Co-reporter:Bernhard Hauer, Stefan Lutz
Current Opinion in Chemical Biology 2017 Volume 37(Volume 37) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.cbpa.2017.04.006
Co-reporter:Leann T. Quertinmont, Stefan Lutz
Tetrahedron 2016 Volume 72(Issue 46) pp:7282-7287
Publication Date(Web):17 November 2016
DOI:10.1016/j.tet.2016.01.007
In protein engineering, cell-free transcription/translation of linear mutagenic DNA templates can tremendously accelerate and simplify the screening of enzyme variants. Using the RApid Parallel Protein EvaluatoR (RAPPER) protocol, we have evaluated the impact of amino acid substitutions and loop truncations on substrate specificity and stereoselectivity of Old Yellow Enzyme 1 from Saccharomyces pastorianus. Our study demonstrates the benefit of systematically assessing amino acid variations including substrate profiling to explore sequence-function space.
Co-reporter:Dr. Pravin Muthu ;Dr. Stefan Lutz
ChemMedChem 2016 Volume 11( Issue 7) pp:660-666
Publication Date(Web):
DOI:10.1002/cmdc.201600096
Abstract
Fast, simple and cost-effective methods for detecting and quantifying pharmaceutical agents in patients are highly sought after to replace equipment and labor-intensive analytical procedures. The development of new diagnostic technology including portable detection devices also enables point-of-care by non-specialists in resource-limited environments. We have focused on the detection and dose monitoring of nucleoside analogues used in viral and cancer therapies. Using deoxyribonucleoside kinases (dNKs) as biosensors, our chemometric model compares observed time-resolved kinetics of unknown analytes to known substrate interactions across multiple enzymes. The resulting dataset can simultaneously identify and quantify multiple nucleosides and nucleoside analogues in complex sample mixtures.
Co-reporter:Ashley B. Daugherty, John R. Horton, Xiaodong Cheng, and Stefan Lutz
ACS Catalysis 2015 Volume 5(Issue 2) pp:892
Publication Date(Web):December 9, 2014
DOI:10.1021/cs501702k
Circular permutation of the NADPH-dependent oxidoreductase Old Yellow Enzyme from Saccharomyces pastorianus (OYE1) can significantly enhance the enzyme’s catalytic performance. Termini relocation into four regions of the protein (sectors I–IV) near the active site has proven effective in altering enzyme function. To better understand the structural consequences and rationalize the observed functional gains in these OYE1 variants, we selected representatives from sectors I–III for further characterization by biophysical methods and X-ray crystallography. These investigations not only show trends in enzyme stability and quaternary structure as a function of termini location but also provide a possible explanation for the catalytic gains in our top-performing OYE variant (new N-terminus at residue 303; sector III). Crystallographic analysis indicates that termini relocation into sector III affects the loop β6 region (amino acid positions: 290–310) of OYE1, which forms a lid over the active site. Peptide backbone cleavage greatly enhances local flexibility, effectively converting the loop into a tether and consequently increasing the environmental exposure of the active site. Interestingly, such an active site remodeling does not negatively impact the enzyme’s activity and stereoselectivity; neither does it perturb the conformation of other key active site residues with the exception of Y375. These observations were confirmed in truncation experiments, deleting all residues of the loop β6 region in our OYE variant. Intrigued by the finding that circular permutation leaves most of the key catalytic residues unchanged, we also tested OYE permutants for possible additive or synergistic effects of amino acid substitutions. Distinct functional changes in these OYE variants were detected upon mutations at W116, known in native OYE1 to cause inversion of diastereoselectivity for (S)-carvone reduction. Our findings demonstrate the contribution of loop β6 toward determining the stereoselectivity of OYE1, an important insight for future OYE engineering efforts.Keywords: biocatalysis; circular permutation; Old Yellow Enzyme; oxidoreductases; protein engineering; X-ray crystallography
Co-reporter:L. T. Quertinmont, R. Orru and S. Lutz
Chemical Communications 2015 vol. 51(Issue 1) pp:122-124
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4CC08240K
Cell-free transcription–translation systems offer an effective and versatile platform to explore the impact of genetic variations on protein function. We have developed a protocol for preparing linear, mutagenic DNA templates for direct use in the PURE system, enabling the fast and semi-quantitative evaluation of amino acid variations on catalytic activity and stereo-selectivity in native and engineered variants of Old Yellow Enzyme.
Co-reporter:Pravin Muthu, Hannah X. Chen, and Stefan Lutz
ACS Chemical Biology 2014 Volume 9(Issue 10) pp:2326
Publication Date(Web):July 31, 2014
DOI:10.1021/cb500463f
Recent advances in nuclear medicine have allowed for positron emission tomography (PET) to track transgenes in cell-based therapies using PET reporter gene/probe pairs. A promising example for such reporter gene/probe pairs are engineered nucleoside kinases that effectively phosphorylate isotopically labeled nucleoside analogues. Upon expression in target cells, the kinase facilitates the intracellular accumulation of radionuclide monophosphate, which can be detected by PET imaging. We have employed computational design for the semi-rational engineering of human 2′-deoxycytidine kinase to create a reporter gene with selectivity for l-nucleosides including l-thymidine and 1-(2′-fluoro-5-methyl-β-l-arabinofuranosyl) uracil. Our design strategy relied on a combination of preexisting data from kinetic and structural studies of native kinases, as well as two small, focused libraries of kinase variants to generate an in silico model for assessing the effects of single amino acid changes on favorable activation of l-nucleosides over their corresponding d-enantiomers. The approach identified multiple amino acid positions distal to the active site that conferred desired l-enantioselectivity. Recombination of individual amino acid substitutions yielded orthogonal kinase variants with significantly improved catalytic performance for unnatural l-nucleosides but reduced activity for natural d-nucleosides.
Co-reporter:Eva Fischereder, Desiree Pressnitz, Wolfgang Kroutil, Stefan Lutz
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 20) pp:5633-5637
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmc.2014.06.023
Strictosidine synthases catalyze the formation of strictosidine, a key intermediate in the biosynthesis of a large variety of monoterpenoid indole alkaloids. Efforts to utilize these biocatalysts for the preparation of strictosidine analogs have however been of limited success due to the high substrate specificity of these enzymes. We have explored the impact of a protein engineering approach called circular permutation on the activity of strictosidine synthase from the Indian medicinal plant Rauvolfia serpentina. To expedite the discovery process, our study departs from the usual process of creating a random protein library, followed by extensive screening. Instead, a small, focused library of circular permutated variants of the six bladed β-propeller protein was prepared, specifically probing two regions which cover the enzyme active site. The observed activity changes suggest important roles of both regions in protein folding, stability and catalysis.
Co-reporter:Ashley B. Daugherty ; Sridhar Govindarajan
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14425-14432
Publication Date(Web):August 29, 2013
DOI:10.1021/ja4074886
Members of the old yellow enzyme (OYE) family are widely used, effective biocatalysts for the stereoselective trans-hydrogenation of activated alkenes. To further expand their substrate scope and improve catalytic performance, we have applied a protein engineering strategy called circular permutation (CP) to enhance the function of OYE1 from Saccharomyces pastorianus. CP can influence a biocatalyst’s function by altering protein backbone flexibility and active site accessibility, both critical performance features because the catalytic cycle for OYE1 is thought to involve rate-limiting conformational changes. To explore the impact of CP throughout the OYE1 protein sequence, we implemented a highly efficient approach for cell-free cpOYE library preparation by combining whole-gene synthesis with in vitro transcription/translation. The versatility of such an ex vivo system was further demonstrated by the rapid and reliable functional evaluation of library members under variable environmental conditions with three reference substrates ketoisophorone, cinnamaldehyde, and (S)-carvone. Library analysis identified over 70 functional OYE1 variants with several biocatalysts exhibiting over an order of magnitude improved catalytic activity. Although catalytic gains of individual cpOYE library members vary by substrate, the locations of new protein termini in functional variants for all tested substates fall within the same four distinct loop/lid regions near the active site. Our findings demonstrate the importance of these structural elements in enzyme function and support the hypothesis of conformational flexibility as a limiting factor for catalysis in wild type OYE.
Co-reporter:Ashley B. Daugherty, Pravin Muthu, and Stefan Lutz
Biochemistry 2012 Volume 51(Issue 41) pp:
Publication Date(Web):September 25, 2012
DOI:10.1021/bi300832v
The propeptide domain of subtilisin BPN′ functions as a molecular chaperone for its cognate protease yet quickly assumes a predominantly unfolded structure following cleavage by the mature protease. In contrast, structural stabilization of the propeptide domain has been proposed to competitively inhibit protease self-cleavage, suggesting the possibility for the generation of novel proteinaceous subtilisin inhibitors. Using a Rosetta fixed backbone design, we have redesigned the subtilisin BPN′ propeptide structure to generate synthetic peptide sequences with increased and tunable structural stability. Molecular dynamics simulations provide supporting evidence that the artificial sequences retain structure without its protease cognate unlike the inherently disordered wild-type propeptide. Experimental evaluation of two designer domains by spectroscopic methods verified their structural integrity. Furthermore, the novel propeptide domains were shown to possess significantly enhanced thermostability. Nevertheless, their modest functional performance as protease inhibitors raises doubt that propeptide stability alone is sufficient for effective inhibitor design.
Co-reporter:Lingfeng Liu, Paul Murphy, David Baker and Stefan Lutz
Chemical Communications 2010 vol. 46(Issue 46) pp:8803-8805
Publication Date(Web):19 Oct 2010
DOI:10.1039/C0CC02961K
We report the computational enzyme design of an orthogonal nucleoside analog kinase for 3′-deoxythymidine. The best kinase variant shows an 8500-fold change in substrate specificity, resulting from a 4.6-fold gain in catalytic efficiency for the nucleoside analog and a 2000-fold decline for the native substrate thymidine.
Co-reporter:Pinar Iyidogan and Stefan Lutz
Biochemistry 2008 Volume 47(Issue 16) pp:
Publication Date(Web):March 25, 2008
DOI:10.1021/bi800157e
Human deoxycytidine kinase (dCK) is responsible for the phosphorylation of a number of clinically important nucleoside analogue prodrugs in addition to its natural substrates, 2′-deoxycytidine, 2′-deoxyguanosine, and 2′-deoxyadenosine. To improve the low catalytic activity and tailor the substrate specificity of dCK, we have constructed libraries of mutant enzymes and tested them for thymidine kinase (tk) activity. Random mutagenesis was employed to probe for amino acid positions with an impact on substrate specificity throughout the entire enzyme structure, identifying positions Arg104 and Asp133 in the active site as key residues for substrate specificity. Kinetic analysis indicates that Arg104Gln/Asp133Gly creates a “generalist” kinase with broader specificity and elevated turnover for natural and prodrug substrates. In contrast, the substitutions of Arg104Met/Asp133Thr, obtained via site-saturation mutagenesis, yielded a mutant with reversed substrate specificity, elevating the specific constant for thymidine phosphorylation by over 1000-fold while eliminating activity for dC, dA, and dG under physiological conditions. The results illuminate the key contributions of these two amino acid positions to enzyme function by demonstrating their ability to moderate substrate specificity.
Co-reporter:Zhen Qian Dr.;Christina J. Fields Dr.
ChemBioChem 2007 Volume 8(Issue 16) pp:
Publication Date(Web):17 SEP 2007
DOI:10.1002/cbic.200700373
The engineering of lipase B from Candida antarctica (CALB) by circular permutation has yielded over sixty hydrolase variants, and several show significantly improved catalytic performance. Here we report a detailed characterization of ten selected enzyme variants by kinetic and spectroscopic methods to further elucidate the impact of circular permutation on the structure and function of CALB. Our experiments identify lipase variants with up to 175-fold enhanced kcat/KM values over wild-type. In addition, circular permutation does not change the enzymes’ enantiopreference and preserves or even improves their enantioselectivity compared to that of the wild-type enzyme. Finally, our spectroscopic analyses suggest that the structural effects of circular permutation on CALB are mostly local, concentrating on regions near the native and new protein termini. The observed changes in secondary structure and protein thermostability vary among enzyme variants but directly correlate with the locations of the new termini, a first step towards a predictive framework.
Co-reporter:Stefan Lutz
Tetrahedron: Asymmetry 2004 Volume 15(Issue 18) pp:2743-2748
Publication Date(Web):20 September 2004
DOI:10.1016/j.tetasy.2004.06.031
The lipase B from Candida antarctica (CAL-B) displays high enantioselectivity on a broad range of substrates, making it an accepted biocatalyst for asymmetric organic chemistry. In recent years, a number of rational and combinatorial protein engineering projects have focused on extending and tailoring CAL-B’s catalytic and physical properties. Beyond generating customized catalysts, these studies have helped to elucidate the enzyme’s structure–function relationship and illuminate its enantioselectivity. Potential directions for future studies, taking into consideration results from engineering efforts on related lipases, are discussed.
Co-reporter:Stefan Lutz
Current Opinion in Biotechnology (December 2010) Volume 21(Issue 6) pp:734-743
Publication Date(Web):1 December 2010
DOI:10.1016/j.copbio.2010.08.011
Over the past two decades, directed evolution has transformed the field of protein engineering. The advances in understanding protein structure and function, in no insignificant part a result of directed evolution studies, are increasingly empowering scientists and engineers to device more effective methods for manipulating and tailoring biocatalysts. Abandoning large combinatorial libraries, the focus has shifted to small, functionally rich libraries and rational design. A critical component to the success of these emerging engineering strategies are computational tools for the evaluation of protein sequence datasets and the analysis of conformational variations of amino acids in proteins. Highlighting the opportunities and limitations of such approaches, this review focuses on recent engineering and design examples that require screening or selection of small libraries.
Co-reporter:Monica L. Gerth, Stefan Lutz
Journal of Molecular Biology (20 July 2007) Volume 370(Issue 4) pp:742-751
Publication Date(Web):20 July 2007
DOI:10.1016/j.jmb.2007.05.021
In antiviral and cancer therapy, deoxyribonucleoside kinases (dNKs) are often the rate-limiting step in activating nucleoside analog (NA) prodrugs into their cytotoxic, phosphorylated forms. We have constructed libraries of hybrid enzymes by non-homologous recombination of the pyrimidine-specific human thymidine kinase 2 and the broad-specificity dNK from Drosophila melanogaster; their low sequence identity has precluded engineering by conventional, homology-dependent shuffling techniques. From these libraries, we identified chimeras that phosphorylate nucleoside analogs with higher activity than either parental enzyme, and that possess new activity towards the anti-HIV prodrug 2′,3′-didehydro-3′-deoxythymidine (d4T). These results demonstrate the potential of non-homologous recombination within the dNK family for creating enzymes with new and improved activities towards nucleoside analogs. In addition, our results exposed a previously unknown role for the C-terminal regions of these dNKs in determining substrate selectivity.
Co-reporter:Monica L. Gerth, Stefan Lutz
Journal of Molecular Biology (20 July 2007) Volume 370(Issue 4) pp:742-751
Publication Date(Web):20 July 2007
DOI:10.1016/j.jmb.2007.05.021
In antiviral and cancer therapy, deoxyribonucleoside kinases (dNKs) are often the rate-limiting step in activating nucleoside analog (NA) prodrugs into their cytotoxic, phosphorylated forms. We have constructed libraries of hybrid enzymes by non-homologous recombination of the pyrimidine-specific human thymidine kinase 2 and the broad-specificity dNK from Drosophila melanogaster; their low sequence identity has precluded engineering by conventional, homology-dependent shuffling techniques. From these libraries, we identified chimeras that phosphorylate nucleoside analogs with higher activity than either parental enzyme, and that possess new activity towards the anti-HIV prodrug 2′,3′-didehydro-3′-deoxythymidine (d4T). These results demonstrate the potential of non-homologous recombination within the dNK family for creating enzymes with new and improved activities towards nucleoside analogs. In addition, our results exposed a previously unknown role for the C-terminal regions of these dNKs in determining substrate selectivity.
Co-reporter:Zhen Qian, John R. Horton, Xiaodong Cheng, Stefan Lutz
Journal of Molecular Biology (16 October 2009) Volume 393(Issue 1) pp:191-201
Publication Date(Web):16 October 2009
DOI:10.1016/j.jmb.2009.08.008
Circular permutation of Candida antarctica lipase B yields several enzyme variants with substantially increased catalytic activity. To better understand the structural and functional consequences of protein termini reorganization, we have applied protein engineering and x-ray crystallography to cp283, one of the most active hydrolase variants. Our initial investigation has focused on the role of an extended surface loop, created by linking the native N- and C-termini, on protein integrity. Incremental truncation of the loop partially compensates for observed losses in secondary structure and the permutants' temperature of unfolding. Unexpectedly, the improvements are accompanied by quaternary-structure changes from monomer to dimer. The crystal structures of one truncated variant (cp283Δ7) in the apo-form determined at 1.49 Å resolution and with a bound phosphonate inhibitor at 1.69 Å resolution confirmed the formation of a homodimer by swapping of the enzyme's 35-residue N-terminal region. Separately, the new protein termini at amino acid positions 282/283 convert the narrow access tunnel to the catalytic triad into a broad crevice for accelerated substrate entry and product exit while preserving the native active-site topology for optimal catalytic turnover.
Co-reporter:Dario Segura-Peña, Joseph Lichter, Manuela Trani, Manfred Konrad, ... Stefan Lutz
Structure (13 December 2007) Volume 15(Issue 12) pp:1555-1566
Publication Date(Web):13 December 2007
DOI:10.1016/j.str.2007.09.025
The human cytosolic thymidine kinase (TK) and structurally related TKs in prokaryotes play a crucial role in the synthesis and regulation of the cellular thymidine triphosphate pool. We report the crystal structures of the TK homotetramer from Thermotoga maritima in four different states: its apo-form, a binary complex with thymidine, as well as the ternary structures with the two substrates (thymidine/AppNHp) and the reaction products (TMP/ADP). In combination with fluorescence spectroscopy and mutagenesis experiments, our results demonstrate that ATP binding is linked to a substantial reorganization of the enzyme quaternary structure, leading to a transition from a closed, inactive conformation to an open, catalytic state. We hypothesize that these structural changes are relevant to enzyme function in situ as part of the catalytic cycle and serve an important role in regulating enzyme activity by amplifying the effects of feedback inhibitor binding.
Co-reporter:Lingfeng Liu, Paul Murphy, David Baker and Stefan Lutz
Chemical Communications 2010 - vol. 46(Issue 46) pp:NaN8805-8805
Publication Date(Web):2010/10/19
DOI:10.1039/C0CC02961K
We report the computational enzyme design of an orthogonal nucleoside analog kinase for 3′-deoxythymidine. The best kinase variant shows an 8500-fold change in substrate specificity, resulting from a 4.6-fold gain in catalytic efficiency for the nucleoside analog and a 2000-fold decline for the native substrate thymidine.
Co-reporter:L. T. Quertinmont, R. Orru and S. Lutz
Chemical Communications 2015 - vol. 51(Issue 1) pp:NaN124-124
Publication Date(Web):2014/11/04
DOI:10.1039/C4CC08240K
Cell-free transcription–translation systems offer an effective and versatile platform to explore the impact of genetic variations on protein function. We have developed a protocol for preparing linear, mutagenic DNA templates for direct use in the PURE system, enabling the fast and semi-quantitative evaluation of amino acid variations on catalytic activity and stereo-selectivity in native and engineered variants of Old Yellow Enzyme.
.jpg)

![6-METHYL-3-(BETA-D-2-DEOXYFURANOSYL)PYRROLO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/382200/382137-74-8.png)
![6-METHYL-3-(BETA-D-2-DEOXYFURANOSYL)PYRROLO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/382200/382137-74-8_b.png)


![2-Buten-1-ol, 4-[[tris(1-methylethyl)silyl]oxy]-, (2Z)-](http://img.cochemist.com/ccimg/179000/178905-41-4.png)
![2-Buten-1-ol, 4-[[tris(1-methylethyl)silyl]oxy]-, (2Z)-](http://img.cochemist.com/ccimg/179000/178905-41-4_b.png)

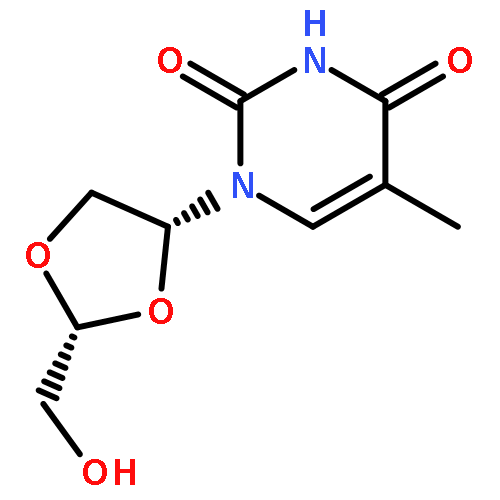
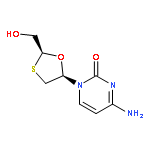
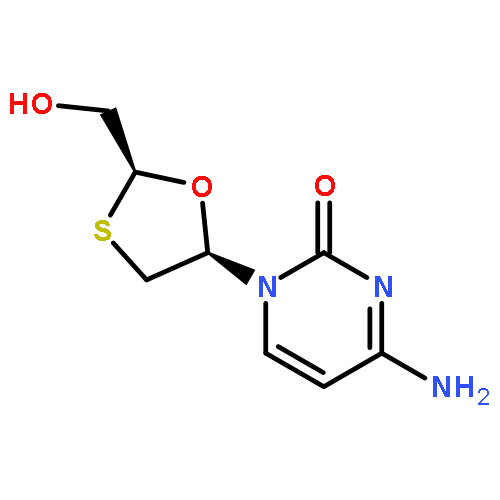
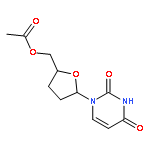




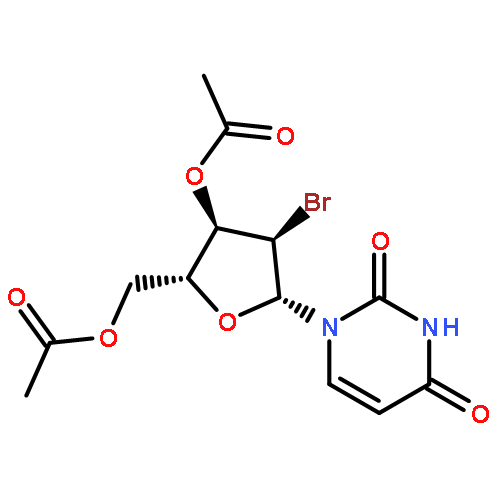
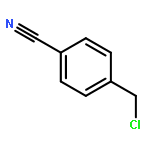
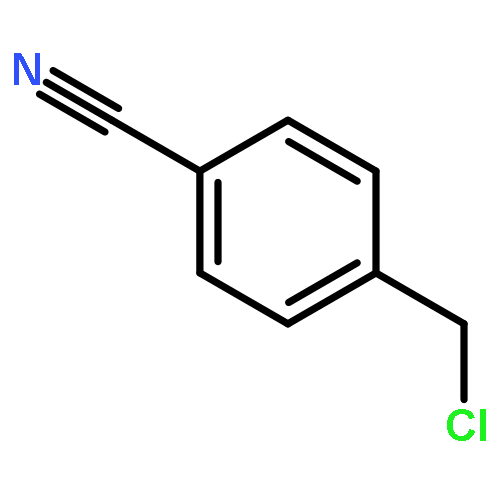

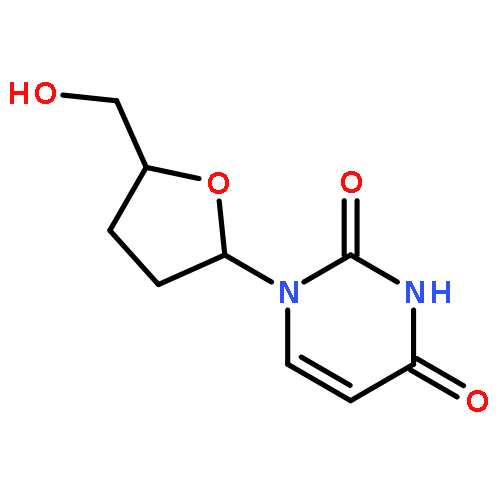




![6-METHYL-3-(BETA-D-2-DEOXY-RIBOFURANOSYL)FURANO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/383900/383897-60-7.png)
![6-METHYL-3-(BETA-D-2-DEOXY-RIBOFURANOSYL)FURANO[2,3-D]PYRIMIDIN-2-ONE](http://img.cochemist.com/ccimg/383900/383897-60-7_b.png)