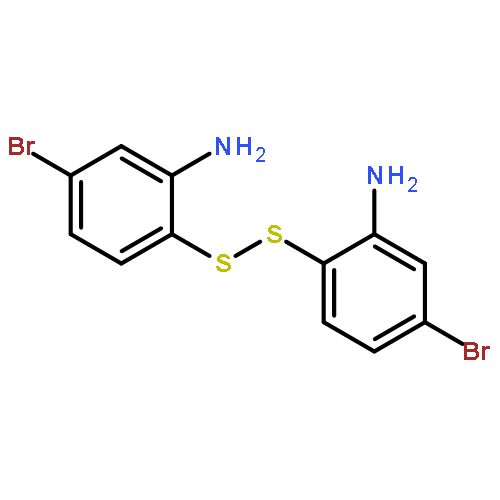




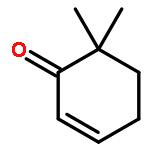
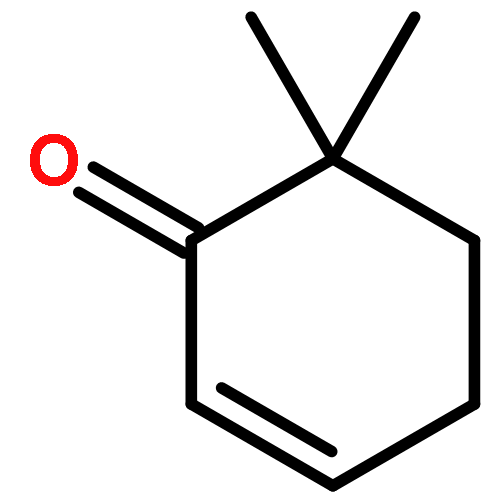
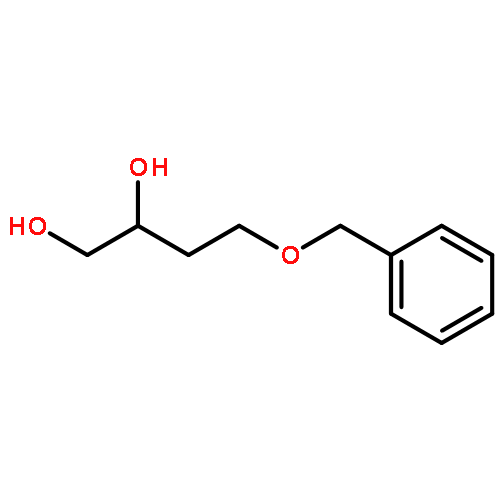
![3-[2-(3-HYDROXYPROPYL)-1,3-DIOXOLAN-2-YL]PROPAN-1-OL](http://img.cochemist.com/ccimg/5700/5694-96-2.png)
![3-[2-(3-HYDROXYPROPYL)-1,3-DIOXOLAN-2-YL]PROPAN-1-OL](http://img.cochemist.com/ccimg/5700/5694-96-2_b.png)
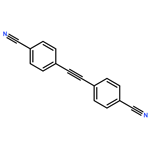
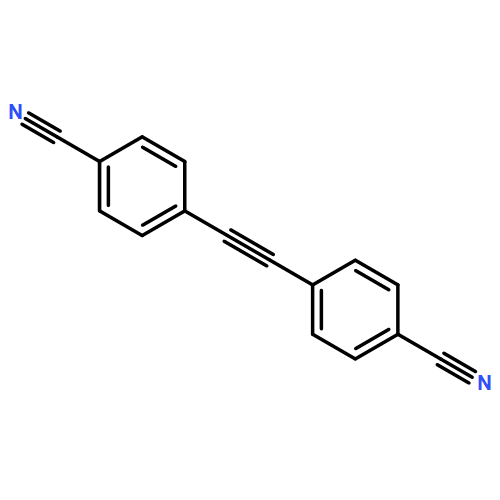
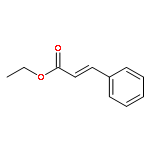
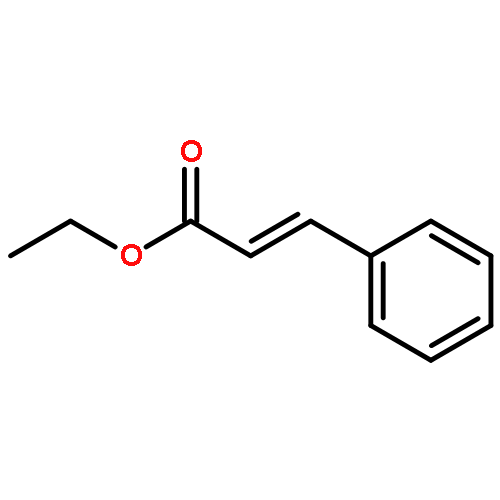


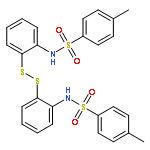
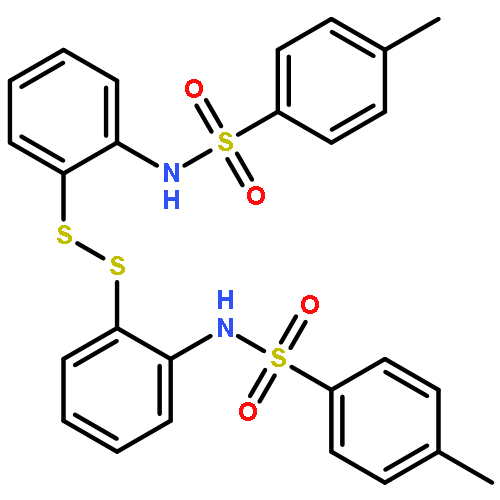


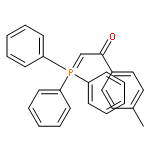
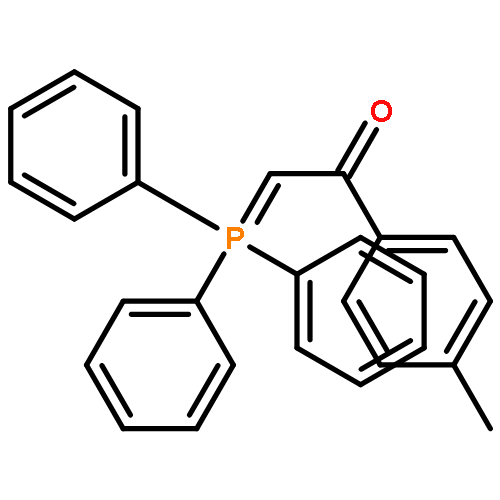
![Ethanone, 1-[1,1'-biphenyl]-4-yl-2-(triphenylphosphoranylidene)-](http://img.cochemist.com/ccimg/3000/2959-60-6.png)
![Ethanone, 1-[1,1'-biphenyl]-4-yl-2-(triphenylphosphoranylidene)-](http://img.cochemist.com/ccimg/3000/2959-60-6_b.png)


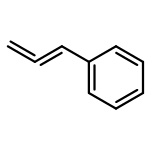


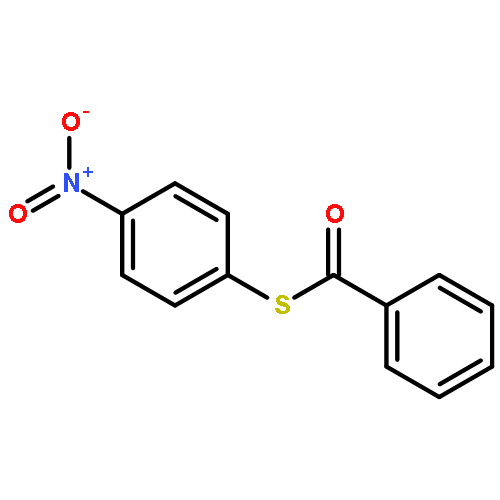






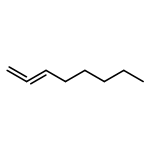
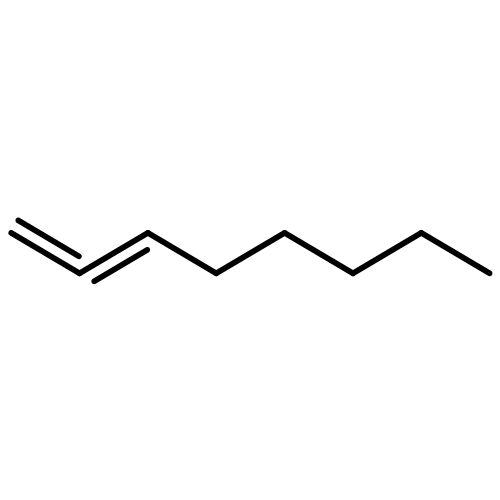
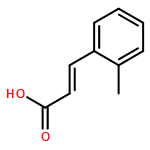


![Benzo[b]thiophene-2,3-dione](http://img.cochemist.com/ccimg/500/493-57-2.png)
![Benzo[b]thiophene-2,3-dione](http://img.cochemist.com/ccimg/500/493-57-2_b.png)






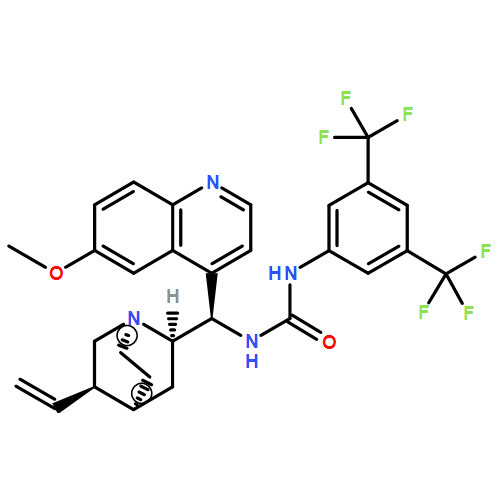





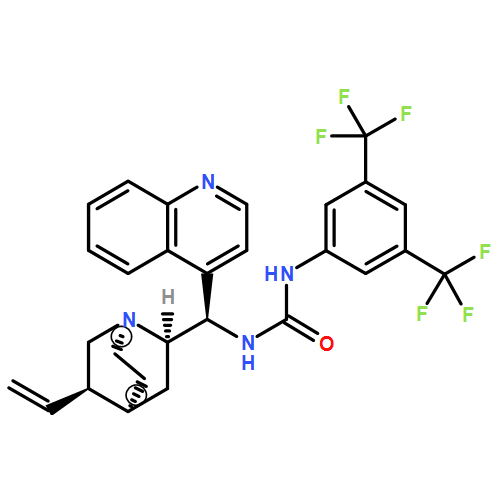
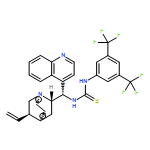
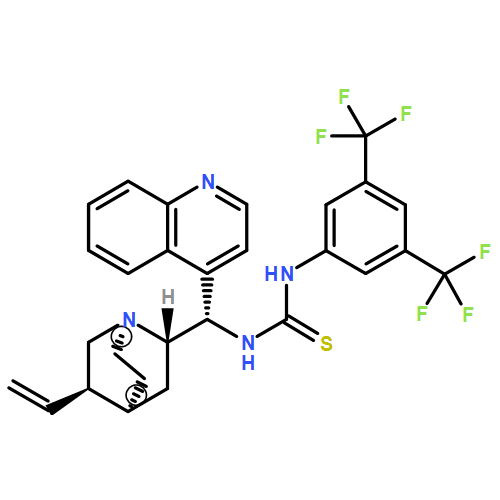
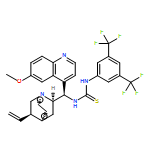
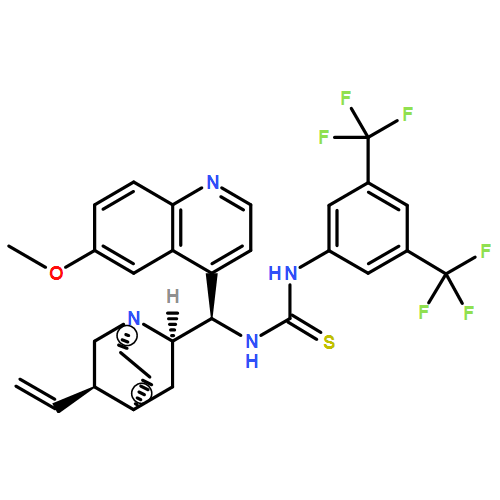
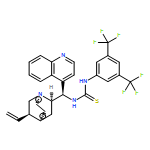
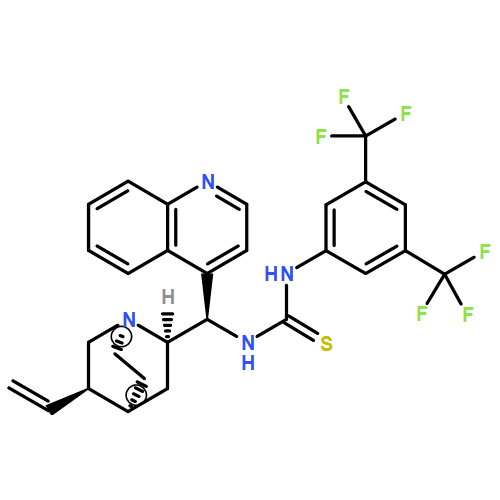
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)
![Ethanone, 1-[4-(trifluoromethyl)phenyl]-2-(triphenylphosphoranylidene)-](/data/chemimg/3765500/56893-05-1.png)
![Ethanone, 1-[4-(trifluoromethyl)phenyl]-2-(triphenylphosphoranylidene)-](/data/chemimg/3765500/56893-05-1_b.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9.png)
![BENZENE, 4-[(4-METHOXYPHENYL)ETHYNYL]-4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/40500/40474-01-9_b.png)